
Abstract
In the present study, a diverse panel of 96 accessions of lentil germplasm was used to study flowering time over environments and to identify simple sequence repeat markers associated with flowering time through association mapping. The study showed high broad sense heritability estimate (h 2 bs=0.93) for flowering time in lentil. Screening of 534 SSR markers resulted in an identification of 75 SSR polymorphic markers (13.9%) across studied genotypes. These markers amplified 266 loci and generated 697 alleles ranging from two to 16 alleles per locus. Model-based cluster analysis used for the determination of population structure resulted in the identification of two distinct subpopulations. Distribution of flowering time was ranged from 40 to 70 days in subpopulation I and from 54 to 69 days in subpopulation II and did not skew either late or early flowering time within a subpopulation. No admixture was observed within the subpopulations. Use of the most accepted maximum likelihood model (P3D mixed linear model with optimum compression) of MTA analysis showed significant association of 26 SSR markers with flowering time at <0.05 probability. The percent of phenotypic explained by each associated marker with flowering time ranged from 2.1 to 21.8% and identified QTLs for flowering time explaining high phenotypic variation across the environments (10.7-21.8%) or in a particular environment (10.2-21.4%). In the present study, 13 EST-SSR showed significant association with flowering time and explained large phenotypic variation (2.3-21.8%) compared to genomic SSR markers (2.1-10.2%). Hence, these markers can be used as functional markers in the lentil breeding program to develop short duration cultivars.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lentil (Lens culinaris L.) is a rainfed crop. It is cultivated in cool-season under residual soil moisture conditions. Generally, this crop encounters terminal moisture and heat stresses during the grain filling period. As a result, the crop experiences forced maturity and lower yield. Possibilities of forced maturity loom large in the coming years with rising atmospheric temperature due to climate change and the delayed sowing of lentil after harvesting of the rice in the Indian subcontinent. An estimate indicates that in India, about 11.7 million ha area now remains fallow after the harvesting of rice (Subbarao et al. 2001). However, a window of 100-110 days is still available for growing lentil after rice harvest in several countries including India. Therefore, development of early maturing cultivars is required for harvesting the highest yield potential within this short periods of time. Short duration cultivars will not only increase the lentil cultivation in rice fallow areas but also increase the productivity by reducing yield losses due to forced maturity. Therefore, in lentil, manipulation of flowering time can have an important role in the development of high yielding early maturing cultivars that are able to adapt to changing environmental conditions. Since the early 1990s, a single germplasm line Precoz has been widely used to improve earliness, seed size, and rust resistance to South Asia (Asghar et al. 2010; Erskine et al. 1998; Singh et al. 2006; Kumar et al. 2014). Recently, efforts have been made toward the identification of other germplasm sources to broaden the genetic base. In this direction, exotic and indigenous germplasm were screened to identify early flowering genotypes. Recently, 1525 accessions of cultivated species have been characterized on the basis of days to 50% flowering that flowered in 71-107 days (Kumar and Solanki 2014), while its range was from 27 to 141 days among 405 accessions of wild species (Singh et al. 2014).
Flowering time is highly influenced by temperature and photoperiod (Summerfield et al. 1985). Therefore, it is essentially required to study flowering time under different environments and also to know its genetics before initiating a breeding program for earliness. Genetic and molecular analysis revealed that several genes control natural variation for flowering time in crop plants (Jung and Muller 2009). However, both qualitative and quantitative inheritance of flowering time has been reported in lentil (Sarker et al. 1999; Emami 1996). One of the studies identified that a single recessive gene (sn) controls the early flowering in lentil (Sarker et al. 1999). The variants of early flowering at this locus could be useful for development of early flowering cultivars for water-limited environments and help to diversify the lentil gene pool in South Asia (Sarker and Erskine 2006). Other studies identified several quantitative trait loci (QTLs) using bi-parental mapping populations that control flowering time in lentil (Tahir et al. 1994; Fratini et al. 2007; Tullu et al. 2008). More recently, a major stable QTL across environments has been reported for flowering time in lentil (Kahriman et al. 2014). In these studies, markers are not very closely linked to QTLs for flowering time due to poor marker density. However, a high-resolution marker-trait association can be established through association mapping by using a diverse natural population because an association mapping panel is developed from several historical recombination events that are able to break apart linked markers. It also has the potential to pick up markers for genes that did not segregate in the bi-parental population. It has been used widely in several crop plants to study the association of molecular markers with agronomically important traits (Wang et al. 2012; Visioni et al. 2013; Mandel et al. 2013; Zhao et al. 2014).
In lentil, use of molecular markers has been accelerated due to the enrichment of genomic resources in the recent past (Kumar et al. 2015). More than 2000 simple sequence repeat (SSR) markers developed from lentil genome are now available in the public domain (Kaur et al. 2011). Besides, de novo assembly of short sequence reads of lentil has resulted in the development of an expressed gene catalog and molecular markers (Verma et al. 2013). Molecular markers including single nucleotide polymorphism (SNP), simple sequence repeat (SSR), inter simple sequence repeat (ISSR), and direct amplification of minisatellite DNA (DAMD) have been used in assessment of genetic diversity among genotypes of cultivated species, land races, and wild relatives (Reddy et al. 2010; Lombardi et al. 2014; Verma et al. 2014; Basheer-Salimia et al. 2015; Seyedimoradi and Talebi 2014; Jain et al. 2013; Dikshit et al. 2015; Khazaei et al. 2016). These molecular markers are useful genomic resources for studying marker-trait association in lentil. Flowering time is an important trait to lentil breeders for developing short duration lentil cultivars. A tightly linked molecular marker can be used for the precise manipulation of the genetic makeup of individuals along with other yield contributing traits in lentil. Therefore, in the present study, a diverse panel of lentil germplasm was used (i) to study flowering time over locations and years, and (ii) to identify simple sequence repeat markers associated with flowering time through association mapping.
