
Abstract
High plasma levels of vitamin D are associated with a reduced risk of high blood pressure, but whether this association is causal remains to be ascertained. We performed a meta-analysis of randomized clinical trials, to examine the effect of vitamin D supplementation on both systolic blood pressure (SBP) and diastolic blood pressure (DBP) and supplemented these results with a Mendelian randomization analysis to investigate the causal relationship between vitamin D status (25-hydroxyvitamin D [25(OH)D]) and BP. Pooled random effects meta-analysis of weighted mean differences across 16 trials of vitamin D supplementation showed a non-significant reduction in SBP (−0.94, 95 % CI −2.98, 1.10 mmHg) and DBP (−0.52, 95 % CI −1.18, 0.14 mmHg), with evidence of heterogeneity (I2 = 67.9 %, P < 0.001) and publication bias (P = 0.02) among trials of SBP. There was a significant reduction in DBP (−1.31, 95 % CI −2.28, −0.34 mmHg, P = 0.01) in participants with pre-existing cardiometabolic disease. Variants at three published loci (GC, DHCR7, CYP2R1, and CYP24A1) for 25(OH)D, were not significantly associated with BP, but rs6013897 in CYP24A1 gene region had nominally significant associations with both SBP and DBP (P < 0.05). Evidence from the associations of the genetic variants with the risk of vitamin D deficiency (defined as a 25(OH)D level < 50 nmol/L) and BP showed that the causal effects of a doubling of genetically-elevated risk of vitamin D deficiency were 0.14 mmHg (95 % CI −0.19, 0.47, P = 0.42), and 0.12 mmHg (95 % CI −0.09, 0.33, P = 0.25) on SBP and DBP respectively. Additional evidence from genetic data are directionally consistent with clinical trial data, though underpowered to reliably demonstrate a strong causal effect of vitamin D status on BP. Further investigation may be warranted.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vitamin D is pivotal in regulating calcium and bone homeostasis [1] and is associated with several biological processes, including modulation of blood pressure (BP). Amongst the proposed mechanistic pathways for the development of high BP, vitamin D inhibits the renin-angiotensin-aldosterone system [2], alters proliferation of vascular endothelial smooth muscle cells [3], and is essential for insulin secretion [4]. Several prospective studies and meta-analyses have consistently shown an inverse association between vitamin D status (as measured by 25-hydroxyvitamin D [25(OH)D]) and BP [5, 6]. As observational epidemiological studies are beset by residual confounding and reverse causation, it is difficult to infer causality from these findings. From a public health perspective, it is crucial to address this issue as the therapeutic modification of circulating vitamin D levels can be achieved through supplementation or therapy, more so as both vitamin D deficiency and high BP have risen to pandemic proportions, individually affecting over 1 billion people worldwide [7, 8]. High BP has been shown to be continuously and linearly associated with cardiovascular risk over several decades ago [9] and is the most common modifiable risk factor for cardiovascular disease (CVD) [10], which represents a worldwide epidemic and is the leading cause of death globally [11]. Vitamin D deficiency may increase CVD risk by activating an inflammatory cascade, which results in endothelial dysfunction and increased arterial stiffness, both of which contribute to high BP and are risk markers for CVD risk [12–14].
Randomized clinical trials (RCTs) of vitamin D supplementation offer the highest clinical evidence for establishing whether vitamin D deficiency is causally related to high BP. However, findings from previous trials have failed to demonstrate significant reductions in BP and results of prior meta-analyses have been inconclusive [15–17]. In the absence of such trials, Mendelian Randomization (MR) [18] studies utilizing genetic variants which specifically alter levels of vitamin D may provide another route to help judge the causal relevance of vitamin D to BP. An MR study utilises the fact that since the presence of particular genetic variants or alleles are randomly allocated at conception (gamete formation) and such allocation is expected to be independent of any behavioural and environmental factors, the associations of such variants with levels of the exposure (in this case vitamin D status) or with disease outcome (in this case high BP) are not likely to be affected by potential confounding or reverse causation [19]. If lower vitamin D status is causally related to high BP, then a genetic variant associated with lower 25(OH)D levels should be associated with a higher risk of high BP. Since the last previous review [17], several interventions studies evaluating the effects of vitamin D supplementation on BP outcomes have been published and their results have been inconsistent. Against this background, we aimed to assess the potential causal relevance of vitamin D deficiency to high BP by updating the evidence on the effect of vitamin D supplementation on circulating levels of 25(OH)D and its impact on both systolic and diastolic BP. We have also supplemented this with evidence from published genetic association studies of 25(OH)D levels and BP and applied a MR approach [20] using summarized published data on four genetic variants of vitamin D status [as measured by 25(OH)D].
