
Summary
Background Sorafenib is the sole molecular-targeted agent showing a survival benefit in patients with advanced hepatocellular carcinoma (HCC). We evaluated the tolerability and effectiveness of a combination of S-1 with sorafenib in patients with advanced HCC. Methods S-1 was administered during days 1–14 and sorafenib was administered every day. This treatment was repeated every 21 days. In phase I, we determined the maximum tolerated dose (MTD) and dose-limiting toxicities (DLTs). The dose of each drug was planned as follows: cohort 1: S-1 48 mg/m2/day and sorafenib 400 mg/day, cohort 2a: S-1 48 mg/m2/day and sorafenib 800 mg/day, cohort 2b: S-1 64 mg/m2/day and sorafenib 400 mg/day, cohort 3: S-1 64 mg/m2/day and sorafenib 800 mg/day, and cohort 4: S-1 80 mg/m2/day and sorafenib 800 mg/day. In phase II, the patients were treated at the MTD to evaluate safety and efficacy. Results Nineteen patients were enrolled in phase I. One of the six patients in cohort 1 and one of the six patients in cohort 3 experienced DLT. None of the three patients in cohort 2a experienced DLT and three of the four patients in cohort 4 experienced DLT. Therefore, cohort 3 was considered the MTD. Subsequently, 26 patients were enrolled in phase II. The most common grade 3/4 toxicities were an increase of aspartate aminotransferase (38.5 %), thrombocytopenia (23.1 %), neutropenia (19.2 %), hyperbilirubinemia (15.4 %), an increase of alanine aminotransferase (15.4 %), hyponatremia (11.5 %), rash (11.5 %), and hypophosphatemia (11.5 %). Sudden death occurred in one patient (3.8 %). A patient (3.8 %) had a partial response, 15 (57.7 %) had stable disease, and 10 (38.5 %) had progressive disease. The median times to progression and overall survival were 2.4 and 10.5 months, respectively. Conclusion The MTD of S-1 and sorafenib in patients with advanced HCC was 64 mg/m2/day and 800 mg/day, respectively. This dose/regimen demonstrated substantial clinical activity among patients with advanced HCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Because of the poor prognosis, hepatocellular carcinoma (HCC) is the sixth most common cancer worldwide and is the third most common cause of death from cancer [1]. In particular, advanced HCC that cannot be treated by loco-regional therapies has a very poor prognosis. Sorafenib (Nexavar, Bayer Healthcare, Leverkusen, Germany), an oral multikinase inhibitor, blocks tumor cell proliferation by targeting Raf/MEK/ERK signaling at the level of Raf kinase and exerts an antiangiogenic effect by targeting tyrosine kinase receptors, such as vascular endothelial growth factor receptor and platelet derived growth factor receptor [2]. This agent has been utilized as standard medical therapy for patients with advanced HCC. The efficacy of sorafenib in advanced HCC has been proven in two large-scale randomized control trials [3, 4]. However, its survival benefit is modest [3, 4] and still unsatisfactory. Combining targeted agents and cytotoxic drugs is one strategy to improve the effectiveness of targeted molecular therapy. Several phase I/II studies on combined treatment of sorafenib with a cytotoxic agent have shown a tolerable toxicity profile and promising results [5].
S-1 (Taiho Pharmaceutical Co., Ltd., Tokyo, Japan) is an orally administered anticancer drug consisting of a combination of tegafur, 5-chloro-2,4-dihydroxypyridine, and oteracil potassium in a molar concentration ratio of 1:0.4:1, respectively [6]. S-1 is effective against a variety of solid tumors [7, 8], and also has an acceptable toxicity profile and promising antitumor activity against HCC [9]. Moreover, S-1/sorafenib combination therapy results in greater inhibition of tumor growth and remarkable thymidylate synthetase suppression when compared with sorafenib or S-1 alone in nonobese diabetic/severe combined immunodeficiency mice with subcutaneously inoculated HCC [10]. Based on this background, we hypothesized that sorafenib and S-1 combination therapy would be efficacious as first-line therapy in patients with HCC.
In the present study, we conducted a phase I/II study to determine maximum tolerated dose (MTD) and to evaluate efficacy and safety of a combination therapy of S-1 and sorafenib in Japanese patients with advanced HCC.
Patients and methods
We conducted prospective, open-label, non-randomized phase I and phase II trials to evaluate the safety and efficacy of combination chemotherapy with sorafenib and S-1 in patients with advanced HCC. Phase I was performed at Chiba University Hospital, and Phase II was performed at Chiba University Hospital and Kanazawa University Hospital. The trials were approved by the ethics investigation committees of each participating hospital, conducted in accordance with the Declaration of Helsinki, and registered at UMIN Clinical Trials Registry (Phase I: UMIN000002590, Phase II: UMIN000007199). Informed consent was obtained from each patient.
