
Abstract
Purpose
This study reports the ophthalmic and genetic findings of a Cameroonian patient with autosomal recessive retinitis pigmentosa (arRP) caused by a novel Receptor Expression Enhancing Protein 6 (REEP6) homozygous mutation.
Patient and methods
A 33-year-old man underwent comprehensive ophthalmic examinations, including visual acuity measurements, dilated fundus imaging, electroretinography (ERG), and spectral-domain optical coherence tomography (SD-OCT). Short-wavelength fundus autofluorescence (SW-AF) and near-infrared fundus autofluorescence (NIR-AF) were also evaluated. Whole exome sequencing (WES) was used to identify potential pathogenic variants.
Results
Fundus examination revealed typical RP findings with additional temporal ten micron yellow dots. SD-OCT imaging revealed cystoid macular edema and perifoveal outer retinal atrophy with centrally preserved inner segment ellipsoid zone (EZ) bands. Hyperreflective spots were seen in the inner retinal layers. On SW-AF images, a hypoautofluorescent area in the perifoveal area was observed. NIR-AF imaging revealed an irregularly shaped hyperautofluorescent ring. His visual acuity was mildly affected. ERG showed undetectable rod responses and intact cone responses. Genetic testing via WES revealed a novel homozygous mutation (c.295G>A, p.Glu99Lys) in the gene encoding REEP6, which is predicted to alter the charge in the transmembrane helix.
Conclusions
This report is not only the first description of a Cameroonian patient with arRP associated with a REEP6 mutation, but also this particular genetic alteration. Substitution of p.Glu99Lys in REEP6 likely disrupts the interactions between REEP6 and the ER membrane. NIR-AF imaging may be particularly useful for assessing functional photoreceptor cells and show an “avocado” pattern of hyperautofluorescence in patients with the REEP6 mutation.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Retinitis pigmentosa (RP) is the most common inherited retinal disorder, affecting approximately 1 in 4000 people worldwide [1]. The affected individuals are characterized initially by nyctalopia secondary to loss of rods, followed by peripheral visual field constriction and severe vision loss from degeneration of cones and the RPE [2,3,4]. RP includes autosomal recessive (50–60%), autosomal dominant (30–40%), and X-linked recessive (5–15%) inheritance [2, 5]. More than 60 genes associated with RP have been identified, and the relationship between these genes and clinical features has been extensively compiled in Retnet (updated January 4, 2019; https://sph.uth.edu/retnet/).
Mutations in the gene REEP6 (Receptor Expression Enhancing Protein 6; OMIM#609346) have been recently reported to cause syndromic autosomal recessive retinitis pigmentosa (arRP) in eight unrelated families [6,7,8]. It is expressed specifically in rod photoreceptors [9], where it functions in trafficking for a subset of Clathrin-coated vesicles, and interacts with the t-SNARE, Syntaxin3 [6]. REEP6 encodes a putative endoplasmic reticulum (ER) shaping factor, which is highly expressed in rod photoreceptors [10]. The protein encoded by this gene may be involved in the transport of receptors from the ER to the cell surface, and thus, regulation of the ER membrane structure [9, 11].
The phenotype of the REEP6-associated RP is typical of RP: attenuated retinal vessels [7], bone spicules, and progressive photoreceptor cell degeneration [6,7,8]. In past reports, the ages of RP patients with the REEP6 mutation ranged from 18 to 60 years, and most of them reported night blindness starting in early childhood [6,7,8]. The ophthalmic examinations typically showed constricted visual fields; the central vision was often reduced in the fifth decade of life [7]. The rod responses of the electroretinograms (ERGs) were undetectable, and the cone responses were severely reduced at early stages [7].
In this study, we report the case of a Cameroonian patient with a novel homozygous mutation c.295G>A, p.(Glu99Lys) in REEP6. While many of the presenting features are typical for the RP phenotype, the additional small yellow dots in the temporal retina and a parafoveal irregularly shaped hyperautofluorescent ring with near-infrared fundus autofluorescence imaging (NIR-AF) could be a characteristic of the REEP6 mutation.