Methods
Data sources and study selection
This review was conducted using a predefined protocol and in accordance with PRISMA guidelines [21] (Appendix 1). We systematically searched MEDLINE, EMBASE, Web of Science, and Cochrane Central Register of Controlled Trials from inception up to November, 2013 for RCTs of vitamin D supplementation (cholecalciferol [vitamin D3] or ergocalciferol [vitamin D2]) on systolic blood pressure (SBP) and diastolic blood pressure (DBP). The searches combined terms for vitamin D and blood pressure and no language restrictions were imposed (Appendix 2). Additional studies were sought from the reference lists of recovered articles and previous review articles, by hand searching of relevant journals, and by correspondence with authors of included studies. We included only RCTs that aimed to study the effects of oral vitamin D supplementation alone. Studies in which the intervention was calcitriol or one of its analogues and those with participants not receiving an intervention to raise their vitamin D [25(OH)D] levels were excluded. The primary outcome was the difference in office or ambulatory SBP and DBP among treatment and control groups compared with baseline BPs. Additionally, lead single nucleotide polymorphisms (SNPs) exclusively associated with circulating levels of 25(OH)D were identified by searching the original publications of genome-wide association studies (GWASs) for vitamin D that have been indexed by the National Human Genome Research Institute (NHGRI) GWAS catalogue [22]. Single nucleotide polymorphisms were considered for inclusion if they were associated with levels of 25(OH)D at genome-wide significant levels (P < 5 × 10−8 unless otherwise specified) and were uncorrelated.
Data extraction and quality assessment
Data on the following characteristics were extracted independently by two investigators who used standardised protocols: number of participants, sampling population, geographical location (defined as Europe, North America, and the Asia–Pacific region); age range of participants at baseline, gender; duration of intervention; type and formulation of vitamin D supplementation, daily dose of supplementations, composition of placebo, and mean BP and standard deviation, or the mean difference were abstracted. Discrepancies were resolved by discussion and by adjudication of a third reviewer. The Cochrane Collaboration’s tool for assessing risk of bias was used to assess the validity of the trials. This tool uses the following methodological features most relevant to the control of bias: randomization, random allocation concealment, masking of treatment allocation and outcome assessments, incomplete outcome data, selective reporting, and other bias [23]. For each individual domain, studies were classified into low, unclear and high risk of bias.
Data synthesis and analysis
Random-effects models were used to pool the weighted mean differences (WMDs) across trials. Heterogeneity was assessed with the I2 statistic, with I2 > 50 % considered to be important. Study-level characteristics including geographical location, gender, number of participants, baseline population (presence or absence of pre-existing cardiometabolic disease), duration of intervention, daily dose of supplementation, and type of vitamin D supplement were pre-specified as characteristics for assessment of heterogeneity, which was conducted using stratified analysis and random effects meta-regression [24]. We assessed the potential for publication bias through formal tests, namely Begg’s funnel plots [25] and Egger’s regression symmetry test [26]. The associations of exclusive SNPs identified from GWASs of 25(OH)D levels and other published reports [27–30], were queried with both systolic and diastolic BP using data from the International Consortium of Blood Pressure GWAS (ICBPGWAS), which has been described in detail elsewhere [31]. Briefly, the ICBPGWAS involves a meta-analysis of GWAS data evaluating the associations between 2.5 million genotyped or imputed SNPs and SBP and DBP in 69,395 individuals of European ancestry from 29 studies. Mendelian randomization analyses were conducted using a likelihood-based method for combining summarized genetic association estimates [20] into single estimates of the causal effects of vitamin D status on SBP and DBP. All analyses were conducted using Stata version 12 (Stata Corp, College Station, Texas) and R version 2.15.3 (R Foundation, Vienna, Austria).
