
Abstract
Purpose
This study aimed at recommending pediatric dosages of the histone deacetylase (HDAC) inhibitor vorinostat and potentially more effective adult dosing regimens than the approved standard dosing regimen of 400 mg/day, using a comprehensive physiologically based pharmacokinetic/pharmacodynamic (PBPK/PD) modeling approach.
Methods
A PBPK/PD model for vorinostat was developed for predictions in adults and children. It includes the maturation of relevant metabolizing enzymes. The PBPK model was expanded by (1) effect compartments to describe vorinostat concentration–time profiles in peripheral blood mononuclear cells (PBMCs), (2) an indirect response model to predict the HDAC inhibition, and (3) a thrombocyte model to predict the dose-limiting thrombocytopenia. Parameterization of drug and system-specific processes was based on published and unpublished in silico, in vivo, and in vitro data. The PBPK modeling software used was PK-Sim and MoBi.
Results
The PBPK/PD model suggests dosages of 80 and 230 mg/m2 for children of 0–1 and 1–17 years of age, respectively. In comparison with the approved standard treatment, in silico trials reveal 11 dosing regimens (9 oral, and 2 intravenous infusion rates) increasing the HDAC inhibition by an average of 31%, prolonging the HDAC inhibition by 181%, while only decreasing the circulating thrombocytes to a tolerable 53%. The most promising dosing regimen prolongs the HDAC inhibition by 509%.
Conclusions
Thoroughly developed PBPK models enable dosage recommendations in pediatric patients and integrated PBPK/PD models, considering PD biomarkers (e.g., HDAC activity and platelet count), are well suited to guide future efficacy trials by identifying dosing regimens potentially superior to standard dosing regimens.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Histone deacetylases (HDACs) regulate gene transcription and cell signaling pathways. Changes in the structure, activity, or expression of HDACs can result in abnormal gene transcription and cell signaling leading to cancer [1]. HDAC inhibitors (HDACi), such as vorinostat, show antineoplastic effects in vitro and in vivo, and offer therefore a new approach in chemotherapy [2]. Vorinostat is a fast, light-binding inhibitor with short residence time at the target that inhibits the enzymatic activity of Class I (HDACs 1–3) and Class II (HDAC 6) HDACs at nanomolar concentrations (IC50 = 30–86 nM) [3, 4]. It was approved by the U.S. Food and Drug Administration (FDA) for the treatment of cutaneous manifestations in patients with cutaneous T-cell lymphoma (CTCL) who have progressive, persistent, or recurrent disease on, or following two systemic therapies [4, 5].
Vorinostat shows a dose-proportional exposure increase after oral or intravenous doses of 100–800 mg or 75–900 mg/m2, respectively, with an absorption rate-limited drug disposition in the gastrointestinal tract after oral dosing, leading to flip-flop pharmacokinetics (PK) [6]. Elimination primarily comprises metabolism, involving glucuronidation and hydrolysis followed by β-oxidation without the contribution of CYP enzymes [7]. Renal excretion is negligible accounting for ~ 1% of total body clearance [8]. While the UDP-glucuronosyltransferases (UGTs) 1A9, 2B7, and 2B17 are the major enzymes of vorinostat glucuronidation, the enzyme responsible for hydrolysis and β-oxidation remains unidentified [7]. These enzymes exhibit nonlinear age-dependent maturations completed within 10 years after birth [9,10,11,12,13,14]. Genetic polymorphisms of UGT 2B17 might play a role in the clearance of vorinostat and in clinical outcomes [15,16,17]. In general, vorinostat exhibits a short half-life of 1 (intravenous) to 2 h (oral) and multiple-dose PK similar to single-dose administration [7].
Clinical studies of vorinostat in patients with stage Ib and higher CTCL and in patients with refractory CTCL demonstrated overall objective responses of 30 and 31%, respectively [18, 19]. The most common adverse reactions, with an incidence ≥ 10%, associated with vorinostat treatment are anorexia, diarrhea, dysgeusia, fatigue, nausea, and thrombocytopenia, with thrombocytopenia being the most common hematologic event [5, 20]. In pediatric studies, vorinostat was well tolerated at 230 mg/m2/day (alone or in combination with bortezomib) or 300 mg/m2/day (in combination with temozolomide) and showed a drug disposition similar to that observed in adults [21,22,23].
While in vitro experiments suggest that vorinostat concentrations of 2.5 µmol/l lead to the maximum accumulation of acetylated histones, it has also been shown that the HDAC inhibition has to be maintained over a significant period of time to show antitumor activity [24, 25]. Furthermore, vorinostat enhances the effect of other chemotherapeutics such as cisplatin and gemcitabine at concentrations > 2 µmol/l, which is, nevertheless, inconsistently achieved in patients at the approved 400 mg/day (qd) dose [6, 26, 27]. Little is known about the impact of different dosing regimens on its efficacy [6]. Hence, Dickson and co-workers attempted to achieve maximum vorinostat concentrations (C max) of > 2.5 µmol/l by an intermittent oral pulse dosing protocol of vorinostat in combination with the cyclin-dependent kinase inhibitor flavopiridol [28]. In this attempt, the C max of vorinostat could be increased, but unfortunately the incidence of myelosuppression was also increased. However, these promising results demand further assessment of alternative vorinostat dosing strategies that might show more effectiveness and still tolerable toxicity compared to the vorinostat standard treatment.
Here, physiologically based pharmacokinetic and pharmacodynamic (PBPK/PD) modeling and simulation enable the assessment of dosing regimens in in silico trials, while also being able to test clinical trial designs.
Objectives
-
1.
Build and evaluate an adult whole-body PBPK model of vorinostat able to describe and predict the PK of varying doses of intravenously and orally administered vorinostat.
-
2.
Develop and evaluate a pediatric PBPK model for vorinostat and estimate vorinostat doses for children between 0 and 17 years.
-
3.
Build and evaluate a PBPK/PD model incorporating (1) a HDAC inhibition model and (2) a thrombocytopenia model.
-
4.
Identify potentially effective vorinostat dosing regimens while considering HDAC activity and the number of circulating thrombocytes.
-
5.
Perform parameter sensitivity analysis for the developed whole-body PBPK model of vorinostat.
Materials and methods
Model development and evaluation
A schematic overview of the PBPK model development and evaluation steps is shown in supplemental Fig. S1. The detailed steps of the model building are outlined in the following sections. In brief, PBPK modeling was performed in a stepwise procedure. First, an intravenous model of vorinostat was developed for adults. Second, an oral PBPK model for adults was established. Third, age-dependent physiological and anatomical changes were implemented enabling pediatric predictions and dose estimations. Fourth, PD models were incorporated into the PBPK model to describe HDAC inhibition and thrombocytopenia. Fifth, the PBPK/PD model was used to identify new dosing regimens. Finally, a parameter sensitivity analysis was performed.