Materials and methods
Plant material, experimental design, and recording of phenotypic data
In the present study, a diverse panel of 96 accessions was used to characterize flowering time in lentil and evaluated over two years (2014-15 and 2015-16) at two locations [Research Farm of Indian Institute of Pulses Research (IIPR), Kanpur and Regional Station of IIPR, Dharwad]. These two locations were selected on the basis of their differences in photoperiod and temperature during the flowering time for studying photo-thermo-sensitivity among the present genotypes. The present diverse panel included breeding lines, cultivars, landraces, and exotic lines that was constituted by covering the sufficient phenotypic diversity of flowering time available in the cultivated gene pool. The details of these accessions are given in Table 1. Experimental materials were evaluated in an augmented design in three-meter single row plots. Three genotypes namely Precoz, DPL 15, and DPL 62 were used as checks, which were repeated four times. Row to row distance was 25 cm and plant to plant distance was 5-7 cm. Standard agronomic practices were followed to raise a successful crop. Observations were recorded on days to flower initiation on five randomly selected plants. It is measured from date of seed sowing in the field to date of flower initiation in a particular genotype.
SSR markers and PCR amplification
Genomic SSR markers developed in lentil at ICRADA by Dr. A. Hamwieh (personal communication; Kumar et al. 2014) and EST-SSR markers developed by Kaur et al. (2011) were used in the present study. These markers represent both coding and non-coding regions of lentil genome. The primers were custom synthesized from Eurofins Genomics India, and Imperial Biosciences, USA. Initially, 534 SSR markers were used to identify polymorphic markers among the 20 accessions of lentil and then 75 SSR markers identified as polymorphic were used to genotype a total of 96 accessions having early, medium, and late flowering. The CTAB extraction protocol described earlier by Doyle and Doyle (1987) and Abdelnoor et al. (1995) were used to isolate the genomic DNA with certain changes (Kumar et al. 2014). The PCR reaction contains a total volume of 20 μl consisting of 50–100 ng genomic DNA, 1X PCR buffer with 1.5 mM MgCl2, 200 μM each of dNTPs (Bangalore Genei, Bengaluru), 0.5 U Taq DNA Polymerase (Bangalore Genei, Bengaluru), and 40 pmoles each of forward and reverse primers. The PCR amplification was performed in a G-STORM PCR System with a program for an initial denaturation of 94 °C for 4 min followed by 39 cycles of 94 °C for 1 min, annealing 50–55 °C for 1 min, elongation 72 °C for 1 min, and a final extension at 72 °C for 15 min.
Polyacrylamide gel electrophoresis (PAGE) and silver staining
The amplified products were run on 10% polyacrylamide gel along with 1 kb DNA ladder using high throughput vertical gel electrophoresis (CBS, USA). The bands were visualized by following silver staining (Benbouza et al. 2006). Each SSR marker amplified single to multiple bands. The multiple bands amplified in a genotype were considered separate loci and size variation in the amplified product of the targeted locus across the accessions was considered as allelic variants for that loci. Size of amplified product of each genotype was calculated online using fragment calculator (http://biotools.nubic.northwestern.edu/SizeCalc.html).
Statistical analysis
Mean and range of flowering time was analyzed using statistical tools (MS Excel 2007). Analysis of variance was performed by considering the recorded data over the years and locations as replication using R software (R Core Team 2013). A T-test was used to test the significance of a particular genotype in a specific environment for flowering time over the mean value of all studied environments. The mean of square due to error and genotypes was used for environmental variance (σ2e) and phenotypic variance (σ2p), respectively, to calculate the broad sense heritability (h 2 bs) using the following formula.
Population structure analysis
Model-based Bayesian clustering analysis was performed to infer the genetic structure and number of subgroups in the studied germplasm set using the STRUCTURE version 2.1 software (Pritchard et al. 2000a, b). This approach uses multi-locus genotypic data, such as SSRs without prior knowledge of their population affinities and assumes loci in Hardy-Weinberg equilibrium in order to assign individuals to clusters/groups (K). Admixture model with correlated allele frequencies was used to estimate each of the K clusters. For this, number of presumed subpopulations (K) was set from 1 to 10 for each accession along with the percentage of its genome derived from each cluster and used higher levels of length of the burning period (i.e. 1,000,000) and number of Markov chain Monte Carlo (MCMC) replication after burning (i.e., 1,000,000) for getting reliable subgroups. Further, each K was repeated ten times, i.e., iterations/replication in order to get consistent and reliable subgroups. DK method was used to determine the number of subpopulations (Evanno et al. 2005) using STRUCTURE HARVESTER available online (http://taylor0.biology.ucla.edu/structureHarvester/). Those markers had an allelic frequency less than 5%, which were excluded from the analysis (Wilson et al. 2004).
Marker-trait association analysis
The software program TASSEL version 2.1 (http://www.maizegenetics.net) was used to calculate the associations between individual markers and flowering time (over locations and years) following the mixed linear model (MLM, Q + K) approach. A structured association approach could correct false associations using the Q-matrix of population membership estimates. The marker data, Q values derived from the software STRUCTURE, and flowering data along with marker-based kinship matrix (K) obtained from TASSEL were used as covariates during MLM analysis. Further, among different options available within MLM, the widely adopted approach called “optimum levels of compression in combination with P3D” for variance component estimation was used for association analysis. The significance of marker-trait associations is determined at P <0.05 and phenotypic variation explained (r2) by each associated marker is stated (Li et al. 2012; Topal et al. 2004).
