
Summary
Background In the clinical development of oncology drugs, the recommended dose is usually determined using a 3 + 3 dose-escalation study design. However, this phase I design does not always adequately describe dose-toxicity relationships. Methods 125 patients, with either solid tumours or lymphoma, were included in the study and 1217 platelet counts were available over three treatment cycles. The data was used to build a population pharmacokinetic/pharmacodynamic (PKPD) model using a sequential modeling approach. Model-derived Recommended Doses (MDRD) of abexinostat (a Histone Deacetylase Inhibitor) were determined from simulations of different administration schedules, and the higher bound for the probability of reaching these MDRD with a 3 + 3 design were obtained. Results The PKPD model developed adequately described platelet kinetics in both patient populations with the inclusion of two platelet baseline counts and a disease progression component for patients with lymphoma. Simulation results demonstrated that abexinostat administration during the first 4 days of each week in a 3-week cycle led to a higher MDRD compared to the other administration schedules tested, with a maximum probability of 40 % of reaching these MDRDs using a 3 + 3 design. Conclusions The PKPD model was able to predict thrombocytopenia following abexinostat administration in both patient populations. A model-based approach to determine the recommended dose in phase I trials is preferable due to the imprecision of the 3 + 3 design.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The principal aim of a phase I clinical trial in oncology is to determine the recommended dose (RD) and/or the administration schedule of the investigated drug in order to continue drug development in phase II [1] Therefore, phase I clinical trial protocols must be planned rigorously as this initial stage will have considerable influence throughout drug development [2, 3]. Phase I trial designs in current use can be divided into three types: rule-based designs (e.g. the classical 3 + 3 design), model-based designs (e.g. continuous reassessment method) and designs for trials of combined agents [1, 4–8]. Of these, the classical 3 + 3 design is the most widely used [1, 4–6, 8, 9].
For cytotoxic agents, dose escalation is driven by toxicity, as the highest safe dose is assumed to be the most efficacious. This assumption is supported by preclinical models and clinical experiments [10] that have demonstrated that toxicity is a surrogate endpoint for efficacy [1]. Chemotherapy-induced myelosuppression, and subsequent thrombocytopenia (TCP), has become an increasingly major dose-limiting toxicity (DLT) [11]. TCP may lead to dose reduction or dose delay and can also cause major complications (including bleeding and death) [12]. The relationship between haemorrhage and the extent and/or duration of TCP is well known [13], but its significance can differ between solid tumour patients and patients with acute leukemia or lymphoma [14–16]. TCP is a frequent DLT of Histone Deacetylase Inhibitors (HDACi) [17–20], such as abexinostat (S-78454), a new HDACi currently at the phase II stage for the treatment of lymphoma [21]. Our previous research demonstrated that TCP was frequently observed and limited dose-escalation in two different phase I clinical studies including solid tumour patients treated by abexinostat [22]. In this previous study a semi-mechanistic pharmacokinetic/pharmacodynamic (PKPD) model of TCP [22] was built and simulations were performed using this model to define the optimal administration schedule for solid tumour patients. The amended protocol favoured a new administration schedule (4 days ON, 3 days OFF every week of a three week-cycle [4ON3OFF]), which was predicted to decrease the depth of TCP. As a result, the dose-escalation process led to the definition of a higher MTD for this new administration schedule [22]. A clinical study including lymphoma patients was also amended, but the change in administration schedule (in favour of 4ON3OFF) did not lead to a higher MTD in this population. This showed that the previous PKPD model predictions were not reliable in lymphoma patients. One explanation for this difference in the PKPD relationship could be due to pathophysiological differences in lymphoma patients compared to solid tumour patients and/or differences in sensitivity to the design or administration schedules (disease-related explanation). Another explanation could be due to the weakness of the 3 + 3 design to determine the real MTD [23–25], leading to an erroneous observed MTD in the amended study. Investigation into the platelet time-course in patients in both clinical trials (including solid tumour and lymphoma patients) showed differences between the two groups. These included smaller platelet counts at inclusion for lymphoma patients and/or a decrease in the platelet count over time compared to solid tumour patients. The first aim of our study was to refine the previously developed PKPD model describing abexinostat-induced TCP in patients with solid tumours by including lymphoma patients in the analysis, in order to accurately predict their platelet kinetics and determine the safest dosing regimen. The second aim was to simulate from the PKPD model to determine the model-derived recommended doses (MDRD) for both populations under different administration schedules. The third aim was to assess the ability of a 3 + 3 design to accurately determine the MDRD.