Experimental datasets from 13 published and unpublished clinical studies were used to support the PBPK model development and evaluation (Table 1). The experimental datasets were split into an internal development (n = 4 trials) dataset and an external evaluation dataset (n = 9), such that wide dose ranges were covered in both datasets. The PBPK model was fitted to the development dataset and the prediction quality of the model was assessed by predicting the evaluation dataset. Parameter optimizations were carried out using either a Monte-Carlo or Nelder–Mead method [29]. An overview of fitted parameters and the applied optimization method is given in supplemental Table S1. The Monte-Carlo approach was used if no initial information on parameter values was available.
Model evaluation was carried out using (1) comparison of observed and simulated PK parameters and the fraction of vorinostat dose excreted unchanged into urine, (2) mean prediction errors (MPE) of pharmacokinetic profiles [37] and (3) visual predictive checks (VPC), which are tools for evaluating the performance of PK and PD models, where percentiles of experimental and simulated data are compared [38]. PK parameter comparisons were deemed successful when they satisfied the twofold acceptance criterion.
Simulations were carried out using virtual PBPK individuals and populations. Virtual PBPK populations were created as described by Willmann and co-workers using ranges for age, height, and weight reported in the internal and external datasets [39]. In this approach, the organ weights and the cardiac output (CO) of a target individual are allometrically scaled with height and the organ blood flows are scaled to the total CO [39]. To assess the inter-study variability of PK parameters, 10 virtual populations were created for each study. Applied dosing schedules and doses were used as published. The parameters of anatomy and physiology were varied as pre-defined in PK-Sim [40,41,42,43,43], parameters of the pharmacodynamic models were varied with a coefficient of variation (CV) of 20%.
An overview of important assumptions made during model development and evaluation is given in supplemental Table S2.
PBPK simulations were carried out using PK-Sim and MoBi (Version 6.3.2, Bayer Technology Services, Leverkusen, Germany). Statistical analyses of the results and graphics were compiled using MATLAB (Version R2013b, The MathWorks Inc., Natick, MA, USA).
Adult PBPK model—intravenous
Intravenous concentration–time profiles were used to establish a PBPK model for vorinostat. The partition coefficients for vorinostat were calculated using the PK-Sim standard model [44, 45]. To describe the distribution phase correctly, the cellular permeability of vorinostat was estimated. The reference concentrations for UGT 1A9, 2B7, and 2B17 were kept at the PK-Sim default values of 1 µmol/l, respectively. The UGT Vmax values were gathered from the literature. The PK-Sim gene expression database was used to distribute the enzymes to the specific organs [46, 47]. To fit the vorinostat half-life and concentration–time profiles, the unidentified hydrolytic and oxidative enzyme were implemented as a generic enzyme with a reference concentration of 1 µmol/l assuming similar availability across organs. The clearance value for hydrolysis and oxidation was fitted. All metabolic reactions were modeled as first-order processes. The unbound vorinostat was assumed to be filtered and excreted with the glomerular filtration rate (GFR).
Adult PBPK model—oral
The developed intravenous PBPK model of vorinostat was expanded by an oral formulation. The particle dissolution module of PK-Sim was used to describe vorinostat drug release in the intestine. This module calculates the dissolution kinetics of spherical particles with a predefined particle size distribution based on the Noyes–Whitney approach [48]. To describe the absorption of vorinostat correctly, the intestinal permeability coefficient was estimated. To capture the whole-blood-to-plasma ratio of 2.87, the red blood cell permeability was estimated [50].
Model evaluation of the intravenous and oral adult PBPK model was carried out comparing observed and predicted PK parameters and concentration–time profiles of the external evaluation dataset.
Pediatric PBPK model and dosage recommendations
The developed oral PBPK model of vorinostat was expanded by importing available age-dependent anatomical and physiological changes in human development from the PK-Sim databases [40]. The age-dependent maturation of UGT 1A9 and 2B7 was already available in PK-Sim, whereas the UGT 2B17 maturation was implemented using a Hill function (Eq. 1) based on recently published mRNA data [14],
where OFmin denotes the intercept with the y-axis, and OFmax and OF50 represent the maximum adult ontogeny factor (= 1) and the post-menstrual age needed to reach 50% of the maximum ontogeny factor, respectively. Age denotes the post-menstrual age in years and γ denotes the Hill factor. The unidentified enzyme responsible for the hydrolysis and oxidation of vorinostat was assumed to be fully developed from birth on.
Model evaluation of the pediatric PBPK model was carried out comparing observed and predicted PK parameters and concentration–time profiles of the external evaluation dataset that consisted of 13 individual vorinostat plasma concentration–time profiles with individual ages ranging from 4.7 to 22 years.
The developed pediatric PBPK model of vorinostat was used to identify age-stratified mg/m2/day doses for children and adolescents from birth to 17 years of age. Pediatric doses were selected when the 25th, 50th, and 75th percentiles of predicted AUC0 − inf or C max values for the pediatric age groups were comparable to the values of the adult population dosed with the approved oral dose of 400 mg/day. Two virtual populations were generated, first, for adults (age ≥ 18, n = 500) and second, for children (age 0–17, n = 10,000). The body surface areas were calculated using the DuBois equation [49].
Adult PBPK model—HDAC activity
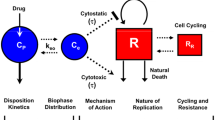
The oral PBPK model of vorinostat was expanded by an indirect response model to describe the HDAC activity in peripheral blood mononuclear cells (PBMCs) during vorinostat treatment [6]. PBMCs are not offered as physiological compartments in the default PK-Sim PBPK model [50]. Therefore, two PBMC effect compartments were implemented in the default model (Fig. 1). First, the rate constants k effect,1, k effect,2, k effect,deep,1, and k effect,deep,2 of the PBMC effect compartmental model were estimated. Second, the HDAC activity in the central PBMC compartment was modeled using an indirect response model, where the HDAC activity R was described by Eq. 2 [51],

Schematic structural PBPK/PD model representation. a PBMC effect compartment model. b Schematic default PBPK model. c Thrombocytopenia model. d HDAC activity indirect response model. PBMC peripheral blood mononuclear effect compartment, PBMC deep peripheral blood mononuclear deep effect compartment, k effect,1/2 and k effect,deep,1/2 concentration transfer rate constants, Q specific organ blood flow, CL clearance, Prol proliferating cells, Transit 1/2/3 cells in transit state, Circ circulating cells, Circ 0 circulating cells baseline, k prol proliferation rate constant, k tr transit rate constant, k circ elimination rate constant, MTT mean transit time, slope slope parameter, Drug conc vorinostat concentration, E drug vorinostat effect on the proliferating cells, FBP circulating cells feedback on proliferating cells, FBM circulating cells feedback on MTT, k in HDAC activity synthesis rate, and k out HDAC activity degradation rate
where kin is the apparent zero-order rate constant for the production of the activity, k out represents the first-order rate constant for activity loss, and R is assumed to be stationary with an initial value of \({R_0}\left( {=\frac{{{k_{{\text{in}}}}}}{{{k_{{\text{out}}}}}}} \right)\). The SC50 value represents the PBMC vorinostat concentration (C V,PBMC) producing 50% of the maximum k out stimulation achieved at the effect site, whereas S max is the maximum k out stimulation achieved at the effect site. Predictions of HDAC activity following multiple oral vorinostat doses were carried out using intracellular vorinostat concentrations of the bone instead of PBMC vorinostat concentrations.