Patient selection
Eligibility criteria for study entry were: Patients who had been diagnosed with HCC by histological examination or typical diagnostic images; no indication for surgical resection and local therapy (percutaneous ethanol injection, radiofrequency ablation, microwave coagulation therapy, transcatheter arterial chemoembolization, radiation therapy); systemic chemotherapy-naive; measurable disease based on Response Evaluation Criteria in Solid Tumors (RECIST) criteria (ver. 1.1) of at least one untreated target lesion; ECOG (Eastern Cooperative Oncology Group) performance status 0–1; age ≥20 years age; neutrophil count ≥1,500/μL; hemoglobin ≥9.0 g/dl; platelets ≥50,000/μL; total bilirubin <3.0 mg/dL; aspartate aminotransferase (AST)/ alanine aminotransferase (ALT) < five times the upper limit of normal (ULN); albumin ≥2.8 g/dL, serum creatinine < within the ULN; prothrombin time ≥40 %; Child-Pugh score A; patients could take food and drugs orally; and life expectancy ≥12 weeks. Exclusion criteria for study entry were: Previous therapy for HCC within 30 days before study entry; major surgery within 30 days before study entry or surgery within 15 days before study entry; portal vein tumor thrombus in the primary trunk; uncontrollable hypertension; pleural effusion, ascites and pericardial fluid requiring drainage or affecting the respiratory and circulating dynamics; patients who received an albumin preparation or a blood transfusion within 30 days before study entry; hepatic encephalopathy or brain lesions with clinical symptoms; central nervous system tumor (including brain metastasis); bone metastasis with clinical symptoms; active infection [except hepatitis B virus and hepatitis C virus (HCV) infection]; evidence of serious gastrointestinal bleeding within 30 days before study entry; gastro-esophageal varices requiring preventive treatment; pregnant or lactating woman; no consent to use contraception during study treatment; second primary malignancy (except in situ carcinoma or prior malignancy treated >5 years ago without recurrence).
Administration and dose escalation
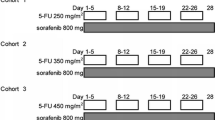
In phase I, we escalated the S-1 and sorafenib dose levels. The S-1 and sorafenib dose levels were as follows: cohort 1, day (D) 1-14 S-1 48 mg/m2/day (60 % of standard dose) + D1-21 sorafenib 400 mg/day; cohort 2a, D1-14 S-1 48 mg/m2/day + D1-21 sorafenib 800 mg/day; cohort 2b, D1-14 S-1 64 mg/m2/day (80 % of standard dose) + D1-21 sorafenib 400 mg/day; cohort 3, D1-14 S-1 64 mg/ m2/day + D1-21 sorafenib 800 mg/day; and cohort 4, D1-14 S-1 80 mg/m2/day + D1-21 sorafenib 800 mg/day. The treatment was repeated every 3 weeks and each treatment cycle was 21 days. The dose was escalated according to the cohort and was not increased in the same patient. If none of the first three patients had dose-limiting toxicities (DLTs) during the first cycle, the dose was increased in the next cohort. If a DLT occurred in one of the three initial patients in a particular cohort, then three additional patients were enrolled in the same cohort for a total of six patients. If a DLT developed in three or more of six patients, it was decided that the dose of this cohort was beyond the MTD. Thus, the preceding cohort’s dose was defined as the MTD and designated as the recommended dose for phase II. If fewer than three of the six patients experienced DLTs in the last cohort, then this cohort’s dose level was recommended for phase II. No intra-patient dose escalation was allowed.
DLT
A DLT was defined as any of following events observed during the first cycle of therapy: grade 4 thrombocytopenia, grade 4 neutropenia lasting >7 days, grade 3 or 4 febrile neutropenia, non-hematological toxicity ≥ grade 3, and AST or ALT ≥20 times the ULN. Safety was assessed every week for the first treatment cycle. Adverse events (AEs) were evaluated according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), ver. 3.0 (URL:http://ctep.cancer.gov/protocolDevelopment/electronic_applications/). All AEs were evaluated until 21 days after the last cycle.
Duration of treatment and follow-up
In the absence of treatment delays due to AEs, treatment continued until one of the following criteria applied: disease progression, intercurrent disease that prevented further administration of treatment, unacceptable AEs, and patient withdrawal. Patients were followed for 6 months after removal from the study or until death, whichever occurred first. Patients removed from study due to an unacceptable AE were followed until the resolution or stabilization of the AE.
Response and toxicity evaluation
Radiological (chest X-ray and computed tomography) studies to assess response were performed after every two cycles of therapy until disease progression. Response definitions were made according to RECIST ver. 1.1. Time to progression (TTP) was defined as the time from the date of registration to the date of the first documentation of radiological or clinical disease progression. Overall survival (OS) was defined as the time from the date of registration to the date of death. Surviving patients were censored at the last confirmed date of survival. The Kaplan–Meier method was used to estimate the median values of time-to-event variables, such as OS and TTP. The severity of all AEs was evaluated according to CTCAE ver. 3.0. The attending physicians initially assessed the duration of all AEs and their relationship to the combination therapy.