Materials and methods
Clinical studies
The results presented here include data from four studies (Table 1) performed in accordance with the ethical principles stated in the Declaration of Helsinki 1964, as revised in Seoul, 2008. Protocols were approved by independent ethic committees. All patients provided written informed consent before inclusion in the study.
Patients with solid tumours were included in the PCYC-402 and CL1-78454-002 phase I clinical studies [26]. Patients with lymphoma were included in the PCYC-403 and CL1-78454-001 phase I/II clinical studies.
Platelets samples and modeling datasets
Blood samples were obtained to assess haematological toxicity and in particular platelet profiles over time (Table 1). Three different datasets were selected from the present study using only data from the first three cycles of treatment (approximately 60 days), as the samples from later cycles were few and provided limited additional information. Of the 125 patients available from the four clinical studies, 95 and 30 patients were randomly selected for inclusion in the “Building dataset” and “Advanced internal evaluation dataset”, respectively; data from all patients constituted the “Final dataset” (Table 1).
Modeling strategy
Platelet-time profiles were analyzed using nonlinear mixed effect modeling (population approach) [27], with a sequential PKPD approach [28], in NONMEM 7.2 (GNU fortran 95 compiler). Empirical Bayesian Estimates (EBE) of individual PK parameters (obtained from the POSTHOC analysis in NONMEM) derived from the previously evaluated PK model [22] of abexinostat were used as the fixed PK parameters during subsequent PD modeling. PD parameters were estimated in NONMEM 7.2 using the ADVAN 6 subroutine and the FOCE-I estimation method.
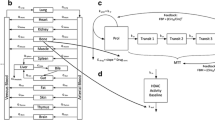
Several drug effects were tested in this model (Fig. 1) such as a linear (E Drug1 = Slope ⋅ Conc), Imax (\( {E}_{Drug2}=\frac{ IMAX\cdot Conc}{I{C}_{50}+ Conc} \)) or full sigmoid Imax model (\( {E}_{Drug3}=\frac{ IMAX\cdot Con{c}^{Hill}}{I{C}_{50}^{Hill}+ Con{c}^{Hill}} \)), where Slope is the patients’ sensitivity to abexinostat hematotoxicity (μg/mL)−1, IMAX the intrinsic activity, IC 50 the potency, Hill the sigmoidicity coefficient, and Conc the concentration in abexinostat (μg/mL). Improvements on the previous PKPD structural model (Fig. 1) were investigated using the Building dataset [22]. A second feedback mechanism affecting the mean transit time (MTT) parameter was also tested in order to quicken/slow down the bone marrow in case of thrombocytopenia/thrombocytosis: the delta feedback parameter (δ): \( MTT=\frac{\left(n+1\right)}{k_{tr}}\cdot {\left(\frac{ BASE}{ CIRC}\right)}^{\delta } \) [29, 30]. To account for the observed differences in platelet count at inclusion (BASE) between both populations, two different baseline parameters (BASE 0 ST for solid tumour patients, and BASE 0 LY for lymphoma patients) were tested. Furthermore, the slight decrease in platelets over treatment cycles observed in some patients was taken into account by the addition of disease progression (impaired bone marrow) on the baseline parameter. Several disease progression models leading to a decrease of the baseline parameter with time were tested: either a linear BASE = BASE 0 − Slope DP ⋅ t or an Imax disease progression model \( BASE= BAS{E}_0-\frac{ IMAT\cdot t}{I{T}_{50}+t} \), where BASE 0 is the baseline value at inclusion (i.e. BASE 0 LY or BASE 0 ST ) and t the time (in days) since the first abexinostat administration.