Model evaluation of the HDAC activity was carried out by comparing the observed and simulated HDAC activity-time profiles of the internal development dataset using a virtual population.
Adult PBPK model—platelet count
Thrombocytopenia was defined as a platelet count < 150 × 109 cells/l [52]. The adult PBPK model of vorinostat was expanded by a thrombocytopenia model as a pharmacodynamic toxicity biomarker. The platelet model was first published and comprehensively described by Friberg and co-workers and refined by du Rieu and co-workers [53, 54]. The refined thrombocytopenia model was used in the presented PBPK modeling approach (Fig. 1).
In brief, the thrombocytopenia model consists of five compartments (cmts) describing proliferating cells (1 cmt), developing thrombocytes (3 transit cmts) and circulating thrombocytes (1 cmt). Both, the mean transit time (MTT) of the developing thrombocytes and the proliferation rate constant are influenced by the number of circulating thrombocytes by a feedback mechanism. Vorinostat reduces the proliferation or induces cell loss of thrombocytes.
Parameter values of the thrombocytopenia model were gathered from the literature (suppl. Table S3), except for the slope parameter of vorinostat, which was estimated using clinical study data of thrombocytosis patients [36].
The thrombocytopenia model was evaluated by comparing the observed and predicted fraction of patients with normal platelet counts that develop thrombocytopenia during a 24-week treatment with 400 mg vorinostat per day.
Identification of new vorinostat dosing regimens
The adult PBPK/PD model of vorinostat, including the HDAC activity and the thrombocytopenia model was used in in silico trials to identify vorinostat doses and dosing schedules that are potentially more effective than the standard dosing regimen of 400 mg qd, where ‘more effective’ is defined as causing a larger decrease in HDAC activity. Tested dosing regimens were chosen from a range of potential dosing frequencies per day (qd, bid, tid) and week (e.g. every 2nd week, 3 consecutive days per week). Intravenous and oral routes of administration were considered.
In silico trials—part 1
25 dosing regimens with varying administration routes, administered doses, and dosing schemes of vorinostat were tested. New regimens were declared potentially more effective when they showed higher maximum (HDACactivity,bone [%]) and longer HDAC inhibition than the standard treatment during a 10-week treatment period without decreasing the thrombocyte count [109 cells/l] more than the standard dosing regimen. A simulation time of 10 weeks was necessary to ensure that the circulating thrombocytes were at steady-state (MTT ~ 95 h).
In silico trials—part 2
Maximum doses were estimated for 15 dosing schedules from in silico trials – part 1. In trial 2, the minimum thrombocyte count was set at 50 × 109 cells/l and the maximum possible single vorinostat dose was set to 3000 mg.
All trial results were compared to the standard treatment based on the change of HDAC activity (%), the change in the accumulated time of HDAC activity < 50% (%), and the change in the thrombocyte count (%).
Parameter sensitivity analysis
A parameter sensitivity analysis of the adult PBPK model was performed to assess the impact of estimated model parameters on the predicted values of AUC0 − inf, C max, half-life, and T max. Assessed parameters were: hydrolysis and β-oxidation clearance, intestinal permeability, cellular permeability, red blood cell permeabilities, and the unstirred layer of the particle dissolution. All parameters were varied individually over a wide range of values (via multiplication by factors between 0.1 and 10.0) and the changes in AUC0 − inf, C max, half-life, and T max was documented [55]. If a 10% change of a single parameter led to a > 1% change in AUC0 − inf, C max, half-life, and T max, the model was considered sensitive to this parameter [56].
Results
A comprehensive PBPK model for the prediction of vorinostat concentrations after different intravenous and oral doses of vorinostat has been successfully developed. The model can be applied in adult and pediatric patients and has been used to recommend doses for children from 0 to 17 years. Furthermore, the PBPK model has been expanded by (1) PBMC effect compartments and (2) pharmacodynamic models of HDAC activity and thrombocytopenia. Finally, the vorinostat PBPK/PD model has been successfully used to identify new vorinostat dosing regimens that are potentially more effective than the approved standard treatment.
The developed vorinostat PBPK model shows good descriptive and predictive performance. In summary, 90 and 96% of the predicted AUC0 − inf and C max values satisfy the twofold acceptance criterion, respectively (Fig. 2, suppl. Fig. S2). The MPE of the simulated and predicted adult and pediatric pharmacokinetic profiles is 44%. Trial-specific MPEs are summarized in supplemental Table S4. The final model parameters on the drug release, absorption, distribution, metabolism, and excretion of vorinostat are shown in Table 2.

Predicted versus observed PK parameters. a AUC0 − inf in adults (n = 52). b Cmax in adults (n = 52). c AUC0 − inf in children (n = 19). Circles indicate adults and triangles children. Dashed line: twofold acceptance limits
Adult PBPK model—intravenous
The intravenous model is able to describe and predict vorinostat concentration–time profiles in close agreement with the observed values over a wide range (75–900 mg/m2) of intravenous single dosages (Fig. 3, suppl. Fig. S3). The mean observed dose-independent half-life of vorinostat across available doses and studies is 0.65 h [4, 30] where the PBPK model predicts 0.75 h, a slight over-prediction of 15%. The C max and the AUC0 − inf have been predicted with mean deviations of 4 and 25% from the observed values, respectively [4, 30]. The simulated fraction of unchanged vorinostat dose excreted in urine (f e) has been consistently below 1% over all doses, which is also in good agreement with observed values [4].

Adult PBPK model—oral
The oral PBPK model also shows a good descriptive and predictive performance (Fig. 3, suppl. Fig. S3). The AUC0 − inf, C max, and T max have been predicted with mean deviations of 5, 1 and 22% from the observed values across available doses, respectively [6, 8, 28, 31,32,33,34,35,35, 50]. The absorption rate-limited PK is well predicted. The observed oral half-life is 1.75 h [6, 8, 28, 31,32,33,34,35,35, 50], where the PBPK model predicts a half-life of 1.66 h. The predicted bioavailability \(\left( {{\text{F}}_{{{\text{pred}}}} {\text{ = }}\frac{{{\text{AUC}}_{{{\text{oral,pred}}}} }}{{{\text{AUC}}_{{{\text{IV,pred}}}} }}{\text{ }} \times {\text{ }}\frac{{{\text{Dose}}_{{{\text{oral,pred}}}} }}{{{\text{Dose}}_{{{\text{IV,pred}}}} }}} \right)\) is comparable to the observed bioavailability (F obs) with F pred = 30% and F obs = 40% [6]. The contribution of the implemented metabolic clearance processes via UGT 1A9, 2B7, 2B17, and hydrolysis/oxidation to the overall metabolized fraction (f m) has been estimated at 3, 56, 11, and 30%, respectively.