Results
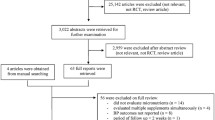
Figure 1 shows the number of studies assessed and excluded through the stages of the meta-analysis. A total of 16 trials (comprising 1,879 participants) reported the effect of vitamin D supplementation on SBP, of which 15 reported on DBP [3, 32–46]. Duration of vitamin D supplementation varied from 5 weeks to 12 months. Risk of bias assessment in each trial is reported in Appendix 3. All trials had low risk of bias for the random sequence generation, blinding of both participants and personnel, and the selective reporting domains. One trial had an unclear risk of bias for allocation concealment and another had a high risk of bias. Eleven trials had an unclear risk of bias for blinding of outcome assessments. One trial had a high risk of bias for incomplete outcome data and risk of other bias was unclear in five trials. There was considerable variability in study populations which included healthy participants as well as participants with pre-existing conditions such as diabetes, hypertension, and cardiovascular disease. All trials used vitamin D supplementation of more than 600 IU per day (which is the US Institute of Health Recommended Dietary Allowance [47]), with the doses varying from 800 to 8,571 IU per day. Comparing follow-up with baseline assessment, circulating levels of 25(OH)D increased substantially in the intervention arms in all the included trials (Table 1).

Trial selection flow diagram. BP blood pressure, RCT randomized clinical trial
Effect of vitamin D supplementation on blood pressure
In pooled random effects meta-analysis of WMDs across eligible trials, vitamin D supplementation showed a non-significant reduction in SBP (−0.94, 95 % CI −2.98, 1.10 mmHg, P = 0.37; I2 = 67.9 %, P for heterogeneity < 0.001) and DBP (−0.52, 95 % CI −1.18, 0.14 mmHg, P = 0.12; I2 = 0.0 %, P for heterogeneity = 0.50) (Fig. 2). The heterogeneity among trials for SBP was not explained by differences in several study level characteristics (Table 2). In sensitivity analysis, we excluded the study by Sugden et al. [3] as it reported a significant decrease in SBP on vitamin D supplementation by about 14 mmHg compared with placebo, which may have unduly influenced our findings. The pooled random effects meta-analysis of WMDs excluding this study also showed a non-significant reduction in SBP (−0.13, 95 % CI −1.89, 1.63 mmHg, P = 0.88; I2 = 55.8 %, P for heterogeneity = 0.004). Subgroup analysis of trials of DBP showed a significant reduction in DBP with vitamin D supplementation in six trials involving participants with pre-existing cardiometabolic disease (−1.31, 95 % CI −2.28, −0.34 mmHg, P = 0.01; I2 = 0.0 %, P for heterogeneity = 0.03) (Table 3). Egger’s test for publication bias among trials for SBP was significant (P = 0.02), consistent with observed funnel plot asymmetry.

Meta-analysis of effects of vitamin D supplementation on blood pressure in eligible randomized controlled trials. The summary estimates presented were calculated using random effects models; CI confidence interval (bars), SD standard deviation, WMD weighted mean difference
Evidence from genome wide association studies
We identified genome-wide significant variants at GC, DHCR7, and CYP2R1and CYP24A1 loci which together explained up to 1–4 % of the variation in 25(OH)D levels (Table 4). Regional association plots within 200 kb window of these vitamin D SNPs showed lack of significant associations (at a Bonferroni corrected P = 1 × 10−4 threshold; Fig. 3) with BP. However, the associations of rs6013897 on chromosome 20q13 in CYP24A1 with SBP and DBP were nominally significant (P < 0.05).