Statistical analysis
The primary endpoint for phase I was to determine the MTD for combination therapy with S-1 and sorafenib for patients with advanced HCC. The primary endpoint in phase II was TTP from time of registration. At the time when this study was designed, the available data about the clinical efficacy of sorafenib in Asia-Pacific patients included a phase III trial of sorafenib vs. placebo reported by Cheng et al. [3]. Sample size was calculated on the assumption of an α error = 0.05, and a β error = 0.30. The null hypothesis and the alternative hypothesis were set at TTP of 2.8 months and 5.6 months, respectively. The reported TTP for sorafenib mono-therapy in Asian patients was 2.8 months [3]. The data were analyzed using SPSS software (IBM Corp., NY, USA). All the statistics were performed for an “intent-to-treat population”, which was defined as patients who received at least one dose of the study drugs. The TTP and OS were analyzed using the Kaplan–Meier method.
Results
Phase I
Patient characteristics
Nineteen eligible patients, consisting of six patients in cohort 1, three patients in cohort 2a, six patients in cohort 3, and four patients in cohort 4, were enrolled in phase I. The patient characteristics are summarized in Table 1.
MTDs and toxicities
One of the six patients in cohort 1 experienced a DLT (grade 4 AST/ALT elevation). No patient in cohort 2a experienced a DLT. One of the six patients in cohort 3 experienced a DLT (grade 3 gastrointestinal bleeding). A DLT occurred in two of the three initial patients and one additional patient in cohort 4 [two with grade 3 hand-foot skin reaction (HFSR) and one with grade 3 infection with normal absolute neutrophil count]. Eventually, cohort 3 (D1-14 S-1 64 mg/m2/day + D1-21 sorafenib 800 mg/day) was considered the MTD.
Phase II
Patient characteristics
Twenty-six patients were accrued into phase II. All patients were eligible for the evaluation of toxicity and efficacy. The characteristics of the patients (23 men and three women; mean age, 65.5 ± 9.4 years (range, 45–85 years)) are summarized in Table 1. At study entry, 19 of 26 (73.1 %) patients were the Barcelona Clinic Liver Cancer (BCLC) stage C and seven of 26 (26.9 %) patients were the BCLC stage B.
Treatment-related toxicity
In the 26 patients who were evaluated for toxicity data, one died by sudden death; the patient was a 76-year-old man with no medical history except that of a chronic HCV infection before inclusion in the study. He had no symptoms and there were no signs before his sudden death. Because an autopsy could not be performed based on the will of his family, an accurate cause of death was not elucidated. In other toxicity data, HFSR, fatigue, hyperbilirubinemia, AST and ALT elevation, neutropenia, thrombocytopenia and anemia occurred in ≥50 % of the patients (Table 2). Summary of ≥ grade 3 AEs is shown in Table 3. Two patients stopped treatment because of AEs.
Treatment efficacy and survival analysis
The phase II response rate is shown in Table 4. Among the 26 patients in whom a response could be evaluated, one had a partial response, 15 had stable disease, and ten had progressive disease. The TTP and OS are shown in Figs. 1 and 2, respectively. The median TTP was 2.4 months [95 % confidence interval (CI), 0.4–3.4 months]. The median OS was 10.5 months (95 % CI, 2.8–18.2 months).

Kaplan-Meier analysis of time to progression (TTP) in phase II (n = 26). The median TTP was 2.4 months

Kaplan-Meier analysis of overall survival (OS) in phase II (n = 26). The median OS was 10.5 months
Discussion
In this study, we determined that the MTDs of S-1 and sorafenib in Japanese patients with advanced HCC were 64 mg/m2/day and 800 mg/day, respectively. Treatment with sorafenib alone is tolerated, but AEs including HFSR, rash, and liver failure occur more frequently in Japanese patients with HCC than those in patients from other regions [11]. This may be the reason that the MTD of S-1 when added to sorafenib was lower in our study than that in a previous study performed in Korea [12].