Pediatric PBPK model and dosage recommendations
The age-dependent maturation of UGT 2B17 has been successfully described and implemented using a Hill function with final parameter values of OFmax = 0.46, OF50 = 6.0 years, and γ = 1.63, and OFmin = 0.58 (suppl. Fig. S4). The maturation time of the primary metabolizing enzymes of vorinostat can be ranked as follows: UGT 2B17 (longest) > 1A9 > 2B7. Given that there is (1) no clear relationship between pediatric doses and exposures and (2) no inter-individual variability estimated, the individually predicted vorinostat concentration–time profiles are in adequate agreement with the observed data in the age range between 4.7 and 22.0 years (suppl. Fig. S5).
Because the, so far, proposed pediatric dosage of 230 mg/m2/day leads to very high exposure in children between 0 and 1 years (suppl. Fig. S6a), the pediatric PBPK model has been used to identify dosages for all age groups from 0 to 17 years leading to Cmax values similar to the ones in adults. The recommended dosages are 80 mg/m2 for children from 0 to 1 year and 230 mg/m2 for children and adolescents between 1 and 17 years (suppl. Fig. S6b).
Adult PBPK model—HDAC activity
The default PK-Sim PBPK model has been successfully expanded by two effect compartments describing PBMC concentrations and by an indirect response model of HDAC activity. The PBMC concentration–time profiles are best described using a two-compartment model (Fig. 4a). A comparison between vorinostat PBMC concentration–time profiles simulated using a 1- or 2-compartmental model is depicted in supplemental Fig. S7. The observed half-life of oral vorinostat in PBMCs is 5.30 h, 3.0-fold longer than observed in plasma, and the predicted half-life in PBMCs (2-cmt) is 5.66 h, 3.4-fold longer than predicted in plasma [60]. The indirect response model is able to accurately describe the HDAC inhibition caused by a single dose of 400 mg vorinostat (Fig. 4b). The minimum observed and predicted HDAC activities are 21.6 and 26.6%, respectively [60]. The final model parameters of both the PBMC effect compartment model and the HDAC indirect response model are given in supplemental Table S5. The structural model alterations of the default PK-Sim PBPK model are depicted in Fig. 1.

PBPK/PD model results. a PBMC concentration–time profiles [50]. b HDAC activity over time [50]. c Thrombocyte count in thrombocytosis patients during a 24-week vorinostat treatment with 400 mg qd [36]. d Thrombocyte count in patients with normal thrombocyte counts. Simulated data are represented as median (black line), 90% prediction interval (gray shaded area) and minimum and maximum values (dotted lines). The red dotted line represents the thrombocyte counts justifying the diagnosis of thrombocytopenia (= 150 × 109 cells/l). Single doses: sd, repeated once daily doses: qd
Adult PBPK model—platelet count
The PBPK model has been successfully expanded by the thrombocytopenia model. The integrated PBPK/PD model is able to capture the thrombocyte count over a 36-week standard vorinostat treatment period in a thrombocytosis population (Fig. 4c). In a population with normal thrombocyte counts, the model has successfully predicted the thrombocytopenia prevalence of 27.9% which is in close agreement with the published observed value of 25.6% [61]. The model parameter values used in the simulations are compiled in supplemental Table S3. The manually estimated slope parameter for vorinostat is 0.22 µM− 1.
Identification of new vorinostat dosing regimens
The adult PBPK/PD model of vorinostat including the HDAC activity and the thrombocytopenia model has been successfully used in in silico trials to identify new vorinostat doses and dosing schedules which are potentially more effective than the standard dosing regimen of 400 mg qd.
Although part 1 suggests that none of the tested 25 dosing regimens offers a higher maximum and/or prolonged HDAC inhibition when the target thrombocyte count is that of the approved standard treatment (~ 193 × 109 cells/l), the alternative treatment D (800 mg, every 2nd day) might be a replacement of the standard treatment showing a 14% higher maximum HDAC inhibition but a 10% shorter HDAC inhibition time (suppl. Table S6).
However, part 2 reveals that 11 (9 oral, and 2 infusions) out of the 15 tested dosing schedules offer higher maximum and prolonged HDAC inhibitions (suppl. Table S7). Compared to the standard treatment, the potentially beneficial dosing regimens exhibit a mean maximum HDAC inhibition that is 27% higher (min: 16%, max: 62%) and the mean accumulated time of the HDAC inhibition being > 50% is 180% longer (min: 6%, max: 509%). Based on the accumulated time of HDAC inhibition, trial C with three daily doses of the maximum 3000 mg dose has shown the best results with a HDAC inhibition time increase of 509% and a platelet decrease of − 72% which is close to the lower thrombocyte limit at − 74%.
Parameter sensitivity analysis
The parameter sensitivity analysis (suppl. Fig. S8) reveals that, among the tested parameters, the intestinal permeability, the clearance via hydrolysis and oxidation, and the unstirred layer of the particle dissolution have the biggest impact on the tested vorinostat PK parameters AUC0 − inf, C max, half-life, and T max.
Discussion
A PBPK model for the prediction of vorinostat concentrations after different intravenous and oral vorinostat doses has been successfully developed. The model was able to accurately predict vorinostat concentrations in adult and pediatric patients and to recommend dosages for different pediatric age groups. After successful implementation of PBMC effect compartments, PD models of HDAC activity (measured in PBMCs) and thrombocytopenia were integrated in the PBPK model. The final integrated PBPK/PD model of vorinostat was successfully applied in in silico trials, thereby identifying potentially more effective vorinostat dosing regimens in comparison with standard treatment.
The cellular permeability drives the permeation rate of vorinostat across the interstitial—intracellular membrane in the PK-Sim PBPK model. The permeability was initially calculated within PK-Sim according to an empirically derived formula, using logMA and molecular weight as input variables, based on the procedure presented by Kawai and co-workers [62]. However, using the calculated cellular permeability value, the plasma concentration–time profiles of vorinostat were under-predicted. With the estimated permeability value, the model successfully predicted the intravenous profiles of the evaluation dataset.
In vitro tests found that UGT 2B17 is the major metabolizing enzyme of vorinostat. However, by compiling all available metabolic data of UGT 1A9, 2B7, and 2B17 in the model, the PBPK analysis suggested that UGT 2B17 is only of minor importance for the vorinostat clearance in vivo. This has important implications for the evaluation of other studies aiming to assess the impact of UGT 2B17 polymorphisms on survival of patients treated with vorinostat. Furthermore, it was reported that the UGT 2B17*2 polymorphism, which leads to reduced vorinostat glucuronidation, was associated with a longer progression-free survival in Asian breast cancer patients [17]. Although, this polymorphism is present in 92% of Asian populations, based on the presented modeling results, it seems unlikely that the reported results are mainly due to the low glucuronidation capacity of UGT 2B17*2 as only a small fraction of the administered vorinostat dose is metabolized by UGT 2B17 (f m = 11%) [63, 64]. The PBPK analysis suggests that UGT 2B7 (f m = 56%) polymorphisms, for instance, could play a vital role in the elimination, which is in accordance with the literature [16]. It is important to point out, that the clinical trial populations used in model development and evaluation were not tested for their UGT genotypes. Hence, additional information on their genotypes might alter the interpretation of these modeling results.