Regional association plots of vitamin D related gene regions. Each panel spans 200 kb around the published vitamin D SNP in the region, which is highlighted with a purple diamond. The SNPs are coloured according to their linkage disequilibrium with the top variant based on the CEU Hap Map population (http://www.hapmap.org). Gene transcripts are annotated in the lower box. The association results for blood pressure were taken from the International Consortium of Blood Pressure Genome Wide Association Studies (ICBPGWAS)
Mendelian randomization analysis using published data
Estimates using genetic variants for the causal effect of vitamin D on BP were −0.11 mmHg (95 % CI −0.31, 0.09, P = 0.27) for systolic BP and −0.10 mmHg (95 % CI −0.22, 0.03, P = 0.13) for diastolic BP, based on a 10 % increase in 25(OH)D levels (Fig. 4). Using published data on the association of the genetic variants with the risk of vitamin D deficiency [defined as a 25(OH)D level < 50 nmol/L] and BP, we estimated the change in BP for an increase in the genetic component of the risk of vitamin D deficiency. The causal effect of a doubling of genetically-determined risk of vitamin D deficiency on systolic BP was 0.14 mmHg (95 % CI −0.19, 0.47, P = 0.42), and on diastolic BP was 0.12 mmHg (95 % CI −0.09, 0.33, P = 0.25).

Estimated effects on blood pressure change plotted against estimated effects on serum vitamin D levels, for four SNPs associated with vitamin D. SNP, single nucleotide polymorphism; Vertical and horizontal solid black lines show 95 % confidence intervals (CIs) for each individual SNP. Estimates of casual effect of vitamin D on blood pressure, by using a likelihood-based method for combining summarized genetic association estimates using all SNPs, are represented by solid black line with gradient. Using all SNPs, multi-SNP risk score analyses identified weak protective causal effects of vitamin D on blood pressure levels, −0.11 mmHg (95 % CI −0.31, 0.09, P = 0.27) for systolic blood pressure and −0.10 mmHg (95 % CI −0.22, 0.03, P = 0.13) for diastolic blood pressure, based on a 10 % increase in 25(OH)D levels
Comment
Pooled results of the available clinical evidence were directionally suggestive of a reduction in both systolic and diastolic BP with vitamin D supplementation, but lacked statistical significance. Subgroup analysis of trials of DBP however, showed a significant reduction in DBP (by 1.3 mmHg) with vitamin D supplementation in participants with pre-existing cardiometabolic disease. In the published literature, [27–30] we identified several genome-wide significant variants at 4 unique loci, involved in 25(OH)D synthesis (DHCR7, CYP2R1) and metabolism (GC, CYP24A1), which have been suggested to be exclusively associated with vitamin D pathways. The variants together, explained up to 1–4 % of the variation in 25(OH)D levels. Utilizing data from ICBP GWAS [31], we demonstrated that vitamin D SNPs had small effects on BP but lacked statistical significance, except for one variant rs6013897 in CYP24A1 gene region. All SNPs showed directionally concordant associations with 25(OH)D levels and BP, which were consistent with the clinical trial results. However, the causal effect estimates based on the available genetic evidence did not achieve statistical significance.