Results
Phenotypic variability and heritability
Analysis of variance (ANOVA) over four environments (two years and two locations) showed highly significant effects of year, locations, and genotypes on flowering time in lentil (Table 2). Besides, genotypes also responded differently over locations and years as interactions of genotype with year and locations were observed highly significant in the present investigation (P< 0.01). The broad sense heritability was high (h 2 bs=0.93) for this trait (Table 2).
In the present study, mean of 96 genotypes for flowering time was 47.8 ± 0.43 days in 2015 and 43.6 ± 0.24 days in 2016 at Dharwad location, while it was 75.1 ± 1.39 days in 2014-15 and 71.1 ± 1.50 days in 2015-16 at Kanpur location (Table 3). These results indicated that the long photoperiod (13 h) probably promoted early flowering at the Dharwad location. In spite of this, genotypic differences were significant at both locations. Among the present 96 accessions, four genotypes (i.e., ILWLS118, IPLS09-17, IPLS09-5, and IPLS 09-34) were identified significantly earlier in flowering (P≤ 0.05) over mean flowering time. However, each genotype did not differ significantly (P<0.05) from the mean flowering time calculated over four environments. These results indicated that these genotypes are not photo-thermo sensitive (Table 3).
Genetic diversity and population structure analysis
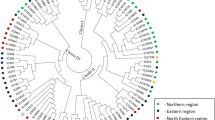
In the present study, out of 96 accessions, 20 accessions were first genotyped with 534 SSR markers in order to use only polymorphic markers for further genotyping of the entire set that led to saving time and money required to genotype 96 accessions of lentil. Only 75 SSR markers (13.9%) were found polymorphic. PIC and gene diversity of these markers ranged from 0.02 to 0.90. Polymorphic SSR markers generated 697 alleles from 266 loci with a range of two to 16 alleles per locus among the 96 accessions. Model based population structure analysis led to the identification of two distinct subpopulations among these accessions. Fifty-seven accessions were grouped in subpopulation I with a range of 40-72 days to flower initiation while the remaining 39 genotypes formed the second subpopulation with a range of 54 to 69 days to flower (Fig. 1). In addition to this, these subpopulations were broadly clustered according to their breeding history and subpopulation I contained breeding lines derived from exotic lines, while the subpopulation II contained most of the cultivars released in India and breeding lines involved these cultivars as one of the parents.

Clustering of 96 accessions of lentil on the basis of flowering time (above) and model based population structure analysis using polymorphic SSR markers; Sub-population I (1-57)- 1) IPL-121; 2) IPL-133; 3) IPL-203; 4) SEHORE-74-3; 5) IPL-218; 6) IPL-219; 7) IPL-221; 8) IPL-325; 9) IPL-324; 10) IPL-519; 11) IC-560291; 12) IC-560322; 13) IPLS-09-06; 14) IC-560113; 15) IPLS-COLD; 16) IPLS-09-23; 17) IPLS-09-32; 18) IC-560117; 19) IPLS-09-03; 20) IPLS-09-11; 21) IPLS-09-10; 22) IPLS-09-38; 23) IPLS-09-22; 24) IC-560150; 25) IPLS-09-37; 26) IPLS-09-36; 27) IPLS-09-14; 28) IPLS-09-33; 29) ILS-09-01; 30) IC-560111; 31) IPLS-09-27; 32) IPLS-99/209; 33) IPLS-09-34; 34) IPLS-09-05; 35) IPL-09-08; 36) IPLS-09-17; 37) ILWLS-118; 38) ILWL-118; 39) L-4603; 40) IPL-220; 41) DPL-58; 42) PRECOZ; 43) PL-02; 44) ILL-7663; 45) DPL-15; 46) IPL-81; 47) IPL-406; 48) DPL-62; 49) ILL-6002; 50) IPL-316; 51) IPL-526; 52) JL-01; 53) DPL-59; 54) LL-864; 55) ILWL-248; 56) IPL-321; 57) IPL-M-08; Sub-population II- (58-96) –58) IPL-98/193; 59) JL-03; 60) K-75; 61) KLS-218; 62) L-4076; 63) L-4147; 64) HUL-57; 65) L-9-12; 66) LH-84-8; 67) LL-147; 68) NARENDRA MASSOOR-1; 69) PANT-L-234; 70) PANT-L-639; 71) PL-04; 72) PL-05; 73) RANJAN; 74) SHALIMAAR MASOOR; 75) T-36; 76) VL-1; 77) VL-103; 78) VL-4; 79) WBL-58; 80) WBL-77; 81) B-77; 82) BR-25-1; 83) EC-208362; 84) IPL-222; 85) IPL-223; 86) IPL-224; 87) IPL-326; 88) IPL-327; 89) IPL-328; 90) IPL-529; 91) IPL-532; 92) IP-534; 93) IPL-230; 94) IPL-231; 95) IPL-10800; 96) IPL-11702
Marker-trait association analysis
In the present study, a total of 26 marker trait associations (MTAs) for the flowering time were identified over the two locations and two years using most accepted maximum likelihood model (P3D mixed linear model with optimum compression) of the MTA analysis. Our results showed a significant association of 13 EST-SSR and 13 genomic SSR markers with flowering time at p-value (P<0.05). These markers explained 2.1 to 21.8% of the total phenotypic variation (R 2) in flowering time (Table 4). Among 13 EST-SSR markers, PBLAC 0250 marker explained 21.8% of the total phenotypic variation (PV) in flowering time. While, another two EST-SSR markers, namely PBALC 0207 and PBALC 0242, showed a significant association at <0.04 and <0.0008 probabilities, respectively, across the locations and explained from 4.6 to 16.8% of the total phenotypic variation in flowering time. However, some MTAs with high PV were detected in a particular environment. For example, EST-SSR markers ‘PBALC 0250’ showed significant association with flowering time (P<0.02) and explained 19.5 to 21.8% of total phenotypic variation at Kanpur location (Table 4). Likewise, EST-SSR markers ‘PBALC 0353’ showed significant association with flowering time (P<0.03) and only explained 21.4% of the total phenotypic variation at the Dharwad location (Table 4). However, none of the 13 MTAs based on genomic SSR for flowering time were observed across all studied environments and these MTAs also explained the low amount of total phenotypic variation that ranged from 1.8 to 10.4% (Table 4). Only one genomic SSR marker ALD 40 showed highly significant (P<0.0007) association with flowering time and explained 10.2% of total phenotypic variation at the Dharward location in 2015 (Table 4).