The PBPK analysis suggests that hydrolysis and oxidation are responsible for 30% of vorinostat metabolism and therefore more important than UGT 2B17 that accounts for 11%. Additionally, the sensitivity analysis underlined that the vorinostat PBPK model is sensitive to changes to the hydrolytic and oxidative clearance. In the model, it was assumed that both hydrolysis and oxidation would be carried out by a single enzyme available in all tissues. Unfortunately, we do not know the contribution of either hydrolysis or oxidation to the metabolized fraction, which means that the tissue-specific expression of hydrolytic or oxidative enzymes cannot be accounted for in the model. It is known that carboxylesterases, responsible for the hydrolysis of other drugs, are not equally expressed in all tissues [65]. In the model, it was further assumed that hydrolysis and oxidation would be fully developed from birth in all tissues, whereas studies have shown that the developmental regulation of esterases occurs in a gene, organ, and age-dependent manner [65,66,68]. As no details on the oxidative enzymes are available, it was decided to assume a scenario in which children would have adult enzyme levels for hydrolysis and oxidation available, and thus the highest possible clearance via this metabolic pathway. This might lead to an over-prediction of hydrolytic and oxidative clearance. Thereby, the recommended dose of 80 mg/m2 for children from 0 to 1 year of age might be over-predicted and should therefore be viewed as a maximum recommended dose.
The maturation of the UGTs should have the biggest impact on the pediatric vorinostat PK between the age 0 and 3 years as the maturation progresses considerably. The youngest observed pediatric patient was 4.7 years old, thus it was not possible to evaluate the model prediction quality in the critical age below 3 years. Although there is uncertainty in the recommended doses for an age < 4.7 years, the PBPK methodology has demonstrated for several other drugs that the pediatric PK can be successfully predicted below the age of 5 [69, 70].
Both, the PBMC concentration–time profiles and the HDAC inhibition could be very well described. Yet, the effect compartment model and the indirect model were based on a single 400 mg dose only. Hence, there is some uncertainty to the in silico trials that were carried out using a wide range of doses. While most parameters of the thrombocytopenia model were gathered from the literature, the slope parameter of vorinostat had to be estimated. Because no other data were available, this parameter was estimated using a thrombocytosis population. It was assumed that all gathered parameters (e.g. MTT) but the baseline cell counts (proliferating (Prol) and circulating (Circ) cells) were similar in patients with and without thrombocytosis, which might not be true. Nevertheless, the integrated PBPK/PD model was able to accurately predict the prevalence of thrombocytopenia in patients with normal thrombocyte counts.
Recommended doses for children were determined by targeting an exposure (AUC0 − inf) in children similar to the one observed in adults assuming the same efficacy and safety in these populations. Although data on the impact of vorinostat on the HDAC activity over time or the platelet count over time was not available in children, the assumption appears reasonable. First, the pediatric dose of 230 mg/m2 and the comparable approved standard dose of 400 mg/day for adults both led to significant accumulation of acetylated H3 histones in PBMCs in children and adults, respectively [21]. Second, dose-limiting toxicities at comparable doses in children and adults are similar [21].
In part 2 of the in silico trials, the lower thrombocyte limit was set to 50 × 109 cells/l (part 1: 193 × 109 cells/l) since platelet counts ≥ 50 × 109 cells/l typically do not lead to clinical interventions; for instance, prophylactic platelet transfusions are recommended for thrombocyte counts of ≤ 10 × 109 cells/l [71]. The upper vorinostat dose limit of 3000 mg was defined by two factors. First, the highest vorinostat dose administered in vivo was 1000 mg (suppl. Fig. S3). As the PBPK model could not be evaluated above doses of 1000 mg, there is some uncertainty in simulations above doses of 1000 mg. Second, vorinostat is typically available in 100 mg capsules, which makes dosing of high doses (> 400 mg) difficult in clinical practice. However, there are already promising new formulations under research that should simplify higher vorinostat dose administrations [72, 73].
Finally, the parameter sensitivity analysis has shown that the vorinostat half-life mainly depends on the intestinal permeability and the unstirred water layer parameter of the particle dissolution, which underlines that the absorption rate-limited flip-flop PK of vorinostat has been correctly implemented. Although the absorption was adequately predicted with the particle dissolution module, there might be other reasons responsible for the limited absorption such as drug-disease interactions or transporters. Vorinostat was found to be a substrate of P-gp and BCRP as well as an inducer of P-gp at the blood–brain barrier [58, 74, 75].
Despite some limitations, the presented work clearly demonstrates the applicability and usefulness of PBPK and PBPK/PD models in pediatric dose identification and in in silico trials. The PBPK modeling approach helps to integrate and leverage all available in vitro, in vivo, and in silico information while potentially reducing the number of clinical trials and study participants. Still, future clinical trials are needed to evaluate efficacy and safety of the newly proposed dosing regimens.
Conclusion
For the first time, a comprehensive PBPK/PD model of vorinostat has been developed which can predict (1) vorinostat concentration–time profiles in adults and (2) the impact of vorinostat on its major PD biomarkers, the HDAC inhibition and the dose-limiting decrease in thrombocytes. The PBPK/PD model was used to recommend (1) pediatric doses from 0 to 17 years and (2) potentially more effective vorinostat dosing regimens for adults. However, further research on (1) the yet unidentified enzymes involved in vorinostat metabolism, (2) the vorinostat PK in pediatric patients < 5 years of age, and (3) the PK/PD relationship in pediatric patients is necessary to increase the confidence in the model predictions. These results exemplify a mechanistic modeling approach that might help researchers to design clinical studies and to potentially improve dosing regimens of newly developed and already established drugs.