The current results argue against a strong causal role of vitamin D pathways in the aetiology of high BP, but cannot rule out a weak causal effect. There was evidence of a significant reduction in DBP in participants with pre-existing cardiometabolic disease on vitamin D supplementation. Several plausible reasons may explain this observation. Whiles, optimal vitamin D status is an excellent marker of good health [48], suboptimal vitamin D status may reflect chronic illnesses [15] such as cardiometabolic diseases. Though our results (Table 1) were not indicative of low baseline vitamin D status [25(OH)D levels] in participants with pre-existing cardiometabolic diseases, there is data to suggest that significant improvements in cardiometabolic outcomes (such as reductions in BP) with vitamin D supplement use may be seen only among those with vitamin D deficiency [15]. Further data are necessary to adjudicate this observation. The inconsistent results reported by the clinical trials have been attributed to several reasons as suggested by previous reviews [15, 16]. These include limited sample sizes to detect incremental differences in BP, heterogeneity in study populations, short follow-up periods, and the fact that majority of trials reported results from post hoc sub-group analyses. If there is a causal relationship between vitamin D deficiency and high BP, then establishing this may require carefully designed RCTs with large-sample sizes and long follow-up durations. The on-going VITamin D and OmegA-3 TriaL (VITAL), with over 20,000 healthy participants randomized to daily dietary supplements of vitamin D3 or omega-3 fatty acids [49] may offer useful insights. Whiles we await results of this trial, MR investigations using individual-level data may provide another efficient method to help establish causality. Such MR investigations have been used to investigate the causal relevance of risk markers, such as C-reactive protein and lipoprotein (a), to risk of coronary heart disease in the absence of interventions that specifically modify levels of these risk markers [50, 51]. Collective evidence from several studies demonstrates that variability in 25(OH)D levels is explained by both genetic and environmental factors. Heritability of 25(OH)D levels has been estimated to be as high as 80 % [52], and given this level of heritability, recent advances have been made in identifying several genetic determinants of 25(OH)D levels. The four genes identified in the present analysis play important roles in the vitamin D metabolic pathway. DHCR7 and CYP2R1 function upstream of the 25(OH)D synthesis pathway, whiles GC and CYP24A1 function downstream of the metabolism pathway [29]. The DHCR7 gene encodes 7-dehydrocholesterol reductase, the enzyme that converts 7-dehydrocholesterol to cholesterol. CYP2R1 is known to encode the enzyme that catalyzes the synthesis of 25(OH)D in the liver [53]. GC, the group-specific component gene (located on chromosome 4q12-q13), which encodes vitamin D-binding protein, harbors a set of SNPs which are associated with circulating levels of 25(OH)D levels at genome-wide significance. The strongest association with 25(OH)D levels has consistently been demonstrated for rs2282679 [28, 29]. The CYP24A1 gene encodes the enzyme which plays an important role in calcium homeostasis and the vitamin D endocrine system, where it acts at the initial stage of 25(OH)D catabolism [54]. Informative MR studies on vitamin D and BP are likely to be feasible given the potential specificity of the associations of these genetic variants with vitamin D. However, given the small fractions of the variances in vitamin D levels explained by these common variants, MR studies would require large sample sizes (~ 80,000 participants) to have sufficient power to establish causality [29]. Fine mapping and exome sequencing of the common gene regions involved in vitamin D pathways may help uncover rarer genetic variations with larger effects on vitamin D levels and may be better instrumental variables for MR.
The strengths and limitations of this study merit careful consideration. This study has provided a comprehensive systematic synthesis of available evidence by including data from different sources, evaluated the impact of vitamin D supplementation for several relevant subgroups in a consistent way, and has utilized genetic data to assess the causal relevance of vitamin D to high BP. The majority of trials included in this review appear to have low risk of bias; however, the current findings should be interpreted with some caution, owing to the potential differences in design and population characteristics of each trial. There was substantial heterogeneity among trials of SBP. Given this, it was debatable whether pooled estimates should be presented rather than reporting estimates in relevant subgroups, as the presence of heterogeneity makes pooling of risk estimates data somewhat controversial. We however systematically explored possible sources of heterogeneity using stratified analyses, metaregression techniques, and sensitivity analyses and also presented pooled estimates for the relevant subgroups. In general, there was a consistent trend of reduction in BP in subgroups assessed. Our findings for studies of SBP may have been over-estimated somewhat due to preferential publication of extreme findings, or, analogously, by selective reporting of striking results. Furthermore, in the current analyses, we employed an MR approach using summarized published data on multiple genetic variants for 25(OH)D levels, as individual-level data on large numbers of participants was unavailable. Though it has been reported that causal estimates from summarized data are almost as precise as those obtained from individual-level data [20], there are several limitations to the use of summarized data. These include inability to (1) fully assess instrumental variable assumptions; (2) address population stratification; (3) test for the attenuation of genetic associations with the outcome on adjustment for the exposure of interest; and (4) assess parametric assumptions required by instrumental variable methods for effect estimation [20]. The results should therefore be interpreted in context of the limitations available.