Semi-mechanistic PKPD model of thrombocytopenia, based on Friberg et al. [40]. PROL, Tr and CIRC represent the proliferation, transit and circulation compartment. BASE is the platelet value at inclusion. FBP and FBM represent the feedback on proliferation and maturation respectively. MTT is the mean transit time. the drug effect (Edrug) affects the constant rate on proliferation (kprol). ktr and kEl are the rate constants of transit and elimination respectively. to avoid identifiability issues, kprol was set to be equal to ktr and kEl. t denotes the time, in days
Inter-individual variability (IIV) was considered to have a log-normal distribution for all parameters except the IMAT parameter, for which a normal distribution was assumed. Finally, as patients were treated over several treatment cycles, inter-occasion variability (IOV) was tested on each of the PD parameters to improve the quality of the statistical model. The residual variability was modelled using a combined additive and proportional model, as this was the best description of the residual error in the original PKPD model [22].
Model selection
Discrimination between hierarchical models was based on the objective function value (OFV) of NONMEM using the Likelihood-Ratio-Test (LRT). An OFV difference greater than 3.84 corresponding to a significance level of 5 % was used to discriminate between two nested models with one parameter difference. Model development was guided by precision in parameter estimates, visual inspection of the classic goodness-of-fit plots (GOFs), and normalized prediction discrepancy errors (NPDE) [31, 32]. Five hundred simulations of the Building dataset, based on the dose regimen and sampling time points of each patient were performed using the PD parameter estimates in order to compute NPDE. These were plotted versus time and versus population prediction (PRED) in order to identify a possible bias in the model. The best model according to the LRT, acceptable parameter precision supported by the GOFs and NPDE was finally selected and named “Intermediate PKPD model” in the present work.
Advanced internal model evaluation
Individual Visual Predictive Checks (VPC) [33, 34] and NPDE [31, 32] were used to evaluate the ability of the Intermediate PKPD model to describe and predict external data. Five hundred simulations, based on the dosing regimen and sampling time points of the 30 patients included in the Advanced internal evaluation dataset, were performed using both the PK and PD parameter estimates of the Intermediate PKPD model. By definition, NPDEs follow a standard normal distribution, consequently a Kolmogorov-Smirnov test was performed [31, 32].
Final PKPD model
The Final dataset, including all patients, was analysed to re-estimate the PD parameters without modifying the structural or statistical models. NPDE versus time and versus PRED plots for each patient population and a Kolmogorov-Smirnov test were performed to evaluate the Final PKPD model.
Model-derived recommended doses (MDRD) determination for lymphoma and solid tumour patients
In a standard 3 + 3 design, if two or more patients in a cohort of maximum six patients exhibit a DLT at the dose level studied, the dose-escalation is stopped. This dose level corresponds to the maximum tolerated dose (MTD) and the dose just below is defined as being the RD for subsequent phases [1, 5, 7]. Consequently, this threshold of 33.33 % (2/6 patients) of DLT was used to determine the MDRD for different administration schedules and for each type of patient (lymphoma and solid tumours) during the simulations.
Simulations were performed using the Final PKPD model. The platelet-time course from 3000 patients with solid tumours and 3000 patients with lymphoma, treated with abexinostat over three cycles, were simulated (i.e. 500 cohorts of six patients for each population) every 20 mg starting from 20 mg up to 500 mg per day (once daily). The following administration schedules were tested:
-
14 days ON (treatment), 7 days OFF in a three-week cycle (14ON7OFF),
-
4 days ON, 3 days OFF every week of a three-week cycle (4ON3OFF),
-
5 days ON, 2 days OFF the first 2 weeks of a three-week cycle (5ON2OFF).
The sampling schedule for the platelet count was identical to that of the CL1-78454-001 clinical study (Table 1).
From the simulations the percentage of grade 4 of TCP (i.e. platelet count less than 25 × 109 platelets per litre) was computed. The MDRD was defined as the dose just below the one exceeding the 33.33 % threshold of toxicity.