References
Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK (2001) Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 1(3):194–202. doi:10.1038/35106079
Falkenberg KJ, Johnstone RW (2014) Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov 13(9):673–691. doi:10.1038/nrd4360
Lauffer BE, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R, Ortwine DF, Gunzner J, Modrusan Z, Neumann L, Koth CM, Lupardus PJ, Kaminker JS, Heise CE, Steiner P (2013) Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J Biol Chem 288(37):26926–26943. doi:10.1074/jbc.M113.490706
U.S. Food and Drug Administration (2006) Center for Drug Evaluation and Research. Vorinostat NDA 021991 Clinical Pharmacology and Biopharmaceutics Review. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021991s000_Zolinza_ClinPharmR.pdf. Accessed 11 Nov 2016
Merck Sharp & Dohme Corp.. A subsidiary of Merck & Co., Inc. Full prescribing information: Zolinza. http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021991s002lbl.pdf. Accessed 22 Feb 2017]
Kelly WK (2005) Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol 23(17):3923–3931. doi:10.1200/jco.2005.14.167
Iwamoto M, Friedman EJ, Sandhu P, Agrawal NG, Rubin EH, Wagner JA (2013) Clinical pharmacology profile of vorinostat, a histone deacetylase inhibitor. Cancer Chemother Pharmacol 72(3):493–508. doi:10.1007/s00280-013-2220-z
Fujiwara Y, Yamamoto N, Yamada Y, Yamada K, Otsuki T, Kanazu S, Iwasa T, Hardwick JS, Tamura T (2009) Phase I and pharmacokinetic study of vorinostat (suberoylanilide hydroxamic acid) in Japanese patients with solid tumors. Cancer Sci 100(9):1728–1734. doi:10.1111/j.1349-7006.2009.01237.x
Strassburg CP, Strassburg A, Kneip S, Barut A, Tukey RH, Rodeck B, Manns MP (2002) Developmental aspects of human hepatic drug glucuronidation in young children and adults. Gut 50(2):259–265
Zaya MJ, Hines RN, Stevens JC (2006) Epirubicin glucuronidation and UGT2B7 developmental expression. Drug Metab Dispos 34(12):2097–2101. doi:10.1124/dmd.106.011387
Pacifici GM, Sawe J, Kager L, Rane A (1982) Morphine glucuronidation in human fetal and adult liver. Eur J Clin Pharmacol 22(6):553–558
McRorie TI, Lynn AM, Nespeca MK, Opheim KE, Slattery JT (1992) The maturation of morphine clearance and metabolism. Am J Dis Child (1960) 146(8):972–976
Edginton AN, Schmitt W, Willmann S (2006) Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin Pharmacokinet 45(10):1013–1034. doi:10.2165/00003088-200645100-00005
Neumann E, Mehboob H, Ramirez J, Mirkov S, Zhang M, Liu W (2016) Age-dependent hepatic UDP-glucuronosyltransferase gene expression and activity in children. Front Pharmacol 7:437. doi:10.3389/fphar.2016.00437
Balliet RM, Chen G, Gallagher CJ, Dellinger RW, Sun D, Lazarus P (2009) Characterization of UGTs active against SAHA and association between SAHA glucuronidation activity phenotype with UGT genotype. Cancer Res 69(7):2981–2989. doi:10.1158/0008-5472.CAN-08-4143
Kang SP, Ramirez J, House L, Zhang W, Mirkov S, Liu W, Haverfield E, Ratain MJ (2010) A pharmacogenetic study of vorinostat glucuronidation. Pharmacogenet Genomics 20(10):638–641. doi:10.1097/FPC.0b013e32833e1b37
Wong NS, Seah E, Wang LZ, Yeo WL, Yap HL, Chuah B, Lim YW, Ang PC, Tai BC, Lim R, Goh BC, Lee SC (2011) Impact of UDP-gluconoryltransferase 2B17 genotype on vorinostat metabolism and clinical outcomes in Asian women with breast cancer. Pharmacogenet Genom 21(11):760–768. doi:10.1097/FPC.0b013e32834a8639
Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, Frankel SR, Chen C, Ricker JL, Arduino JM, Duvic M (2007) Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol 25(21):3109–3115. doi:10.1200/jco.2006.10.2434
Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, Frankel SR (2007) Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 109(1):31–39. doi:10.1182/blood-2006-06-025999
Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL (2010) Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals 3(9):2751–2767. doi:10.3390/ph3092751
Fouladi M, Park JR, Stewart CF, Gilbertson RJ, Schaiquevich P, Sun J, Reid JM, Ames MM, Speights R, Ingle AM, Zwiebel J, Blaney SM, Adamson PC (2010) Pediatric phase I trial and pharmacokinetic study of vorinostat: a Children’s Oncology Group phase I consortium report. J Clin Oncol 28(22):3623–3629. doi:10.1200/JCO.2009.25.9119
Muscal JA, Thompson PA, Horton TM, Ingle AM, Ahern CH, McGovern RM, Reid JM, Ames MM, Espinoza-Delgado I, Weigel BJ, Blaney SM (2013) A phase I trial of vorinostat and bortezomib in children with refractory or recurrent solid tumors: a Children’s Oncology Group phase I consortium study (ADVL0916). Pediatr Blood Cancer 60(3):390–395. doi:10.1002/pbc.24271
Hummel TR, Wagner L, Ahern C, Fouladi M, Reid JM, McGovern RM, Ames MM, Gilbertson RJ, Horton T, Ingle AM, Weigel B, Blaney SM (2013) A pediatric phase 1 trial of vorinostat and temozolomide in relapsed or refractory primary brain or spinal cord tumors: a Children’s Oncology Group phase 1 consortium study. Pediatr Blood Cancer 60(9):1452–1457. doi:10.1002/pbc.24541
Munshi A, Tanaka T, Hobbs ML, Tucker SL, Richon VM, Meyn RE (2006) Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol Cancer Ther 5(8):1967–1974. doi:10.1158/1535-7163.mct-06-0022
Wilson PM, Labonte MJ, Martin SC, Kuwahara ST, El-Khoueiry A, Lenz HJ, Ladner RD (2013) Sustained inhibition of deacetylases is required for the antitumor activity of the histone deactylase inhibitors panobinostat and vorinostat in models of colorectal cancer. Invest New Drugs 31(4):845–857. doi:10.1007/s10637-012-9914-7
Arnold NB, Arkus N, Gunn J, Korc M (2007) The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances gemcitabine-induced cell death in pancreatic cancer. Clin Cancer Res 13(1):18–26. doi:10.1158/1078-0432.ccr-06-0914
Shen J, Huang C, Jiang L, Gao F, Wang Z, Zhang Y, Bai J, Zhou H, Chen Q (2007) Enhancement of cisplatin induced apoptosis by suberoylanilide hydroxamic acid in human oral squamous cell carcinoma cell lines. Biochem Pharmacol 73(12):1901–1909. doi:10.1016/j.bcp.2007.03.009
Dickson MA, Rathkopf DE, Carvajal RD, Grant S, Roberts JD, Reid JM, Ames MM, McGovern RM, Lefkowitz RA, Gonen M, Cane LM, Dials HJ, Schwartz GK (2011) A phase I pharmacokinetic study of pulse-dose vorinostat with flavopiridol in solid tumors. Invest New Drugs 29(5):1004–1012. doi:10.1007/s10637-010-9447-x
Nelder JA, Mead R (1965) A simplex method for function minimization. Comput J 7(4):308–313. doi:10.1093/comjnl/7.4.308