Mining of the elite alleles
The phenotypic allele effect of each SSR that significantly associated with flowering time was shown in Suppl. Table S1. Among the markers associated with flowering time, EST-SSR marker PBALC 0207 was associated with flowering time across locations. Its allele PBALC0207-144 had the most positive phenotypic effect and ability to increase the flowering time by 7.28 and 9.26 days, whereas allele PBALC 0207-169 had the most negative phenotypic effect (−5.39 and -4.44 days) in 2014-15 and 2015-16, respectively at Kanpur location. These alleles (PBALC 0207-144 and PBALC 0207-169) also had a positive phenotypic effect (0.81 days) and negative phenotypic effect (-0.09 days) in 2014 at the Dharward location. For EST SSR marker PBALC 0250 that showed significant association with flowering at Kanpur location, the allele PBALC 0250-200 had a positive phenotypic effect and the ability to increase the flowering time by 16.92 days, while another allele had the most negative effect (-11.38 days) in 2014-15. Similarly, these alleles also had the most positive (10.33 days) and negative (-9.85 days) phenotypic effects in 2015-16. In addition to this, other alleles (i.e., PBALC 0250-160 and PBALC 0250-150) also had separately almost similar positive (10.54 days) and negative phenotypic effects (-9.89 days) in this year (Suppl. Table S1). The allele PBALC 0353-97 had the most positive phenotypic effect and was able to increase the flowering time by 3.71 days, whereas two alleles, namely PBALC 0353-125 and PBALC 0353-107, were able to decrease the flowering time of 1.78 and 1.36 days, respectively, only at the Dharwad location in 2015.
Alignment of EST sequences to identify their role in controlling the flowering time
EST-SSR markers used in the present study have been developed from gene sequences of lentil, i.e., express sequence tags (ESTs). Hence, EST sequences of those SSR markers that explained >10% of the total phenotypic variance across environments or in a particular environment were aligned with the nucleotide database of NCBI in order to know their similarity with gene sequences available in the public domain. The results showed close similarity between lentil EST sequences and the genes of other legumes (Table 5).
Discussion
Flowering time (FT) contributes significantly to crop adaptation and grain yield (Kumar and Van Rheenen 2000; Anbessa et al. 2006). However, early flowering in lentil generally leads to poor biomass (Kumar et al. 2014). Therefore, lentil breeders target the development of short duration cultivars having high biomass. For this purpose, identification of diverse genetic resources for earliness, their use for hybridization, and selection of recombinant having high biomass along with earliness are the logical steps that a breeder takes. Flowering time is highly influenced by day length (photoperiod) and temperatures (Welch et al. 2005). Therefore, flowering in crop plants, including lentil responds differently at different latitudes under different climate regimes, seasons, sowing dates, and altitudes (Summerfield and Roberts 1988). In our study, analysis of variance showed significant effects of locations and years on flowering time because two locations used in this study differed for photoperiod and temperature at the time of flowering (Table 3). For example, high temperature (25.5°C and 26.5°C) resulted in early flowering at the Kanpur location in 2014-15 and Dharwad location in 2015-16, respectively, in comparison to 2013-14 at Kanpur (22.4°C) and Dharwad (20.5°C) among the four selected genotypes (see Table 3). This shows that warm conditions promote early flowering in lentil. Erskine et al. (1990) also reported that Indian germplasm is more sensitive to temperature and less sensitive to photoperiod compared to the genetic materials belonging to West Asia. Other studies reported that multiple signaling pathways, including the components of the photoperiod pathway, are involved in promoting flowering under warm conditions (Thines et al. 2014). The genes sensitive to high temperature might be a part of genes that are involved in the photoperiod pathways as reported earlier (Jung and Muller 2009). The photoperiod pathways have the two important genes, i.e., CONSTANS (CO) and FLOWERING LOCUS T (FT), which regulate flowering time (Imaizumi 2010). It has also been reported in Arabidopsis and cereals that these genes are controlled by several positive and negative regulators (Jung and Muller 2009). In previous studies, the allelic variation has been reported to influence flowering time among the genotypes of crop plants. For example, in rice, the CO ortholog Hd1 has allelic variation, which controls much of the phenotypic variation in flowering time (Takahashi et al. 2009). In the present study, genotypes differ in flowering time under different environmental conditions. This could be the outcome of the photo-thermo sensitivity genes present in the background of these genotypes. However, non-significant differences in flowering time over the locations and years among the selected early flowering genotypes indicated their photo-thermo insensitivity.
Association mapping is widely used in crop plants to identify genomic regions associated with a trait of interest that would not be picked up by analysis of a bi-parental population. However, a high frequency of spurious associations is also detected through association mapping in those cases where population structure is not to be taken into consideration (Rosenberg and Nordborg 2006). Therefore, we used model-based analysis for determining population structure of studied genotypes and its information was used in association mapping analysis to reduce the chance of false positive association. In our study, the present genotypes were structured into two distinct subpopulations, and early, medium, and late flowering genotypes were distributed in each subpopulation. Moreover, genotypes in these subpopulations were broadly clustered according to their breeding history. One subpopulation contained breeding lines that derived from exotic lines, while another one comprised the Indians’ cultivars and those breeding lines which have these cultivars as one of the parents in their breeding history. Overall, the nature of population structure in the present study suggests that flowering time has no bias within a specific sub-population. It shows that the population is appropriate for association analysis and association of marker loci with flowering time recorded in the present study should be real and the chance of obtaining many false positives would be less (Pritchard et al. 2000a; Cardon and Palmer 2003; Gupta et al. 2005; Malysheva-Otto et al. 2006; Ostrowski et al. 2006; Jaiswal et al. 2012).