DLT percentages at the MDRD in a 3 + 3 dose escalation design
Asymptotically (i.e. with an infinite number of patients) at the RD dose level, the probability of escalating to a higher dose (PRD) level is 100 %. For each administration schedule tested (i.e. 14ON7OFF, 4ON3OFF and 5ON2OFF), 500 cohorts of 6 patients with either solid tumours or lymphoma were simulated at the MDRD using the Final PKPD model. The PRD was calculated as the percentage of the 6-patient cohorts in which only zero or one patient underwent a DLT (i.e. thus leading to dose escalation).
Again, the sampling schedule for the platelet count was identical to that of the CL1-78454-001 clinical study (Table 1).
Results
Intermediate PKPD model
The Building dataset contained 925 platelet counts over the first 3 cycles of treatment in 95 patients (49 and 46 solid tumour and lymphoma patients, respectively) (Table 1). Several improvements were made to the previous PKPD model [22]. First, a feedback mechanism affecting the MTT was added to quicken/slow down the bone marrow maturation in case of thrombocytopenia/thrombocytosis (Fig. 1) [29, 30, 35, 36]. Secondly, the drug effect on the proliferation rate was best described using an Imax model, as previously described by Quartino et al. [29]. Two different baseline parameters of 203 × 109 and 274 × 109 platelets per litre for lymphoma (BASE 0 LY ) and solid tumour (BASE 0 ST ) patients were estimated, respectively. Finally, the addition of a disease progression model on the baseline parameter improved the description of the platelet-time profiles in lymphoma patients. Disease progression was best described by an Imax model; characterised by IT 50 , the time necessary to obtain half the maximum decrease (IMAT). IIV was estimated on all parameters except on Imax and IT 50 . BASE 0 ST and BASE 0 LY shared the same IIV. IOV significantly improved the model only when estimated on δ and IC 50 . The Intermediate PKPD model parameter estimates are presented in Table 2. GOFs and NPDE were satisfactory (figures not shown). The p-value of the Kolmogorov-Smirnov test was approximately 0.13 indicating that the null hypothesis of a standard normal distribution for NPDE could not be rejected.
The Intermediate PKPD model was evaluated using data from 30 additional patients (Table 1). Individual VPC were satisfactory and NPDE showed the absence of any bias in the structural model (results not shown) indicating the ability of the Intermediate PKPD model to predict the individual platelet time-course of patients treated by abexinostat. However, the p-value of the Kolmogorov-Smirnov test was 3.8 × 10−5, rejecting the null-hypothesis of normality.
Final PKPD model
Finally, the PD parameter estimates were re-estimated with the Final dataset (Table 1) containing 1217 platelet samples from 125 patients (56 and 69 lymphoma and solid tumour patients respectively) over three treatment cycles (Table 2). The Final PKPD model described the data well as shown in Fig. 2a and d NPDE versus TIME or versus PRED showed that the Final PKPD model had a good ability to describe and predict platelet time-course of both lymphoma (Fig. 2b and c) and solid tumour (Fig. 2e and f) patients treated with abexinostat. The p-value of the Kolmogorov-Smirnov test was 0.12, indicating that the null hypothesis of a standard normal distribution for the NPDE could not be rejected.

Internal evaluation of the Final PKPD model. individual platelet count predictions are shown over time for two representative lymphoma a and solid tumour d patients. open circles represent the observations and the solid lines the individual predictions. NPDE versus time and versus PRED are shown for lymphoma b, c) and solid tumour e, f) patients respectively. the dashed lines represent the 95 % prediction interval. the solid curve shows the trend curve
Model-derived recommended doses (MDRD) determination for both lymphoma and solid tumour patients
For the lymphoma patients, the MDRD were 80, 200 and 140 mg/day for the 14ON7OFF, 4ON3OFF and 5ON2OFF administration schedules, respectively (Fig. 3a). These MDRDs led to a cumulated dose of 1,120, 2,400 and 1,400 mg per cycle for the 14ON7OFF, 4ON3OFF and 5ON2OFF administration schedules, respectively. For the solid tumour patients the MDRD were 180, 440 and 280 mg/day for the 14ON7OFF, 4ON3OFF and 5ON2OFF administration schedules respectively (Fig. 3b). These MDRDs led to a cumulated dose of 2,520, 5,280 and 2,800 mg per cycle for the 14ON7OFF, 4ON3OFF and 5ON2OFF administration schedules, respectively.