Patients and methods
Clinical assessment
This study was approved by the Institutional Review Board of Columbia University Medical Center and adhered to the tenets of the Declaration of Helsinki. Informed consent was obtained from the patient. The 33-year-old Cameroonian male was examined at the Columbia University Medical Center. Ophthalmic evaluations, including best-corrected visual acuity (BCVA) measurements, slit-lamp biomicroscopy, fundus examination and photography after pupillary dilation (> 7 mm), spectral-domain optical coherence tomography (SD-OCT), short-wavelength fundus autofluorescence (SW-AF, 488 nm), and near-infrared fundus autofluorescence (NIR-AF), were performed (Spectralis HRA + OCT; Heidelberg Engineering, Heidelberg, Germany). Full-field electroretinography (ff-ERGs) was performed using Dawson, Trick, and Litzkow (DTL) recording electrodes and Ganzfeld stimulation according to standards from the International Society for Clinical Electrophysiology of Vision [12]. The ERG recordings in the normal controls were performed at Columbia University Medical Center.
DNA analyses
Whole exome sequencing (WES) was obtained from the patient’s peripheral blood. It was performed with SureSelectXT Human All Exon V5 + UTRs (Agilent Technologies) capture and HiSeq2500 (Illumina) sequencing technology. Sequence reads obtained were analyzed for the presence of pathogenic mutations using the NextGENe software (Softgenetics) and our own analytical pipeline at the Laboratory of Personalized Genomic Medicine at Columbia University [13]. Identified variants were assessed for clinical phenotypic match and American College of Medical Genetics and Genomics (ACMG) guidelines for the interpretation of sequence variants [14]. The variants were confirmed by Sanger sequencing by using primers 5′- GCCTGTATCTGCTGTTCGGC-3′ (forward) and 5′-CACCATCAGACGTCCTAC TG-3′ (reverse).
Measurement of hyperautofluorescent ring on NIR-AF
The fovea was identified to be the center of the elliptical-shaped hyperautofluorescent ring. A vertical line was drawn through the fovea. The intersection of the vertical line and the outer border of the ring were used as the start and end points for measuring of nasal and temporal circumferences. The circumferences and distances of the ring were analyzed by ImageJ (https://imagej.nih.gov/ij/; provided in the public domain by the NIH, Bethesda, MD, USA).
Structural bioinformatics analysis
A BLAST search for human REEP6 against the protein database (PDB) did not return sequences producing significant alignments of greater than 31% coverage. The lack of usable template structures prevented homology-based modeling of the full-length REEP6 3-dimensional structure. Instead, we analyzed the biochemical features of the REEP6 protein using the primary sequence as input. Prediction of membrane-spanning segments in the REEP6 sequence was performed using TMHMM 2.0 [15] under default parameters. This analysis predicted residues 87-140 containing the p.Glu99Lys mutation to contain a putative transmembrane helix. This putative REEP6 transmembrane helix (residues 87-105) was then modeled in MODELLER 9.14 [16] using the ASH1L histone methyltransferase structure as a template (PDB: 4YPA; 30% sequence identity). Sequence alignments were performed with ClustalW [17] and visualized using Espript 3.0 (http://espript.ibcp.fr/). PyMOL (http://www.pymol.org/) generated all structural figures.
Results
Clinical findings
A 33-year-old man was referred for RP evaluation. Around the age of 18, he noticed problems with night vision. He was of Cameroonian descent, and family history was negative for consanguinity. One of the patient’s cousins was also affected with RP (Fig. 1), but he did not undergo genetic testing. There were no other similarly affected individuals in the family.

Segregation analysis of the REEP6 variant in a pedigree. The arrow indicates the proband described in this pedigree. Family history was negative for consanguinity
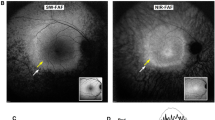
The anterior segment appeared normal and quiet on slit-lamp examination in both eyes. His best-corrected visual acuity (BCVA) was 20/30−2 in the right eye and 20/20−2 in the left eye. Fundus examination revealed attenuated retinal vessels and intra-retinal bone spicule pigmentary migration in the periphery bilaterally. Temporal tiny yellow dots and waxy-pale optic disks were also found (Fig. 2a–c′). On short-wavelength fundus autofluorescence (SW-AF) images, a hypoautofluorescent ring in the perifoveal area was observed (Fig. 2d, d′). The NIR-reflectance (NIR-R) image corresponding to the SD-OCT exhibited darkening within the macula, with a slightly brighter elliptical ring in the parafovea (Fig. 2e, e′). SD-OCT imaging revealed cystoid macular edema (CME) and perifoveal outer retinal atrophy with centrally preserved ellipsoid zone (EZ). Hyperreflective spots (HRS) were seen in the SD-OCT image within the ganglion cell layer (GCL), inner plexiform layer (IPL), and inner nuclear layer (INL) (Fig. 2f, f′). NIR-AF showed an irregular-shaped hyperautofluorescence ring in this patient (Fig. 2g, g′). The hyperautofluorescent ring had an elliptical shape. The ring extended further on the nasal side of the fovea; the nasal one was 9933 μm in the right eye and 9375 μm in the left eye, while the temporal circumference of the ring was 8290 μm in the right eye and 7995 μm in the left eye. The horizontal diameter (right eye: 3509 μm, left eye: 3370 μm) was longer than the vertical diameter (right eye: 2117 μm, left eye: 1975 μm). The location of the outer border of the ring was not well defined in either SW-AF or NIR-R images. However, in the NIR-AF image, the location of the outer border corresponded to the position in the SD-OCT scans where the ellipsoid zone (EZ) band was at least partially intact. Scotopic rod-specific and maximal responses on ff-ERG were extinguished in both eyes. Photopic cone-specific and 30 Hz-flicker amplitudes were significantly abnormal bilaterally, but there was approximately 3 μV on the cone response (Fig. 3).