In the present study, 13.9% of SSR markers were polymorphic, while a high level of polymorphism (66% of genomic SSR and 47.5% of EST-SSR markers) has been observed in earlier studies (Kaur et al. 2011; Kumar et al. 2014). This low-level polymorphism of SSR markers probably resulted due to the use of elite breeding lines in the present study. Previously, a small population size having <100 individuals was used in many association mapping studies to identify QTLs in crop plants (Szalma et al. 2005; Salvi et al. 2007). However, a population of 100 individuals can be sufficient to identify the QTL(s) for a trait of interest, if we use breeding lines in association mapping (Bradbury et al. 2011). Also, use of diverse breeding lines makes association mapping more cost-effective because these are routinely recorded for phenotypic traits. Moreover, elite breeding materials are generally narrow in the genetic base. If QTLs for a trait are identified in the background of such materials, they can be used directly in marker-assisted selection (Kumar et al. 2017). Previously, molecular marker-trait association analysis was conducted for a number of economically important traits in the legumes as well other crops (Sun et al. 2011; Niu et al. 2013; Zhang et al. 2014; Cheng et al. 2015; Bajaj et al. 2016; Contreras Soto et al. 2017). In rice, association mapping detected 76 significant (P<0.5) trait—marker associations between agronomic traits and SSR markers, and among them 11 significant MTAs explained >10% of the total genetic variation (Zhang et al. 2014). Similarly, in barley, marker loci associated with plant height and stem diameter explained 4.59-17.48% of the total variation (Sun et al. 2011). In our study, association mapping detected 26 MTAs that are significantly associated with flowering time. Both genic and genomic SSR markers were found to be associated with flowering time. The percentage of total phenotypic variation explained by these MTAs ranged from 2.1 to 21.8%. Interestingly, EST-SSR markers showed highly significant association with flowering time along with high phenotypic variance. These markers can be closely associated with the genes that control flowering time in lentil and hence can be used as functional markers in a breeding program. Thus, our results show that a number of genes/QTLs are responsible for controlling flowering time in lentil. However, previously, a single major gene or QTL was reported for flowering time and a recessive allele of a major Mendelian locus Sn conferred an early flowering under non-inductive short-day conditions (Sarker et al. 1999; Kahriman et al. 2014). While other genetic studies reported that flowering time in lentil is controlled by two to four QTLs. These QTLs can be distributed on two or more than two linkage groups (Tahir et al. 1994; Fratini et al. 2007; Tullu et al. 2008). More recently, a number of QTLs having a main effect and interactions between QTLs have also been reported for days to 50% flowering in a bi-parental mapping population (Saha et al. 2013). However, association mapping has an ability to detect novel QTLs because it uses diverse germplasm that has allelic diversity. Therefore, marker locus associated with flowering time in the present study might be different from QTL/genes identified in earlier studies. One of the reasons for this is many QTLs are not segregated in the bi-parental population because only two alternative alleles are present at a limited number of loci for a trait in such kind of mapping population (Jaiswal et al. 2012). In our study, markers associated with the flowering time explained phenotypic variation from 2.1 to 21.8%, which is comparatively lower than the phenotypic variation observed by analysis of bi-parental populations. However, it is not surprising to observe lower phenotypic variation in association mapping due to several reasons, including low LD and low allele frequency (Yan et al. 2011). In earlier studies, a marker associated significantly with a trait of interest explained low phenotypic variation in association mapping studies (Roy et al. 2010; Pasam et al. 2012).
The longer reproductive duration provides enough time to synthesize greater sink size leading to higher grain yield in lentil (Erskine et al. 1990). In another study, a reproductive period of 35-40 days has been reported as enough to develop high yielding genotypes (Kumar and Srivastava 2014). Thus, combining the early flowering (<50 days) with 40 days of reproductive periods can help in the development of high yielding super early genotypes in lentil (i.e., maturing <100 days). In South Asian countries, these super early genotypes can lead to an expansion of lentil in rice fallow areas and increase the yield productivity of lentil by avoiding the terminal heat, which has become a major challenge under changing environments (Choudhary et al. 2016). As, in the present study, several alleles of the associated markers have contributed significantly to reducing flowering time. Moreover, the sequence of express sequence tags (ESTs) belonging to EST-SSR markers associated with flowering time showed high-level similarity with the gene sequences of closely related food legumes. The roles of these genes in controlling flowering time have also been reported in earlier studies (Ito et al. 2000; Roxrud et al. 2007; Zhang et al. 2009, 2013; Kandasamy et al. 2002; Šlajcherová et al. 2012). Therefore, in the present study, EST-SSR markers could be associated with those genes that are involved in controlling flowering time in lentil. These EST-SSR markers can be used as functional markers and their allelic variants, responsible for reducing flowering time, can provide an opportunity to use them in lentil breeding programs for precise introgression of early flowering QTL in the background of high biomass genotypes to develop high yielding super early genotypes in lentil.