Determination of the model-derived recommended doses (MRD) for both lymphoma a and solid tumour b patients. the dashed lines represent the threshold of 33.33 % (2/6 patients) of dose-limiting toxicity (DLT), which is a grade 4 thrombocytopenia. open circle, triangle and dash stand for the 5ON2OFF, 4ON3OFF and 14ON7OFF administration schedules respectively. shaded areas represent the 95 % prediction intervals
DLT percentages at the MDRD in a 3 + 3 dose escalation design
Asymptotically (i.e. with an infinite number of patients) at the RD dose level, the probability of escalating to a higher dose level is 100 %. However, simulations of a 3 + 3 design in lymphoma patients (i.e. 500 cohorts of six patients) showed that these probabilities were only 45.6, 36.6 and 36.2 % for the 14ON7OFF, 4ON3OFF and 5ON2OFF administration schedules, respectively (Fig. 4a–c). For solid tumour patients these probabilities were only 40, 39.8 and 39.6 % for the 14ON7OFF, 4ON3OFF and 5ON2OFF administration schedules, respectively (Fig. 4d–f).

DLT percentages at the MDRD for both population (lymphoma and solid tumour patients in the white and black panels respectively), for each administration schedule (14ON7OFF a. and d./4ON3OFF b. and e./5ON2OFF c. and f.) in a 3 + 3 dose-escalation. GO is for the probability to escalate the dose and STOP is the probability to stop the dose escalation
Discussion
TCP is the major DLT of HDACi such as abexinostat [17, 19, 22]. To determine the dose/toxicity relationship it is vital to both describe and predict the time-course of platelet counts in the entire study population. This study has refined the previously developed PKPD model [22] to adequately describe platelet profiles after administration of abexinostat in both patients with solid tumours and lymphoma (Fig. 1). The extended semi-mechanistic PKPD model was used to determine the MDRD for different administration schedules and patient populations using a simulation study. Finally, the ability of the 3 + 3 escalation design to properly determine the MDRD was evaluated using a simulation study.
Although the Kolmogorov-Smirnov test rejected the null-hypothesis of NPDE normality (p-value of 3.8 × 10−5) in the advanced internal evaluation step, the Intermediate PKPD model showed good predictability features after examining NPDE versus Time, PRED and QQ plots. As suggested by Comets et al. [37], the normality assumption test of the NPDE distribution is very powerful in case of a rich dataset and may be considered conservative. Indeed, the visual inspection of the NPDE graphs showed the descriptive and predictive ability of the model. In this study the Advanced internal evaluation dataset was probably too small (292 platelet counts from 30 patients) to enable a powerful Kolmogorov-Smirnov test. Finally, the PD parameters were re-estimated using the whole dataset (1217 platelet counts from 125 patients treated according to six different administration schedules), leading to the Final PKPD model. GOFs, individual plots and NPDE versus Time or PRED (Fig. 2) showed that the model described the data well. Consequently, this Final PKPD model was able to describe and predict a larger and more heterogeneous population of patients, taking into account meaningful differences between solid tumours and lymphoma patients. Lymphoma patients had a lower baseline platelet count than solid tumour patients (195 versus 273 × 109/L), possibly due to a weakening of their bone marrow. The slight decrease in platelet counts over time observed in some patients was also accounted for by adding the effect of disease progression on this baseline parameter (BASE 0 LY ). Disease progression was only observed and statistically significant in lymphoma patients. IOV from one treatment cycle to the other was added on IC50 and δ, with population estimates of approximately 10 and 5 % respectively (Table 2).
Simulations were performed using the Final PKPD model. The MDRD were determined using a simulation approach for each population and considered different administration schedules (Fig. 3). In comparison with the other administration schedules tested, the 4ON3OFF schedule was considered safer, as a higher dose could be administered to both populations of patients with an acceptable toxicity level. The duration (in days) of the first sequence of dose administration could partly explain these differences, as longer initial exposure to the treatment may lead to a stronger platelet decrease. The MDRD was approximately twice as high for the solid tumour patients compared to the lymphoma patients, despite administration schedule, reflecting the impact of the pathophysiology of lymphoma patients (lower platelet count at inclusion and decrease of platelet count over time).