Multimodal retinal imaging of both eyes in the patient. a, a′ Color fundus photography shows retinal degeneration, macular atrophy, peripheral bone-spicule pigmentation, temporal tiny yellow dots, narrowed retinal vessels, and waxy optic disk pallor. b, b′ Posterior pole (white dot frame of a, a′) is magnified. c, c′ Peripheral features (yellow frame of a, a′) are also magnified. d, d′ Short-wavelength fundus autofluorescence imaging showed diffuse perimacular hypoautofluorescence. e, e′ Near-infrared (NIR) reflectance showed hyporeflectivity around the fovea, which was surrounded by a slightly brighter elliptical ring with unclear border. f, f′ Spectral-domain optical coherence tomography showed cystoid macular edema and perifoveal outer retinal atrophy with centrally preserved inner segment ellipsoid (ISe) bands. Hyperreflective spots were seen under the ganglion cell, inner plexiform and inner nuclear layers. g, g′ NIR autofluorescence showed an elliptical hyperautofluorescence ring resembling an “avocado”

Electroretinography (ERG) of the patient and a normal control. Scotopic rod-specific ERG b-wave amplitudes were 9.744 μV in the right and 10.02 μV in the left. Maximal ERG a- and b-wave amplitudes were 15.75 and 22.49 μV in the right, 15.29 and 21.77 μV in the left. Photopic single-flash responses had a b-wave amplitude and implicit time of 4.005 μV and 26 ms in the right, 4.752 μV and 37 ms in the left. Photopic 30 Hz amplitudes were reduced to 3.174 μV in the right eye and 2.625 μV in the left, with implicit times of 39 and 32 ms in the right and left eye, respectively
Mutation analyses
Genetic testing of the proband via WES revealed a homozygous missense variant c.295G>A in the gene REEP6 (NM_138393.3; OMIM# 609346) (Fig. 4a), and it is confirmed by Sanger sequencing (Fig. 4b). A multiple sequence alignment of REEP6 homologs revealed that the Glu99 residue is 100% conserved among vertebrates and among REEP paralogs (Fig. 4c, d). This change replaces the highly evolutionarily conserved glutamic acid with a lysine, and in silico algorithms predicted it was deleterious to protein structure and/or function (SIFT Score: 0.00; Provean Score: − 3.99). This variant occurred at a very low allele frequency of 8.137E−6 (2 heterozygotes, 0 homozygotes out of 245,790 individuals) in the Genome Aggregation Database (gnomAD) exomes and was absent in genomes (median coverage: 34.0X), indicating it was not a common benign polymorphism represented by these databases. Results obtained with the bioinformatics software Mutation Taster (http://www.mutationtaster.org/) indicated that the mutation was predicted to be “disease causing.” Other rare variants identified in WES analysis were excluded based on clinical assessment of phenotypic fit, or were benign or likely benign, using ACMG guidelines for the interpretation of sequence variants [14]. Based on the prediction of membrane-spanning residues by TMHMM, Glu99 is predicted to lie in a putative transmembrane region, C-terminal to a stretch of six hydrophobic residues (residues 93-98; ALFGLA; Fig. 4e). Substitution of a glutamic acid to a lysine in this region would alter the charge, potentially disrupting interactions between REEP6 and the ER membrane (Fig. 4f).