References
Abdelnoor RV, Barros EG, Moreira (1995) Determination of genetic diversity within Brazilian soybean germplasm using random amplified polymorphic DNA techniques and comparative analysis with pedigree data. Braz J Genet 18:265–273
Anbessa Y, Warkentin T, Vandenberg A, Ball R (2006) Inheritance of time to flowering in chickpea in a short-season temperate environments. J Hered 97:55–61
Asghar MJ, Abbas G, Shah TM (2010) Study of genetic diversity in some local and exotic lentil (Lens culinaris Medik) genotypes. Pak J Bot 42:2681–2690
Bajaj D, Srivastava R, Nath M, Tripathi S, Bharadwaj C, Upadhyaya HD, Tyagi AK, Parida SK (2016) Ecotilling-based association mapping efficiently delineates functionally relevant natural allelic variants of candidate genes governing agronomic traits in chickpea. Front Plant Sci 2016:7. https://doi.org/10.3389/fpls.2016.00450
Basheer-Salimia R, Camilli B, Scacchi S, Noli E, Awad M (2015) Assessment of genetic diversity in lentils (Lens culinaris Medik.) based on SNPs. Genet Mol Res 14:5870–5878
Benbouza H, Jacquemin JM, Baudoin JP, Mergeai G (2006) Optimization of a reliable, fast, cheap and sensitive silver staining method to detect SSR markers in polyacrylamide gels. Biotechnol Agron Soc Environ 10(2):77–81
Bradbury P, Parker T, Hamblin MT, Jannink JL (2011) Assessment of power and false discovery rate in genome-wide association studies using the barley CAP germplasm. Crop Sci 51:52–59
Cardon LR, Palmer LJ (2003) Population stratification and spurious allelic association. Lancet 361:598–604
Cheng P, Holdsworth W, Ma Y, Coyne CJ, Mazourek M, Grusak MA, Fuchs S, McGee RJ (2015) Association mapping of agronomic and quality traits in USDA pea single-plant collection. Mol Breed 35(2):75
Choudhary AK, Kumar J, Gupta S, Sultana R, Singh IS (2016) Breeding for adaptive traits in pulses. In: Singh (ed) Souvenir & Abstract of National Conference on Bringing Self-sufficiency in Pulses for eastern India. 5–6 August 2016. Bihar Agricultural University, Sabour, pp 36–43
Contreras Soto RI, de Oliveira MB, Costenaro da Silva D, Scapim CA, Schuster I (2017) Population structure, genetic relatedness and linkage disequilibrium blocks in cultivars of tropical soybean (Glycine max). Euphytica 213(8):173
Core Team R (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna URL http://www.R-project.org/
Dikshit HK, Singh A, Singh D, Aski MS, Prakash P, Jain N, Meena S, Kumar S, Sarker A (2015) Genetic diversity in Lens species revealed by EST and genomic simple sequence repeat analysis. PLOS One 10:e0138101
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Emami MK (1996) Genetic mapping in lentil (Lens culinaris Medik). Ph.D. Thesis. Division of Genetics, Indian Agricultural Research Institute, New Delhi, India
Erskine W, Ellis RH, Summerfield RJ, Roberts EH, Hussain A (1990) Characterization of responses to temperature and photoperiod for time to flowering in a world lentil collection. Theor Appl Genet 80(2):193–199
Erskine W, Chandra S, Chaudhary M, Malik IA, Sarker A, Sharma B, Tufail M, Tyagi MC (1998) A bottleneck in lentil: widening its genetic base in South Asia. Euphytica 101:207–211
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fratini R, Duran Y, Garcia P, Perez de la Vega M (2007) Identification of quantitative trait loci (QTL) for plant structure, growth habit and yield in lentil. Span J Agric Res 5:348–356
Gupta PK, Rustgi S, Kulwal PL (2005) Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol Biol 57:461–485
Imaizumi T (2010) Arabidopsis circadian clock and photoperiodism: time to think about location. Curr Opin Plant Biol 13:83–89
Ito T, Gyung Tae K, Kazuo S (2000) Disruption of an Arabidopsis cytoplasmic ribosomal protein S13-homologous gene by transposon-mediated mutagenesis causes aberrant growth and development. Plant J 22:257–264
Jain N, Dikshit HK, Singh D, Singh A, Kumar H (2013) Discovery of EST-derived microsatellite primers in the legume Lens culinaris (Fabaceae). Appl Pl Sci 1:1200539. https://doi.org/10.3732/apps.1200539
Jaiswal V, Mir RR, Mohan A, Balyan HS, Gupta PK (2012) Association mapping for pre-harvest sprouting tolerance in common wheat (Triticum aestivum L.) Euphytica 188:89–102
Jung C, Muller AE (2009) Flowering time control and applications in plant breeding. Trends in Plant Science 14:563–573
Kahriman A, Temel HY, Aydoğan A, Tanyolaç MB (2014) Major quantitative trait loci for flowering time in lentil. Turkish J Agric Fores 38:1–8
Kandasamy MK, McKinney EC, Meagher RB (2002) Functional non equivalency of actin isovariants in Arabidopsis. Mol Biol Cell 13:251–261
Kaur S, Cogan NO, Pembleton LW, Shinozuka M, Savin KW, Materne M, Forster JW (2011) Transcriptome sequencing of lentil based on second-generation technology permits large-scale unigene assembly and SSR marker discovery. BMC Genomics 12(1):1
Khazaei H, Caron CT, Fedoruk M, Diapari M, Vandenberg A, Coyne CJ, McGee R, Bett KE (2016) Genetic diversity of cultivated lentil (Lens culinaris Medik) and its relation to the world’s agro-ecological zones. Front Plant Sci 7:1093
Kumar J, Solanki RK (2014) Evaluation of germplasm accessions for agro-morphological traits in lentil. J Food Leg 27:275–280