The Observed Recommended Dose (ORD) associated with the 14ON7OFF administration schedule in CL1-78454-002 for patients with solid tumours was lower than the ORD associated with 4ON3OFF [22], which is consistent with Modeling & Simulations (M & S) predictions regarding their respective MDRDs (Fig. 3). In CL1-78454-001, involving lymphoma patients, the ORD were the same despite administration schedule, whereas M & S demonstrated that the MDRD associated with the 14ON7OFF administration schedule was lower than that of the 4ON3OFF (Fig. 3). The fact that this difference was not observed in a clinical setting could partly be explained by the weakness and lack of sensitivity of the 3 + 3 design to discriminate across administration schedules. Changing dose schedule from 14ON7OFF to 4ON3OFF led to an increase of the MDRD of 260 mg/day in solid tumours versus 120 mg/day lymphoma, making it easier to detect.
Although the 3 + 3 design is predominantly used in phase I clinical trials, the determination of the MTD and also the RD tends to be erroneous as a result of using this design [23–25]. Our simulation study showed that for each administration schedule and for each population (Fig. 4), the maximum probability of reaching the MDRD, assuming that the actual RD was known, was approximately 40 % (only a single step of the 3 + 3 dose-escalation design was simulated). This leads to a 60 % minimum risk of stopping the dose-escalation process just before the actual RD is reached. Consequently, with the 3 + 3 design the probability of stopping the dose-escalation at each step is high, and therefore the probability of reaching the real RD is poor. Conversely, the small number of patients included at each step can lead to an erroneously high RD if these patients happen to have a high tolerance profile. In a 3 + 3 rule-based design, the decision to go up or to stop the dose escalation is only based on the information extracted from 3 to 6 patients at a particular dose level [1, 5, 38, 39]. Therefore, M & S is a useful tool to support the classical 3 + 3 design to determine the dose-toxicity relationship and guide the clinical development of a drug by taking into account all the information obtained throughout the dose-escalation process. The determination of the MDRD through simulations can be performed only after having built a PKPD model, and thus relies on the availability of PK and PD data. Nevertheless, such a model-based approach could be used at the end of a classical 3 + 3 dose escalation in order to determine the MDRD.
Conclusion
TCP is the major dose-limiting toxicity of abexinostat in phase I clinical trials. The previous semi-mechanistic PKPD model of TCP in solid tumour patients treated by abexinostat [22] has been refined to predict the dose-toxicity relationship in a larger and more heterogeneous population including lymphoma patients. This PKPD model resulted in the development of an optimal administration schedule (4 days ON, 3 days OFF every week of a three week-cycle) associated with a model-based recommended dose of abexinostat of 200 and 440 mg per day for lymphoma and solid tumour patients, respectively. The weakness of the 3 + 3 design to properly determine the MTD, and therefore the RD, was demonstrated. The results from this study have demonstrated the value of a model-based approach in the dose-escalation process to investigate the RD for future phase II trials.