The common AEs in phase II were AST elevation (92.3 %), hyperbilirubinemia (92.3 %), thrombocytopenia (80.8 %), anemia (73.1 %), ALT elevation (69.2 %), HFSR (61.5 %), fatigue (57.7 %) and neutropenia (50.0 %). The toxic profiles are very different between sorafenib and S-1 monotherapies [3, 4, 9, 11, 13]. Common AEs of sorafenib alone are HFSR, rash, and elevation of liver enzymes [11] and those of S-1 in patients with advanced HCC include bone marrow suppression [9]. In our study, the frequencies of HFSR and rash were similar to that of sorafenib monotherapy, and the frequency of bone marrow suppression is similar to that of S-1 monotherapy. The frequency of elevated liver enzymes was higher than that of both monotherapies. The toxicity profile of the S-1 and sorafenib combination appeared similar to the added toxic profiles of S-1 and sorafenib. In addition, three severe AEs occurred (sudden death, G4 hyponatremia, bleeding) in our study. The patient who died suddenly met the inclusion and exclusion criteria, and he had no major medical history, no symptoms, and no obvious AEs before death. The exact cause of death was not determined because no autopsy was performed. From these findings, it can be inferred that the toxicity of combination therapy was more severe than that of S-1 and sorafenib monotherapies.
Only one patient had a partial response in phase II. The tumor response rate and disease control rate of the combination therapy in our study did not increase compared with those of sorafenib alone reported previously [3, 4, 11, 13]. Median TTP was only 2.4 months, and it was not longer than that of sorafenib monotherapy [3, 4, 13]. However, median OS in our study was modest at 10.5 months, and it appeared to be dissociated from the result of the median TTP. Dissociation of TTP and OS in Japanese patients with advanced HCC was reported in several studies [9, 13], and the reason is attributable to various treatments after progressive disease, including hepatic arterial infusion chemotherapy, and palliative care.
In conclusion, the MTD of S-1 and sorafenib in Japanese patients with advanced HCC was S-1 64 mg/m2/day D1-14 and sorafenib 800 mg/day D1-21 every 3 weeks. Although the toxicities were slightly severe, similar results were seen in the therapeutic effects of the combination therapy compared with those observed in sorafenib monotherapy. Other new drugs or combination therapy with sorafenib is needed to improve the prognosis of patients with advanced HCC.
References
Ferlay J, Shin HR, Bray F et al (2010) Estimates of worldwide burden of cancer in 2008: Globocan 2008. Int J Cancer 127:2893–2917
Wilhelm SM, Carter C, Tang L et al (2004) Bay 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64:7099–7109
Cheng AL, Kang YK, Chen Z et al (2009) Efficacy and safety of sorafenib in patients in the Asia-pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 10:25–34
Llovet JM, Ricci S, Mazzaferro V et al (2008) Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359:378–390
Takimoto CH, Awada A (2008) Safety and anti-tumor activity of sorafenib (nexavar) in combination with other anti-cancer agents: a review of clinical trials. Cancer Chemother Pharmacol 61:535–548
Shirasaka T, Shimamato Y, Ohshimo H et al (1996) Development of a novel form of an oral 5-fluorouracil derivative (S-1) directed to the potentiation of the tumor selective cytotoxicity of 5-fluorouracil by two biochemical modulators. Anticancer Drugs 7:548–557
Shirasaka T (2009) Development history and concept of an oral anticancer agent S-1 (TS-1): its clinical usefulness and future vistas. Jpn J Clin Oncol 39:2–15
Saif MW, Syrigos KN, Katirtzoglou NA (2009) S-1: a promising new oral fluoropyrimidine derivative. Expert Opin Investig Drugs 18:335–348
Furuse J, Okusaka T, Kaneko S et al (2010) Phase I/II study of the pharmacokinetics, safety and efficacy of S-1 in patients with advanced hepatocellular carcinoma. Cancer Sci 101:2606–2611
Zhai JM, Yin XY, Lai YR (2013) Sorafenib enhances the chemotherapeutic efficacy of S-1 against hepatocellular carcinoma through downregulation of transcription factor E2F-1. Cancer Chemother Pharmacol 71:1255–1264
Ogasawara S, Kanai F, Obi S et al (2011) Safety and tolerance of sorafenib in Japanese patients with advanced hepatocellular carcinoma. Hepatol Int 5:850–856
Lee SJ, Lee J, Park SH et al (2012) Phase 1 trial of S-1 in combination with sorafenib for patients with advanced hepatocellular carcinoma. Investig New Drugs 30:1540–1547
Furuse J, Ishii H, Nakachi K et al (2008) Phase I study of sorafenib in Japanese patients with hepatocellular carcinoma. Cancer Sci 99:159–165
Conflict of interest
Prof. Osamu Yokosuka received research grants from Bayer Healthcare. Yoshihiko Ooka, Tetsuhiro Chiba, Sadahisa Ogasawara, Kuniaki Arai, Eiichiro Suzuki, Akinobu Tawada, Tatsuya Yamashita, Fumihiko Kanai, and Shuichi Kaneko declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ooka, Y., Chiba, T., Ogasawara, S. et al. A phase I/II study of S-1 with sorafenib in patients with advanced hepatocellular carcinoma. Invest New Drugs 32, 723–728 (2014). https://doi.org/10.1007/s10637-014-0077-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-014-0077-6