Identification of a variant in REEP6 gene and protein structural modeling of the mutation. a Alignment of WES data in the REEP6 gene, with the reference sequence above, the patient’s arrangement below, and the changes highlighted in blue. In the reads, there is an G to A change at base pair 295 of the coding sequence resulting in a missense change from glutamine to lysine at position 99. b The homozygous mutation of c.295G>A in REEP6 is confirmed by Sanger sequencing. c Based on protein alignment, the affected amino acid is highly conserved in vertebrates and located in a conserved region of the protein. d The phylogenetic tree of REEP6 with other REEP paralogs. e Based on the prediction of membrane-spanning residues by TMHMM, Glu99 is predicted to lie in a putative transmembrane region. f Substitution of a glutamic acid to a lysine in the putative transmembrane region would alter the charge
Discussion
REEP6 is essential for the sorting of certain proteins involved in phototransduction. In particular, the absence of REEP6 impacts the synthesis, stability, and trafficking phototransduction proteins such as phosphodiesterase-6 (PDE6) and guanylate cyclases (GCs) in the outer segment of rods [6, 18]. The patient described in this study was found to harbor a homozygous missense variant mutation (c.295G>A, p.Glu99Lys) in the putative membrane-spanning region in REEP6. Alteration of a charge in this transmembrane region likely disrupts interactions between REEP6 and the ER membrane. Based on these results, we predict that the p.Glu99Lys mutation may affect REEP6 function by disrupting phototransduction protein localization.
The patient in this study had a phenotype typical of autosomal recessive RP. Past studies have shown severely contracted retinal vessels, mid-peripheral RPE atrophy, diffuse retinal atrophy, and intra-retinal pigmented bone spicules in individuals with homozygous mutations in the REEP6 gene of exon 1, 3, 4, and 5 [7]. In comparison, the phenotype of our patient was not severe, as only slight vascular attenuation and minor pigmentary changes in the form of bone spicules. These differences may be attributed to the relatively early stage of the disease process in our patient. A homozygous missense variant in REEP6 (c.279_280del, p.Leu94Valfs*320) was recently been reported in a 29-year-old man [6], whose mutation was also in exon 3 with a phenotype (and age) similar to the patient in this study. We also found HRS located in the GCL, IPL, and INL. According to Vujosevic et al.’s study [19], HRS may represent a biomarker of microglial activation in the retina. Microglial cells are activated in neurodegenerative diseases and migrate beyond the retinal layers where degeneration is occurring [20]. Some HRS have been proposed as indicators of a prevalent inflammatory condition [21]. With progressing retinopathy, HRS reaches the outer retinal layer. The HRS found in the outer retina of diabetic macular edema patients are associated with disrupted external limiting membrane and IS/OS anatomy and are closely related to decreased visual acuity, suggesting they are derived from degraded photoreceptors or macrophages that phagocytose them [22]. We suspect that HRS in patients with RP and diabetic macular edema have similar mechanisms, but whether or not the location of HRS in these patients is different is currently unknown.
The apparent hyperautofluorescence in the NIR-AF images may be indicative of the preservation of central retina. It is notable that the patient in this study showed an “avocado-shaped” elliptical, hyperautofluorescent ring around the fovea on the NIR-AF image, which is more clearly distinguishable than the ring on the SW-AF image. The outer border of the ring corresponded to the location in SD-OCT scans where the EZ band was intact. The EZ is now thought to be formed mainly by mitochondria within the EZ band of the outer portion of the inner segments of the photoreceptors [23]. Loss of REEP6 function leads to ER stress and abnormal mitochondrial proliferation, eventually leading to photoreceptor degeneration [18]. The NIR-AF may be a viable choice for identifying the shape and position of the EZ band, thus allowing us to indirectly infer the function of photoreceptor cells in patients with REEP6 mutations.
Although it is still not clear why the patient’s rod ERG signal was reduced, it is noteworthy that a previous study [7] also showed rod dysfunction in patients with a homozygous REEP6 mutation. Veleri S et al. [6] reported similar ERG findings in their patients. There was generalized retinal dysfunction in our patient affecting the rods more than the cones. Thus, homozygous or compound heterozygous mutations in the REEP6 gene may cause typical RP with retinal dysfunction of various degrees. Further study is needed, however, as we do not know with certainty that the mutations of REEP6 in exon 3 truly have slower progression than mutation sites in other exons.
The patient is currently undergoing treatment of macular edema. Although no effective therapy is available on human REEP6-associated RP, attempts at replacing REEP6 by adeno-associated virus vectors have been reported in mouse models of RP, which showed significant improvements in retinal function and morphology after a prolonged period [18]. Further, gene therapy-related research and signaling pathway studies for REEP6 are expected.