References
Eisenhauer EA, O’Dwyer PJ, Christian M, Humphrey JS (2000) Phase I clinical trial design in cancer drug development. J Clin Oncol 18:684–692
Le Tourneau C, Faivre S, Raymond E, Dieras V (2007) Phase I cancer trials methodology. Bull Cancer 94:943–951
Ratain MJ, Mick R, Schilsky RL, Siegler M (1993) Statistical and ethical issues in the design and conduct of phase I and II clinical trials of new anticancer agents. J Natl Cancer Inst 85:1637–1643
Cannistra SA (2008) Challenges and pitfalls of combining targeted agents in phase I studies. J Clin Oncol 26:3665–3667
Le Tourneau C, Lee JJ, Siu LL (2009) Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst 101:708–720
Simon R, Freidlin B, Rubinstein L, Arbuck SG, Collins J, Christian MC (1997) Accelerated titration designs for phase I clinical trials in oncology. J Natl Cancer Inst 89:1138–1147
Storer BE (1989) Design and analysis of phase I clinical trials. Biometrics 45:925–937
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van GM, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst 92:205–216
Storer BE (2001) An evaluation of phase I clinical trial designs in the continuous dose–response setting. Stat Med 20:2399–2408
Von Hoff DD, Turner J (1991) Response rates, duration of response, and dose response effects in phase I studies of antineoplastics. Invest New Drugs 9:115–122
Bhatia M, Davenport V, Cairo MS (2007) The role of interleukin-11 to prevent chemotherapy-induced thrombocytopenia in patients with solid tumors, lymphoma, acute myeloid leukemia and bone marrow failure syndromes. Leuk Lymphoma 48:9–15
Elting LS, Rubenstein EB, Martin CG, Kurtin D, Rodriguez S, Laiho E, Kanesan K, Cantor SB, Benjamin RS (2001) Incidence, cost, and outcomes of bleeding and chemotherapy dose modification among solid tumor patients with chemotherapy-induced thrombocytopenia. J Clin Oncol 19:1137–1146
Elting LS, Martin CG, Kurtin DJ, Cantor SB, Rubenstein EB, Rodriguez S, Kanesan K, Vadhan-Raj S, Benjamin RS (2002) The bleeding risk index: a clinical prediction rule to guide the prophylactic use of platelet transfusions in patients with lymphoma or solid tumors. Cancer 94:3252–3262
Belt RJ, Leite C, Haas CD, Stephens RL (1978) Incidence of hemorrhagic complications in patients with cancer. JAMA 239:2571–2574
Dutcher JP, Schiffer CA, Aisner J, O’Connell BA, Levy C, Kendall JA, Wiernik PH (1984) Incidence of thrombocytopenia and serious hemorrhage among patients with solid tumors. Cancer 53:557–562
Gaydos, L. A., Freireich, E. J., Mantel, N. (3-5-1962) The quantitative relation between platelet count and hemorrhage in patients with acute leukemia. N Engl J Med 266:905–909
Bishton MJ, Harrison SJ, Martin BP, McLaughlin N, James C, Josefsson EC, Henley KJ, Kile BT, Prince HM, Johnstone RW (2011) Deciphering the molecular and biologic processes that mediate histone deacetylase inhibitor-induced thrombocytopenia. Blood 117:3658–3668
Marks PA, Xu WS (2009) Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem 107:600–608
Matsuoka H, Unami A, Fujimura T, Noto T, Takata Y, Yoshizawa K, Mori H, Aramori I, Mutoh S (2007) Mechanisms of HDAC inhibitor-induced thrombocytopenia. Eur J Pharmacol 571:88–96
Prince HM, Bishton MJ, Harrison SJ (2009) Clinical studies of histone deacetylase inhibitors. Clin Cancer Res 15:3958–3969
Ali A, Bluteau O, Messaoudi K, Palazzo A, Boukour S, Lordier L, Lecluse Y, Rameau P, Kraus-Berthier L, Jacquet-Bescond A, Lelievre H, Depil S, Dessen P, Solary E, Raslova H, Vainchenker W, Plo I, Debili N (2013) Thrombocytopenia induced by the histone deacetylase inhibitor abexinostat involves p53-dependent and -independent mechanisms. Cell Death Dis 4:e738
Chalret du Rieu Q, Fouliard S, Jacquet-Bescond A, Robert R, Kloos I, Depil S, Chatelut E, Chenel M (2013) Application of hematological toxicity modeling in clinical development of abexinostat (S-78454, PCI-24781). a new histone deacetylase inhibitor. Pharm Res 30:2640–2653
Faries D (1994) Practical modifications of the continual reassessment method for phase I cancer clinical trials. J Biopharm Stat 4:147–164