In conclusion, pooled results of relevant clinical trials provide non-significant reductions in both SBP and DBP on vitamin D supplementation, with evidence of considerable heterogeneity and publication bias among studies of SBP. Subgroup analysis however showed evidence of a significant reduction in DBP in participants with pre-existing cardiometabolic disease. Additional evidence from genetic data are directionally consistent with clinical trial data, though underpowered to reliably demonstrate a strong causal effect of vitamin D status on BP. Since vitamin D remains a promising though unproven strategy in the prevention of high BP (hypertension), further evaluation may be warranted to assess any causal association. Further research is also warranted to assess the evidence with more refined indices of BP such as heart rate, pulse pressure, and cardiac output.
References
DeLuca HF. Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr. 2004;80(6 Suppl):1689S–96S.
Lee JH, O’Keefe JH, Bell D, Hensrud DD, Holick MF. Vitamin D deficiency an important, common, and easily treatable cardiovascular risk factor? J Am Coll Cardiol. 2008;52(24):1949–56.
Sugden JA, Davies JI, Witham MD, Morris AD, Struthers AD. Vitamin D improves endothelial function in patients with Type 2 diabetes mellitus and low vitamin D levels. Diabet Med. 2008;25(3):320–5.
Chiu KC, Chu A, Go VLW, Saad MF. Hypovitaminosis D is associated with insulin resistance and beta cell dysfunction. Am J Clin Nutr. 2004;79(5):820–5.
Burgaz A, Orsini N, Larsson SC, Wolk A. Blood 25-hydroxyvitamin D concentration and hypertension: a meta-analysis. J Hypertens. 2011;29(4):636–45.
Kunutsor SK, Apekey TA, Steur M. Vitamin D and risk of future hypertension: meta-analysis of 283,537 participants. Eur J Epidemiol. 2013;28(3):205–21.
Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266–81.
Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365(9455):217–23.
Kannel WB, Schwartz MJ, McNamara PM. Blood pressure and risk of coronary heart disease: the Framingham study. Dis Chest. 1969;56(1):43–52.
Lawes CM, Bennett DA, Feigin VL, Rodgers A. Blood pressure and stroke: an overview of published reviews. Stroke. 2004;35(3):776–85.
Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013;127(1):e6–245.
Geleijnse JM. Vitamin D and the prevention of hypertension and cardiovascular diseases: a review of the current evidence. Am J Hypertens. 2011;24(3):253–62.
Laurent S, Cockcroft J, Van Bortel L, et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. 2006;27(21):2588–605.
Corretti MC, Anderson TJ, Benjamin EJ, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39(2):257–65.
Pittas AG, Chung M, Trikalinos T, et al. Systematic review: Vitamin D and cardiometabolic outcomes. Ann Intern Med. 2010;152(5):307–14.
Witham MD, Nadir MA, Struthers AD. Effect of vitamin D on blood pressure: a systematic review and meta-analysis. J Hypertens. 2009;27(10):1948–54.
Elamin MB, Abu Elnour NO, Elamin KB, et al. Vitamin D and cardiovascular outcomes: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2011;96(7):1931–42.
Davey Smith G, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22.
Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–63.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–65.
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097.
Hindorff LAMJ, Morales J, Junkins HA, Hall PN, Klemm AK, Manolio TA: A Catalog of Published Genome-Wide Association Studies. www.genome.gov/gwastudies. Accessed 18 Sep 2013.
Higgins JP, Altman DG, Gotzsche PC, et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ. 2011;343:d5928.
Thompson SG, Sharp SJ. Explaining heterogeneity in meta-analysis: a comparison of methods. Stat Med. 1999;18(20):2693–708.
Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics. 1994;50(4):1088–101.
Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315(7109):629–34.
Wang TJ, Zhang F, Richards JB, et al. Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet. 2010;376(9736):180–8.
Ahn J, Yu K, Stolzenberg-Solomon R, et al. Genome-wide association study of circulating vitamin D levels. Hum Mol Genet. 2010;19(13):2739–45.
Berry DJ, Vimaleswaran KS, Whittaker JC, Hingorani AD, Hypponen E. Evaluation of genetic markers as instruments for mendelian randomization studies on vitamin D. PLoS ONE. 2012;7(5):e37465.
Vimaleswaran KS, Berry DJ, Lu C, et al. Causal relationship between obesity and vitamin D status: bi-directional Mendelian randomization analysis of multiple cohorts. PLoS Med. 2013;10(2):e1001383.
Ehret GB, Munroe PB, Rice KM, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478(7367):103–9.
Scragg R, Khaw KT, Murphy S. Effect of winter oral vitamin D3 supplementation on cardiovascular risk factors in elderly adults. Eur J Clin Nutr. 1995;49(9):640–6.
Pfeifer M, Begerow B, Minne HW, Nachtigall D, Hansen C. Effects of a short-term vitamin D(3) and calcium supplementation on blood pressure and parathyroid hormone levels in elderly women. J Clin Endocrinol Metab. 2001;86(4):1633–7.
Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr. 2006;83(4):754–9.
Nagpal J, Pande JN, Bhartia A. A double-blind, randomized, placebo-controlled trial of the short-term effect of vitamin D3 supplementation on insulin sensitivity in apparently healthy, middle-aged, centrally obese men. Diabet Med. 2009;26(1):19–27.
Jorde R, Sneve M, Torjesen P, Figenschau Y. No improvement in cardiovascular risk factors in overweight and obese subjects after supplementation with vitamin D3 for 1 year. J Intern Med. 2010;267(5):462–72.
Witham MD, Dove FJ, Dryburgh M, Sugden JA, Morris AD, Struthers AD. The effect of different doses of vitamin D(3) on markers of vascular health in patients with type 2 diabetes: a randomised controlled trial. Diabetologia. 2010;53(10):2112–9.
Shab-Bidar S, Neyestani TR, Djazayery A, et al. Regular consumption of vitamin D-fortified yogurt drink (Doogh) improved endothelial biomarkers in subjects with type 2 diabetes: a randomized double-blind clinical trial. BMC Med. 2011;9:125.
Larsen T, Mose FH, Bech JN, Hansen AB, Pedersen EB. Effect of cholecalciferol supplementation during winter months in patients with hypertension: a randomized, placebo-controlled trial. Am J Hypertens. 2012;25(11):1215–22.
Witham MD, Dove FJ, Sugden JA, Doney AS, Struthers AD. The effect of vitamin D replacement on markers of vascular health in stroke patients—a randomised controlled trial. Nutr Metab Cardiovasc Dis. 2012;22(10):864–70.
Gepner AD, Ramamurthy R, Krueger DC, Korcarz CE, Binkley N, Stein JH. A prospective randomized controlled trial of the effects of vitamin D supplementation on cardiovascular disease risk. PLoS ONE. 2012;7(5):e36617.
Wood AD, Secombes KR, Thies F, et al. Vitamin D3 supplementation has no effect on conventional cardiovascular risk factors: a parallel-group, double-blind, placebo-controlled RCT. J Clin Endocrinol Metab. 2012;97(10):3557–68.
Forman JP, Scott JB, Ng K, et al. Effect of vitamin D supplementation on blood pressure in blacks. Hypertension. 2013;61(4):779–85.
Witham MD, Price RJ, Struthers AD, et al. Cholecalciferol treatment to reduce blood pressure in older patients with isolated systolic hypertension: The VitDISH Randomized Controlled Trial. JAMA Intern Med. 2013.
Witham MD, Adams F, Kabir G, Kennedy G, Belch JJ, Khan F. Effect of short-term vitamin D supplementation on markers of vascular health in South Asian women living in the UK: a randomised controlled trial. Atherosclerosis. 2013;230(2):293–9.