Kumar J, Srivastava E (2014) Impact of reproductive duration on yield and its component traits in lentil. Leg Res 38(2):139–148
Kumar J, van Rheenen HA (2000) A major gene for time of flowering in chickpea. J Hered 91:67–68
Kumar J, Srivastva E, Singh M, Kumar S, Nadarajan N, Sarker A (2014) Diversification of indigenous gene-pool by using exotic germplasm in lentil (Lens culinaris Medikus subsp. culinaris). Physiol Mol Biol Plants 20(1):125–132
Kumar J, Gupta DS, Gupta S, Dubey S, Gupta P, Kumar S (2017) Quantitative trait loci from identification to exploitation for crop improvement. Plant Cell Rep 21:1–27
Kumar S, Rajendran K, Kumar J, Hamwieh A, Baum M (2015) Current knowledge in lentil genomics and its application for crop improvement. Front Plant Sci 6:78
Li XJ, Xu X, Yang XM, Li XQ, Liu WH, Gao AN, Li LH (2012) Genetic diversity among a founder parent and widely grown wheat cultivars derived from the same origin based on morphological traits and microsatellite markers. Crop Pasture Sci 63(4):303–310
Lombardi M, Materne M, Cogan NO, Rodda M, Daetwyler HD, Slater AT, Forster JW, Kaur S (2014) Assessment of genetic variation within a global collection of lentil (Lens culinaris Medik.) cultivars and landraces using SNP markers. BMC Genet 15:150
Malysheva-Otto LV, Ganal MW, Roder MS (2006) Analysis of molecular diversity, population structure and linkage disequilibrium in a worldwide survey of cultivated barley germplasm (Hordum vulgare L.) BMC Genet 7:6
Mandel JR, Nambeesan S, Bowers JE, Marek LF, Ebert D, Rieseberg LH, Knapp SJ, Burke JM (2013) Association mapping and the genomic consequences of selection in sunflower. PLoS Genet 9(3):e1003378. https://doi.org/10.1371/journal.pgen.1003378
Niu Y, Xu Y, Liu XF, Yang SX, Wei SP, Xie FT, Zhang YM (2013) Association mapping for seed size and shape traits in soybean cultivars. Mol Breed 31(4):785–794
Ostrowski MF, David A, Santoni S, Mckhann H, Reboud X, Corre VL, Camilleri C, Brunel D, Bouchez D, Faure B, Bataillon T (2006) Evidence for a large-scale population structure among accessions of Arabidopsis thaliana: possible causes and consequences for the distribution of linkage disequilibrium. Mol Ecol 15:1507–1517
Pasam RK, Sharma R, Malosetti M, van Eeuwijk FA, Haseneyer G, Kilian B, Graner A (2012) Genome-wide association studies for agronomical traits in a worldwide spring barley collection. BMC Plant Biol 12:16
Pritchard JK, Stephens M, Donnelly P (2000a) Inference of population structure using multi-locus genotype data. Genetics 155:945–959
Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000b) Association mapping in structured populations. Am J Hum Genet 67:170–181
Reddy MRK, Rathour R, Kumar N, Katoch P, Sharma TR (2010) Cross-genera legume SSR markers for analysis of genetic diversity in Lens species. Plant Breed 129:514–518
Rosenberg N, Nordborg M (2006) A general population-genetic model for the production by population structure of spurious genotype–phenotype associations in discrete, admixed, or spatially distributed populations. Genetics 173:1665–1678
Roxrud I, Lid SE, Fletcher JC, Schmidt ED, Opsahl-Sorteberg HG (2007) GASA4, one of the 14-member Arabidopsis GASA family of small polypeptides, regulates flowering and seed development. Plant Cell Physiol 48:471–483
Roy JK, Smith KP, Muehlbauer GJ, Chao S, Close TJ, Steffenson BJ (2010) Association mapping of spot blotch resistance in wild barley. Mol Breed 26:243–256
Saha GC, Sarker A, Chen W, Vandemark GJ, Muehlbauer FJ (2013) Inheritance and linkage map positions of genes conferring agromorphological traits in Lens culinaris Medik. Intl J Agron. http://dx.doi.org/10.1155/2013/618926
Salvi S, Sponza G, Morgante M, Tomes D, Niu X, Fengler KA, Meeley R, Ananiev EV, Svitashev S, Bruggemann E, Li B (2007) Conserved noncoding genomic sequences associated with a flowering-time quantitative trait locus in maize. Proc Natl Acad Sci 104(27):11376–11381
Sarker A, Erskine W, Sharma B, Tyagi MC (1999) Inheritance and linkage relationships of days to flower and morphological loci in lentil (Lens culinaris Medikus subsp. culinaris). J Hered 90:270–275
Sarker A, Erskine W, Sharma B, Tyagi MC (1999) Inheritance and linkage relationships of days to flower and morphological loci in lentil (Lens culinaris Medikus subsp. culinaris). J Hered 90:270–275
Seyedimoradi H, Talebi R (2014) Detecting DNA polymorphism and genetic diversity in lentil (Lens culinaris Medik.) germplasm: comparison of ISSR and DAMD marker. Physiol Mol Biol Plants 20:495–500
Singh BB, Mishra SK, Sardana S, Dixit GP (2006) Lentil and pea. In: Dhillon BS, Saxena S, Agarwal A, Tyagi RK (eds) Plant genetic resources: food grain crops. Narosa, New Delhi, pp 240–254
Singh M, Bisht IS, Kumar S, Dutta M, Bansal KC, Karale M, Sarker A, Amri A, Kumar S, Datta SK (2014) Global wild annual Lens collection: a potential resource for lentil genetic base broadening and yield enhancement. PLoS One 9(9):e107781
Šlajcherová K, Fišerová J, Fischer L, Schwarzerová K (2012) Multiple actin isotypes in plants: diverse genes for diverse roles? Front Plant Sci 2012:3