He W, Liu J, Binkowitz B, Quan H (2006) A model-based approach in the estimation of the maximum tolerated dose in phase I cancer clinical trials. Stat Med 25:2027–2042
Smith TL, Lee JJ, Kantarjian HM, Legha SS, Raber MN (1996) Design and results of phase I cancer clinical trials: three year experience at M.D. anderson cancer center. J Clin Oncol 14:287–295
Fouliard S, Robert R, Jacquet-Bescond A, du Rieu QC, Balasubramanian S, Loury D, Loriot Y, Hollebecque A, Kloos I, Soria JC, Chenel M, Depil S (2013) Pharmacokinetic/pharmacodynamic modelling-based optimisation of administration schedule for the histone deacetylase inhibitor abexinostat (S78454/PCI-24781) in phase I. Eur J Cancer 49:2791–2797
Sheiner LB, Beal SL (1980) Evaluation of methods for estimating population pharmacokinetics parameters. I. michaelis-menten model: routine clinical pharmacokinetic data. J Pharmacokinet Biopharm 8:553–571
Zhang L, Beal SL, Sheiner LB (2003) Simultaneous vs. sequential analysis for population PK/PD data I: best-case performance. J Pharmacokinet Pharmacodyn 30:387–404
Quartino AL, Friberg LE, Karlsson MO (2012) A simultaneous analysis of the time-course of leukocytes and neutrophils following docetaxel administration using a semi-mechanistic myelosuppression model. Invest New Drugs 30:833–845
Van Kesteren C, Zandvliet AS, Karlsson MO, Mathot RA, Punt CJ, Armand JP, Raymond E, Huitema AD, Dittrich C, Dumez H, Roche HH, Droz JP, Ravic M, Yule SM, Wanders J, Beijnen JH, Fumoleau P, Schellens JH (2005) Semi-physiological model describing the hematological toxicity of the anti-cancer agent indisulam. Invest New Drugs 23:225–234
Brendel K, Comets E, Laffont C, Laveille C, Mentre F (2006) Metrics for external model evaluation with an application to the population pharmacokinetics of gliclazide. Pharm Res 23:2036–2049
Brendel K, Comets E, Laffont C, Mentre F (2010) Evaluation of different tests based on observations for external model evaluation of population analyses. J Pharmacokinet Pharmacodyn 37:49–65
Lavielle M, Bleakley K (2011) Automatic data binning for improved visual diagnosis of pharmacometric models. J Pharmacokinet Pharmacodyn 38:861–871
Post TM, Freijer JI, Ploeger BA, Danhof M (2008) Extensions to the visual predictive check to facilitate model performance evaluation. J Pharmacokinet Pharmacodyn 35:185–202
Hitchcock, I. S., Kaushansky, K. (2014) Thrombopoietin from beginning to end. Br J Haematol.
Kaushansky K (2003) Thrombopoietin: a tool for understanding thrombopoiesis. J Thromb Haemost 1:1587–1592
Comets E, Brendel K, Mentre F (2008) Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput Methods Programs Biomed 90:154–166
Ji Y, Li Y, Nebiyou BB (2007) Dose-finding in phase I clinical trials based on toxicity probability intervals. Clin Trials 4:235–244
Ji Y, Wang SJ (2013) Modified toxicity probability interval design: a safer and more reliable method than the 3 + 3 design for practical phase I trials. J Clin Oncol 31:1785–1791
Friberg LE, Henningsson A, Maas H, Nguyen L, Karlsson MO (2002) Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J Clin Oncol 20:4713–4721
Acknowledgments and disclosures
Authors would like to thank Pharmacyclics for providing data from the PCYC-402 and the PCYC-403 clinical studies.
Author’s disclosures of potential conflicts of interest: Quentin Chalret du Rieu, Sylvain Fouliard, Ioana Kloos and Marylore Chenel are employed by Institut de Recherches Internationales Servier. The other author (s) indicated no potential conflicts of interest.
This work was integrated in a Ph. D. project (Quentin Chalret du Rieu), granted by Institut de Recherches Internationales Servier.
We thank Ms Katie Owens for her editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chalret du Rieu, Q., Fouliard, S., White-Koning, M. et al. Pharmacokinetic/Pharmacodynamic modeling of abexinostat-induced thrombocytopenia across different patient populations: application for the determination of the maximum tolerated doses in both lymphoma and solid tumour patients. Invest New Drugs 32, 985–994 (2014). https://doi.org/10.1007/s10637-014-0118-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-014-0118-1