In conclusion, we report the clinical features of an African patient with arRP caused by a novel homozygous nonsense variant c.295G>A, (p. Glu99Lys) in the REEP6 gene. Future studies will more accurately determine the clinical course of REEP6-related RP patients as well as the precise boundaries of REEP6 mutations.
References
Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, Hoyng CB, Roepman R, Klevering BJ (2018) Non-syndromic retinitis pigmentosa. Prog Retin Eye Res 66:157–186. https://doi.org/10.1016/j.preteyeres.2018.03.005
Hartong DT, Berson EL, Dryja TP (2006) Retinitis pigmentosa. Lancet 368(9549):1795–1809. https://doi.org/10.1016/S0140-6736(06)69740-7
Hamel C (2006) Retinitis pigmentosa. Orphanet J Rare Dis 1:40. https://doi.org/10.1186/1750-1172-1-40
Bhatia S, Goyal S, Singh IR, Singh D, Vanita V (2018) A novel mutation in the PRPF31 in a North Indian adRP family with incomplete penetrance. Doc Ophthalmol 137(2):103–119. https://doi.org/10.1007/s10633-018-9654-x
Bunker CH, Berson EL, Bromley WC, Hayes RP, Roderick TH (1984) Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol 97(3):357–365
Veleri S, Nellissery J, Mishra B, Manjunath SH, Brooks MJ, Dong L, Nagashima K, Qian H, Gao C, Sergeev YV, Huang XF, Qu J, Lu F, Cideciyan AV, Li T, Jin ZB, Fariss RN, Ratnapriya R, Jacobson SG, Swaroop A (2017) REEP6 mediates trafficking of a subset of Clathrin-coated vesicles and is critical for rod photoreceptor function and survival. Hum Mol Genet 26(12):2218–2230. https://doi.org/10.1093/hmg/ddx111
Arno G, Agrawal SA, Eblimit A, Bellingham J, Xu M, Wang F, Chakarova C, Parfitt DA, Lane A, Burgoyne T, Hull S, Carss KJ, Fiorentino A, Hayes MJ, Munro PM, Nicols R, Pontikos N, Holder GE, Ukirdc AC, Raymond FL, Moore AT, Plagnol V, Michaelides M, Hardcastle AJ, Li Y, Cukras C, Webster AR, Cheetham ME, Chen R (2016) Mutations in REEP6 cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet 99(6):1305–1315. https://doi.org/10.1016/j.ajhg.2016.10.008
Mejecase C, Mohand-Said S, El Shamieh S, Antonio A, Condroyer C, Blanchard S, Letexier M, Saraiva JP, Sahel JA, Audo I, Zeitz C (2018) A novel nonsense variant in REEP6 is involved in a sporadic rod-cone dystrophy case. Clin Genet 93(3):707–711. https://doi.org/10.1111/cge.13171
Hao H, Veleri S, Sun B, Kim DS, Keeley PW, Kim JW, Yang HJ, Yadav SP, Manjunath SH, Sood R, Liu P, Reese BE, Swaroop A (2014) Regulation of a novel isoform of Receptor Expression Enhancing Protein REEP6 in rod photoreceptors by bZIP transcription factor NRL. Hum Mol Genet 23(16):4260–4271. https://doi.org/10.1093/hmg/ddu143
Keeley PW, Luna G, Fariss RN, Skyles KA, Madsen NR, Raven MA, Poche RA, Swindell EC, Jamrich M, Oh EC, Swaroop A, Fisher SK, Reese BE (2013) Development and plasticity of outer retinal circuitry following genetic removal of horizontal cells. J Neurosci 33(45):17847–17862. https://doi.org/10.1523/JNEUROSCI.1373-13.2013
Bjork S, Hurt CM, Ho VK, Angelotti T (2013) REEPs are membrane shaping adapter proteins that modulate specific g protein-coupled receptor trafficking by affecting ER cargo capacity. PLoS ONE 8(10):e76366. https://doi.org/10.1371/journal.pone.0076366
McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, Bach M (2015) ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol 130(1):1–12. https://doi.org/10.1007/s10633-014-9473-7
Wang Y, Lichter-Konecki U, Anyane-Yeboa K, Shaw JE, Lu JT, Ostlund C, Shin JY, Clark LN, Gundersen GG, Nagy PL, Worman HJ (2016) A mutation abolishing the ZMPSTE24 cleavage site in prelamin A causes a progeroid disorder. J Cell Sci 129(10):1975–1980. https://doi.org/10.1242/jcs.187302
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305(3):567–580. https://doi.org/10.1006/jmbi.2000.4315