Subbarao GV, Kumar Rao JVDK, Kumar J, Johansen C, Deb UK, Ahmed I, Krishna Rao MV, Venkataratnam L, Hebber KR, Sai MVSR, Harris D (2001) Spatial distribution and quantification of rice-fallows in South Asia-potential for legumes. International Crops Research Institute for the Semi-Arid Tropics, Patancheru, p 316
Summerfield RJ, Roberts EH (1988) Photo-thermal regulation of flowering in pea, lentil, faba bean and chickpea. In: Summerfield RJ (ed) World crops: cool season food legumes. Current plant science and biotechnology in agriculture, vol 5. Springer, Dordrecht, pp 911–922
Summerfield RJ, Muehlbauer FJ, Roberts EH (1985) Lens culinaris. In: Halvey AH (ed) Handbook of flowering, vol 1. CRC, Boca Raton, pp 118–124
Sun D, Ren W, Sun G, Peng J (2011) Molecular diversity and association mapping of quantitative traits in Tibetan wild and worldwide originated barley (Hordeum vulgare L.) germplasm. Euphytica 178(1):31–43
Szalma SJ, Buckler IVES, Snook ME, McMullen MD (2005) Association analysis of candidate genes for maysin and chlorogenic acid accumulation in maize silks. Theor Appl Genet 110:1324–1333
Tahir M, Muehlbauer FJ, Spaeth SC (1994) Association of isozyme markers with quantitative trait loci in random single seed descent derived lines of lentil (Lens culinaris Medik.) Euphytica 75:111–119
Takahashi Y, Teshima KM, Yokoi S, Innan H, Shimamoto K (2009) Variations in Hd1 proteins, Hd3a promoters, and Ehd1 expression levels contribute to diversity of flowering time in cultivated rice. Proc Natl Acad Sci USA 106:4555–4560
Thines BC, Youn Y, Duarte MI, Harmon FG (2014) The time of day effects of warm temperature on flowering time involve PIF4 and PIF5. J Exp Bot 65:1141–1151
Topal A, Aydın C, Akgün N, Babaoglu M (2004) Diallel cross analysis in durum wheat (Triticum durum Desf.): identification of best parents for some kernel physical features. Field Crops Research 87(1):1–2
Tullu A, TarTa B, Warkentin T, Vandenberg A (2008) Construction of an intraspecific linkage map and QTL analysis for earliness and plant height in lentil. Crop Sci. 48:2254–2264
Verma P, Shah N, Bhatia S (2013) Development of an expressed gene catalogue and molecular markers from the de novo assembly of short sequence reads of the lentil (Lens culinaris Medik.) transcriptome. Plant Biotechnol J 11:894–905
Verma P, Sharma TR, Srivastava PS, Abdin MZ, Bhatia S (2014) Exploring genetic variability within lentil (Lens culinaris Medik.) and across related legumes using a newly developed set of microsatellite markers. Mol Biol Rep 41:5607–5625
Visioni A, Tondelli A, Francia E, Pswarayi A, Malosetti M, Russell J, Thomas W, Waugh R, Pecchioni N, Romagosa I, Comadran J (2013) Genome-wide association mapping of frost tolerance in barley (Hordeum vulgare L.) BMC Genomics 14:424
Wang YH, Bible P, Loganantharaj R, Upadhyaya HD (2012) Identification of SSR markers associated with height using pool-based genome-wide association mapping in sorghum. Mol Breed 30:281–292
Welch SM, Dong Z, Roe JL, Das S (2005) Flowering time control: gene network modelling and the link to quantitative genetics: modelling complex traits for plant improvement. Aust J Agr Res 56:919–936
Wilson LM, Whitt SR, Ibáñez AM, Rocheford TR, Goodman MM, Buckler ES (2004) Dissection of maize kernel composition and starch production by candidate gene association. Plant Cell 16:2719–2733
Yan J, Warburton M, Crouch J (2011) Association mapping enhancing maize (Zea mays L.) genetic improvement. Crop Sci 51:433–449
Zhang P, Liu X, Tong H, Lu Y, Li J (2014) Association mapping for important agronomic traits in core collection of rice (Oryza sativa L.) with SSR markers. PloS One 9(10):e111508
Zhang S, Yang C, Peng J, Sun S, Wang X (2009) GASA5, a regulator of flowering time and stem growth in Arabidopsis thaliana. Plant Mol Biol 69:745–759
Zhang S, Cheng F, Wang C, Zhang L, An Y (2013) Cloning and tissue-specific expression of predicted Pisum sativum actin isoform PEAc14-1. Bioch Genet 51:722–727
Zhao Y, Wang H, Chen W, Li Y (2014) Genetic structure, linkage disequilibrium and association mapping of verticillium wilt resistance in elite cotton (Gossypium hirsutum L.) germplasm population. PLoS One 9(1):e86308. https://doi.org/10.1371/journal.pone.0086308
Acknowledgements
The authors thank the Indian Council of Agricultural Research, New Delhi for research support. This work is partially funded by the Department of Agriculture Corporation and Framers Welfare (DAC & FW), Government of India, New Delhi and Department of Biotechnology (BT/PR10921/AG11/106/943/2014), Govt. of India.
Author information
Authors and Affiliations
Contributions
JK designed the work, analyzed and interpreted the results, and drafted the manuscript. RSB, SG, SD, and PG helped in collection and tabulation of genotypic and phenotypic data. NPS helped to edit the revised manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by: Barbara Naganowska
Electronic supplementary material
ESM 1
(DOCX 24 kb)
Rights and permissions
About this article

Cite this article
Kumar, J., Gupta, S., Biradar, R.S. et al. Association of functional markers with flowering time in lentil. J Appl Genetics 59, 9–21 (2018). https://doi.org/10.1007/s13353-017-0419-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-017-0419-0