Wamberg L, Kampmann U, Stodkilde-Jorgensen H, Rejnmark L, Pedersen SB, Richelsen B. Effects of vitamin D supplementation on body fat accumulation, inflammation, and metabolic risk factors in obese adults with low vitamin D levels: results from a randomized trial. Eur J Intern Med. 2013;24(7):644–9.
Pramyothin P, Holick MF. Vitamin D supplementation: guidelines and evidence for subclinical deficiency. Curr Opin Gastroenterol. 2012;28(2):139–50.
Prentice A, Goldberg GR, Schoenmakers I. Vitamin D across the lifecycle: physiology and biomarkers. Am J Clin Nutr. 2008;88(2):500S–6S.
Manson JE, Bassuk SS, Lee IM, et al. The VITamin D and OmegA-3 TriaL (VITAL): rationale and design of a large randomized controlled trial of vitamin D and marine omega-3 fatty acid supplements for the primary prevention of cancer and cardiovascular disease. Contemp Clin Trials. 2012;33(1):159–71.
Hingorani AD, Shah T, Casas JP. Linking observational and genetic approaches to determine the role of C-reactive protein in heart disease risk. Eur Heart J. 2006;27(11):1261–3.
Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518–28.
Dastani Z, Li R, Richards B. Genetic Regulation of Vitamin D Levels. Calcif Tissue Int. 2012.
Cheng JB, Levine MA, Bell NH, Mangelsdorf DJ, Russell DW. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci USA. 2004;101(20):7711–5.
Sakaki T, Kagawa N, Yamamoto K, Inouye K. Metabolism of vitamin D3 by cytochromes P450. Front Biosci J Virt Lib. 2005;10:119–34.
Acknowledgments
We thank the International Consortium of Blood Pressure Genome Wide Association Studies for providing data on request.
Conflict of interest
PBM is supported by the National Institutes for Health Research Biomedical Research Unit (NIHRBRU) at Barts. SB is supported by the Wellcome Trust (Grant number 100114). The NIHRBRU or Wellcome Trust had no role in study design, in data collection, analysis or interpretation, in writing the report, or in the decision to submit for publication. There was no external funding for this work.
Author information
Authors and Affiliations
Corresponding authors
Appendices
Appendix 1
See Table 5.
Appendix 2: Literature search strategy
Relevant studies, published before November 30, 2013 (date last searched), were identified through electronic searches not limited to the English language using MEDLINE, EMBASE, and Cochrane databases. Electronic searches were supplemented by scanning reference lists of articles identified for all relevant studies (including review articles), by hand searching of relevant journals and by correspondence with study investigators. The computer-based searches combined search terms related to vitamin D supplementation and blood pressure without language restriction.
-
(i)
MEDLINE strategy to identify relevant exposures: (“Vitamin D”[Mesh] OR “vitamin d”[All Fields] OR “25-hydroxyvitamin D”[All Fields] OR “25(OH)D”[All Fields] OR “calcidiol”[All Fields] OR “ergocalciferols”[Mesh] OR “ergocalciferols”[All Fields] OR “Vitamin D Supplementation”[Mesh])
-
(ii)
MEDLINE strategy to identify relevant outcomes: (“Hypertension”[Mesh] OR “hypertension”[All Fields] OR “blood pressure”[Mesh])
-
(iii)
MEDLINE strategy to identify relevant population: (“humans”[MeSH Terms])
Parts i, ii and iii were combined using ‘AND’ to search MEDLINE. Each part was specifically translated for searching alternative databases.
Appendix 3
See Table 6.
Rights and permissions
About this article
Cite this article
Kunutsor, S.K., Burgess, S., Munroe, P.B. et al. Vitamin D and high blood pressure: causal association or epiphenomenon?. Eur J Epidemiol 29, 1–14 (2014). https://doi.org/10.1007/s10654-013-9874-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10654-013-9874-z