Kelly WK, Richon VM, O’Connor O, Curley T, MacGregor-Curtelli B, Tong W, Klang M, Schwartz L, Richardson S, Rosa E, Drobnjak M, Cordon-Cordo C, Chiao JH, Rifkind R, Marks PA, Scher H (2003) Phase I clinical trial of histone deacetylase inhibitor suberoylanilide hydroxamic acid administered intravenously. 9 (10):3578–3588
Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, Mita AC, Curiel TJ, Espitia CM, Nawrocki ST, Giles FJ, Carew JS (2014) Combined autophagy and HDAC inhibition. Autophagy 10(8):1403–1414. doi:10.4161/auto.29231
Munster PN, Rubin EH, Van Belle S, Friedman E, Patterson JK, Van Dyck K, Li X, Comisar W, Chodakewitz JA, Wagner JA, Iwamoto M (2009) A single supratherapeutic dose of vorinostat does not prolong the QTc interval in patients with advanced cancer. Clin Cancer Res 15(22):7077–7084. doi:10.1158/1078-0432.CCR-09-1214
Ramalingam SS, Parise RA, Ramanathan RK, Lagattuta TF, Musguire LA, Stoller RG, Potter DM, Argiris AE, Zwiebel JA, Egorin MJ, Belani CP (2007) Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin Cancer Res 13(12):3605–3610. doi:10.1158/1078-0432.CCR-07-0162
Rubin EH, Agrawal NG, Friedman EJ, Scott P, Mazina KE, Sun L, Du L, Ricker JL, Frankel SR, Gottesdiener KM, Wagner JA, Iwamoto M (2006) A study to determine the effects of food and multiple dosing on the pharmacokinetics of vorinostat given orally to patients with advanced cancer. Clin Cancer Res 12(23):7039–7045. doi:10.1158/1078-0432.CCR-06-1802
Wada H, Tsuboi R, Kato Y, Sugaya M, Tobinai K, Hamada T, Shimamoto T, Noguchi K, Iwatsuki K (2012) Phase I and pharmacokinetic study of the oral histone deacetylase inhibitor vorinostat in Japanese patients with relapsed or refractory cutaneous T-cell lymphoma. J Dermatol 39(10):823–828. doi:10.1111/j.1346-8138.2012.01554.x
Andersen CL, McMullin MF, Ejerblad E, Zweegman S, Harrison C, Fernandes S, Bareford D, Knapper S, Samuelsson J, Lofvenberg E, Linder O, Andreasson B, Ahlstrand E, Jensen MK, Bjerrum OW, Vestergaard H, Larsen H, Klausen TW, Mourits-Andersen T, Hasselbalch HC (2013) A phase II study of vorinostat (MK-0683) in patients with polycythaemia vera and essential thrombocythaemia. Br J Haematol 162(4):498–508. doi:10.1111/bjh.12416
Wu G, Baraldo M, Furlanut M (1995) Calculating percentage prediction error: a user’s note. Pharmacol Res 32(4):241–248
Post TM, Freijer JI, Ploeger BA, Danhof M (2008) Extensions to the visual predictive check to facilitate model performance evaluation. J Pharmacokinet Pharmacodyn 35(2):185–202. doi:10.1007/s10928-007-9081-1
Willmann S, Hohn K, Edginton A, Sevestre M, Solodenko J, Weiss W, Lippert J, Schmitt W (2007) Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J Pharmacokinet Pharmacodyn 34(3):401–431. doi:10.1007/s10928-007-9053-5
Claassen K, Thelen K, Coboeken K, Gaub T, Lippert J, Allegaert K, Willmann S (2015) Development of a physiologically-based pharmacokinetic model for preterm neonates: evaluation with in vivo data. Curr Pharm Des 21(39):5688–5698
Willmann S, Becker C, Burghaus R, Coboeken K, Edginton A, Lippert J, Siegmund H-U, Thelen K, Mück W (2014) Development of a paediatric population-based model of the pharmacokinetics of rivaroxaban. Clin Pharmacokinet 53(1):89–102. doi:10.1007/s40262-013-0090-5
Thelen K, Coboeken K, Willmann S, Burghaus R, Dressman JB, Lippert J (2011) Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part 1: oral solutions. J Pharm Sci 100(12):5324–5345. doi:10.1002/jps.22726
Thelen K, Coboeken K, Willmann S, Dressman JB, Lippert J (2012) Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part II: extension to describe performance of solid dosage forms. J Pharm Sci 101(3):1267–1280. doi:10.1002/jps.22825
Willmann S, Lippert J, Schmitt W (2005) From physicochemistry to absorption and distribution: predictive mechanistic modelling and computational tools. Expert Opin Drug Metab Toxicol 1(1):159–168. doi:10.1517/17425255.1.1.159
Willmann S, Lippert J, Sevestre M, Solodenko J, Fois F, Schmitt W (2003) PK-Sim®: a physiologically based pharmacokinetic ‘whole-body’ model. Biosilico 1(4):121–124. doi:10.1016/S1478-5382(03)02342-4
Meyer M, Schneckener S, Ludewig B, Kuepfer L, Lippert J (2012) Using expression data for quantification of active processes in physiologically based pharmacokinetic modeling. Drug Metab Dispos 40(5):892–901. doi:10.1124/dmd.111.043174
Wheeler DL, Church DM, Federhen S, Lash AE, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E, Tatusova TA, Wagner L (2003) Database resources of the National Center for Biotechnology. Nucleic Acids Res 31(1):28–33
Johnson KC (2003) Dissolution and absorption modeling: model expansion to simulate the effects of precipitation, water absorption, longitudinally changing intestinal permeability, and controlled release on drug absorption. Drug Dev Ind Pharm 29(8):833–842. doi:10.1081/ddc-120024179
Du BD, Du BE (1916) Clinical calorimetry: Tenth paper a formula to estimate the approximate surface area if height and weight be known. Archives of Internal Medicine XVII (6_2):863–871. doi:10.1001/archinte.1916.00080130010002
Liu L, Detering JC, Milde T, Haefeli WE, Witt O, Burhenne J (2014) Quantification of vorinostat and its main metabolites in plasma and intracellular vorinostat in PBMCs by liquid chromatography coupled to tandem mass spectrometry and its relation to histone deacetylase activity in human blood. J Chromatogr B Analyt Technol Biomed Life Sci 964:212–221. doi:10.1016/j.jchromb.2014.02.014
Sharma A, Jusko WJ (1998) Characteristics of indirect pharmacodynamic models and applications to clinical drug responses. Br J Clin Pharmacol 45(3):229–239. doi:10.1046/j.1365-2125.1998.00676.x
Izak M, Bussel JB (2014) Management of thrombocytopenia. F1000Prime Reports 6:45. doi:10.12703/P6-45
Friberg LE, Henningsson A, Maas H, Nguyen L, Karlsson MO (2002) Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J Clin Oncol 20(24):4713–4721. doi:10.1200/jco.2002.02.140
Chalret du Rieu Q, Fouliard S, White-Koning M, Kloos I, Chatelut E, Chenel M (2014) Pharmacokinetic/Pharmacodynamic modeling of abexinostat-induced thrombocytopenia across different patient populations: application for the determination of the maximum tolerated doses in both lymphoma and solid tumour patients. Invest New Drugs 32(5):985–994. doi:10.1007/s10637-014-0118-1
Peters SA, Hultin L (2008) Early identification of drug-induced impairment of gastric emptying through physiologically based pharmacokinetic (PBPK) simulation of plasma concentration-time profiles in rat. J Pharmacokinet Pharmacodyn 35(1):1–30. doi:10.1007/s10928-007-9073-1