Webb B, Sali A (2016) Comparative protein structure modeling using MODELLER. Curr Protoc Bioinform 54:561–5637. https://doi.org/10.1002/cpbi.3
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–4680
Agrawal SA, Burgoyne T, Eblimit A, Bellingham J, Parfitt DA, Lane A, Nichols R, Asomugha C, Hayes MJ, Munro PM, Xu M, Wang K, Futter CE, Li Y, Chen R, Cheetham ME (2017) REEP6 deficiency leads to retinal degeneration through disruption of ER homeostasis and protein trafficking. Hum Mol Genet 26(14):2667–2677. https://doi.org/10.1093/hmg/ddx149
Vujosevic S, Bini S, Torresin T, Berton M, Midena G, Parrozzani R, Martini F, Pucci P, Daniele AR, Cavarzeran F, Midena E (2017) hyperreflective retinal spots in normal and diabetic eyes: B-scan and en face spectral domain optical coherence tomography evaluation. Retina 37(6):1092–1103. https://doi.org/10.1097/IAE.0000000000001304
Zhang L, Cui X, Jauregui R, Park KS, Justus S, Tsai YT, Duong JK, Hsu CW, Wu WH, Xu CL, Lin CS, Tsang SH (2018) Genetic rescue reverses microglial activation in preclinical models of retinitis pigmentosa. Mol Ther 26(8):1953–1964. https://doi.org/10.1016/j.ymthe.2018.06.014
Bonfiglio V, Reibaldi M, Pizzo A, Russo A, Macchi I, Faro G, Avitabile T, Longo A (2018) Dexamethasone for unresponsive diabetic macular oedema: optical coherence tomography biomarkers. Acta Ophthalmol. https://doi.org/10.1111/aos.13935
Uji A, Murakami T, Nishijima K, Akagi T, Horii T, Arakawa N, Muraoka Y, Ellabban AA, Yoshimura N (2012) Association between hyperreflective foci in the outer retina, status of photoreceptor layer, and visual acuity in diabetic macular edema. Am J Ophthalmol 153(4):710–717. https://doi.org/10.1016/j.ajo.2011.08.041
Tao LW, Wu Z, Guymer RH, Luu CD (2016) Ellipsoid zone on optical coherence tomography: a review. Clin Exp Ophthalmol 44(5):422–430. https://doi.org/10.1111/ceo.12685
Funding
YCL is supported by the China Scholarship Council (No. 201806320164). SHT and the Jonas Children’s Vision Care is supported by the National Institute of Health 5P30CA013696, U01EY030580, R24EY027285, 5P30EY019007, R01EY018213, R01EY024698, R01EY026682, R21AG050437, the Schneeweiss Stem Cell Fund, New York State [SDHDOH01-C32590GG-3450000], the Foundation Fighting Blindness New York Regional Research Center Grant [C-NY05-0705-0312], Nancy & Kobi Karp, the Crowley Family Funds, The Rosenbaum Family Foundation, Alcon Research Institute, the Gebroe Family Foundation, the Research to Prevent Blindness (RPB) Physician-Scientist Award, unrestricted funds from RPB, New York, NY, USA. JRS is supported by a NIH Grant [R01EY024091]. VBM is supported by NIH Grants [R01EY026682, R01EY024665, R01EY025225, R01EY024698, R21AG050437, and P30EY026877], and Research to Prevent Blindness (RPB), New York, NY. GV is supported by NIH Grants [F30EYE027986 and T32GM007337].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest to disclose for any author.
Ethics approval and consent to participate
All study procedures were defined, and patient consent was obtained as outlined by the protocol #AAAB6560 approved by the Institutional Review Board at Columbia University Medical Center. The study adhered to the tenets of the Declaration of Helsinki.
Informed consent
Informed consent was obtained from the patient.
Statement of human rights
This study was approved by the Institutional Review Board of Columbia University Medical Center and adhered to the tenets of the Declaration of Helsinki.
Statement on the welfare of animals
This article does not contain any studies with animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lin, Y., Xu, C.L., Velez, G. et al. Novel REEP6 gene mutation associated with autosomal recessive retinitis pigmentosa. Doc Ophthalmol 140, 67–75 (2020). https://doi.org/10.1007/s10633-019-09719-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10633-019-09719-1