Moj D, Hanke N, Britz H, Frechen S, Kanacher T, Wendl T, Haefeli WE, Lehr T (2017) Clarithromycin, midazolam, and digoxin: application of PBPK modeling to gain new insights into drug–drug interactions and co-medication regimens. AAPS J 19(1):298–312. doi:10.1208/s12248-016-0009-9
Nakashima S, Yamamoto K, Arai Y, Ikeda Y (2013) Impact of physicochemical profiling for rational approach on drug discovery. Chem Pharm Bull 61(12):1228–1238. doi:10.1248/cpb.c13-00436
Konsoula R, Jung M (2008) In vitro plasma stability, permeability and solubility of mercaptoacetamide histone deacetylase inhibitors. Int J Pharm 361(1–2):19–25. doi:10.1016/j.ijpharm.2008.05.001
Margaillan G, Rouleau M, Klein K, Fallon JK, Caron P, Villeneuve L, Smith PC, Zanger UM, Guillemette C (2015) Multiplexed targeted quantitative proteomics predicts hepatic glucuronidation potential. Drug Metab Dispos 43(9):1331–1335. doi:10.1124/dmd.115.065391
Burhenne J, Liu L, Heilig CE, Meid AD, Leisen M, Schmitt T, Kasper B, Haefeli WE, Mikus G, Egerer G (2017) Intracellular vorinostat accumulation and its relationship to histone deacetylase activity in soft tissue sarcoma patients. Cancer Chemother Pharm 80(2):433–439
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R (2007) FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12(10):1247–1252. doi:10.1634/theoncologist.12-10-1247
Kawai R, Lemaire M, Steimer JL, Bruelisauer A, Niederberger W, Rowland M (1994) Physiologically based pharmacokinetic study on a cyclosporin derivative, SDZ IMM 125. J Pharmacokinet Biopharm 22(5):327–365
Park J, Chen L, Ratnashinge L, Sellers TA, Tanner JP, Lee JH, Dossett N, Lang N, Kadlubar FF, Ambrosone CB, Zachariah B, Heysek RV, Patterson S, Pow-Sang J (2006) Deletion polymorphism of UDP-glucuronosyltransferase 2B17 and risk of prostate cancer in African American and Caucasian men. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research. Cosponsored Am Soc Prev Oncol 15(8):1473–1478. doi:10.1158/1055-9965.epi-06-0141
Xue Y, Sun D, Daly A, Yang F, Zhou X, Zhao M, Huang N, Zerjal T, Lee C, Carter NP, Hurles ME, Tyler-Smith C (2008) Adaptive evolution of UGT2B17 copy-number variation. Am J Hum Genet 83(3):337–346. doi:10.1016/j.ajhg.2008.08.004
Imai T (2006) Human carboxylesterase isozymes: catalytic properties and rational drug design. Drug Metab Pharmacokinet 21(3):173–185
Boberg M, Vrana M, Mehrotra A, Pearce RE, Gaedigk A, Bhatt DK, Leeder JS, Prasad B (2017) Age-dependent absolute abundance of hepatic carboxylesterases (CES1 and CES2) by LC-MS/MS proteomics: application to PBPK modeling of oseltamivir in vivo pharmacokinetics in infants. Drug Metab Dispos 45(2):216–223. doi:10.1124/dmd.116.072652
Chen YT, Trzoss L, Yang D, Yan B (2015) Ontogenic expression of human carboxylesterase-2 and cytochrome P450 3A4 in liver and duodenum: postnatal surge and organ-dependent regulation. Toxicology 330:55–61. doi:10.1016/j.tox.2015.02.007
Zhu HJ, Appel DI, Jiang Y, Markowitz JS (2009) Age- and sex-related expression and activity of carboxylesterase 1 and 2 in mouse and human liver. Drug Metab Dispos 37(9):1819–1825. doi:10.1124/dmd.109.028209
Zhou W, Johnson TN, Xu H, Cheung SYA, Bui KH, Li J, Al-Huniti N, Zhou D (2016) Predictive performance of physiologically based pharmacokinetic and population pharmacokinetic modeling of renally cleared drugs in children. CPT: Pharm Syst Pharmacol 5(9):475–483. doi:10.1002/psp4.12101
Jiang XL, Zhao P, Barrett JS, Lesko LJ, Schmidt S (2013) Application of physiologically based pharmacokinetic modeling to predict acetaminophen metabolism and pharmacokinetics in children. CPT Pharm Syst Pharmacol 2:e80. doi:10.1038/psp.2013.55
Estcourt LJ, Stanworth SJ, Doree C, Hopewell S, Trivella M, Murphy MF (2015) Comparison of different platelet count thresholds to guide administration of prophylactic platelet transfusion for preventing bleeding in people with haematological disorders after myelosuppressive chemotherapy or stem cell transplantation. Cochrane Database Syst Rev (11):Cd010983. doi:10.1002/14651858.CD010983.pub2
Cai YY, Yap CW, Wang Z, Ho PC, Chan SY, Ng KY, Ge ZG, Lin HS (2010) Solubilization of vorinostat by cyclodextrins. J Clin Pharm Ther 35(5):521–526. doi:10.1111/j.1365-2710.2009.01095.x
Jayant RD, Atluri VS, Agudelo M, Sagar V, Kaushik A, Nair M (2015) Sustained-release nanoART formulation for the treatment of neuroAIDS. Int J Nanomed 10:1077–1093. doi:10.2147/ijn.s76517
Sajja RK, Liles TR, Prasad S, Cucullo L (2016) Vorinostat induces blood-brain barrier P-Glycoprotein drug efflux activity: implications for limited brain disposition of co-administered chemotherapeutics. FASEB J 30(1 Supplement):935.933
Hanson JE, La H, Plise E, Chen Y-H, Ding X, Hanania T, Sabath EV, Alexandrov V, Brunner D, Leahy E, Steiner P, Liu L, Scearce-Levie K, Zhou Q (2013) SAHA enhances synaptic function and plasticity in vitro but has limited brain availability in vivo and does not impact cognition. PLoS One 8(7):e69964. doi:10.1371/journal.pone.0069964
Acknowledgements
The analyses of vorinostat were supported by Merck & Co., Inc., USA that provided deuterium-labeled and unlabeled reference standards of vorinostat and its metabolites M1 and M2. Some of the studies were financially supported by MSD Sharp & Dohme GmbH, Germany.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical standards
All procedures performed in studies involving human participants were in accordance with the legal requirements and ethical standards of the responsible institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants before inclusion.
Conflict of interest
Daniel Moj, Hannah Britz, Thorsten Lehr and Clinton F. Stewart have reported no potential conflicts of interest. Walter E. Haefeli and Jürgen Burhenne have received financial and non-financial (analytical standards) support from Merck & Co., Inc., USA for the development and application of the vorinostat assay. Jürgen Burhenne has received travel support and lecture fees from MSD Sharp & Dohme GmbH, Germany.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Moj, D., Britz, H., Burhenne, J. et al. A physiologically based pharmacokinetic and pharmacodynamic (PBPK/PD) model of the histone deacetylase (HDAC) inhibitor vorinostat for pediatric and adult patients and its application for dose specification. Cancer Chemother Pharmacol 80, 1013–1026 (2017). https://doi.org/10.1007/s00280-017-3447-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-017-3447-x