
Abstract
Cervical cancer (CC) is a common gynecological tumor, ranking second in the female reproductive system tumor. The work aims to study the function of miR-17-5p in the occurrence and pathogenesis of CC. We collected 36 cases of CC tissues for clinical analysis, and two CC cell lines (C33a and HCC94) were obtained for cellular analysis. As expected, the up-regulated miR-17-5p and down-regulated TIMP2 were detected in CC tissues and cell lines by RT-qPCR, in contrast with their normal counterparts. Then, overexpression of miR-17-5p significantly increased the CC cells viability and colonies formation abilities. Moreover, the Transwell analysis revealed that miR-17-5p promoted the capability of invasion and migration. Meanwhile, the expression levels of MMP2 and MMP9 was inhibited by the inhibition of miR-17-5p. The luciferase analysis demonstrated that TIMP2 was the target of miR-17-5p. In addition, cell proliferation, invasion and migration in HCC94 cells were repressed by silencing miR-17-5p, which were reversed by TIMP2 knockdown. In summary, all results indicated that miR-17-5p targeted TIMP2 to modulate CC cells’ proliferation, invasion and migration through MMPs signaling pathway; and the miR-17-5p/TIMP2/MMPs signaling pathway had the potential to become a therapeutic target of CC for clinical utilization.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cervical cancer (CC) originates from human papillomavirus infection of normal cervical epithelium, developing to cervical intraepithelial neoplasia and further into invasive squamous cell carcinoma. (Farchoukh et al. 2020; Kumar et al. 2020; Ono et al. 2020). CC is still the world’s second-ranked gynecological tumor and causes high mortality in developing countries’ female (Asgary et al. 2020; Siegel et al. 2020). MicroRNAs (miRNAs) participated in modulating cell cycle disorders of cervical squamous cells, changes in invasion ability and epithelial cell transformation and other processes to promote the occurrence and development of CC, which may become early diagnostic and prognostic biomarker for CC(Banno et al. 2014; Tornesello et al. 2020).
MiRNAs are regulators of post-transcriptional levels, inhibiting the translation of mRNA or catalyze the cleavage of mRNA to prevent the expression of target mRNA (Farazi et al. 2013; He et al. 2020). miR-17-5p is a member of miR-17-92 cluster (Wei et al. 2012; Wong et al. 2010), which usually involved in dysregulated physiological processes in cancer(Shen et al. 2019). miR-17-5p has tissue specificity in CC patients and served as possible marker for early detection, predicting prognosis and monitoring their response to treatment in CC patients (Agrawal et al. 2018; Hasanzadeh et al. 2019; Shukla et al. 2019). Previous research found that miR-17-5p boosted CC cell growth and invasion (Cai et al. 2018). However, the underlying mechanism of miR-17-5p affecting the growth and metastasis of CC cell is not very clear.
Therefore, this study determined miR-17-5p levels in the CC tissues and cells and explored the possible molecules of miR-17-5p on the proliferation, migration and invasion of CC cells through miR-17-5p overexpression and suppression and target gene loss experiments for providing a new enlightenment for miR-17-5p as potential target of CC therapy.
Materials and methods
Clinical sample collection
The 36 patients (age range, 51–75 years) with CC came to the Second People’s Hospital of China Three Gorges University to receive surgical removal of the tumor tissue and had never received radiotherapy or chemotherapy before the operation. According to the International Federation of Obstetrics and Gynecology, the cases of patients’ tumor stage I–II and III–IV were 14 and 22, respectively. Total of 36 pairs CC samples were obtained and stored in liquid nitrogen. This study had been approved by the Research Ethics Committee of the Second People’s Hospital of China Three Gorges University. All patients signed a written informed consent form.
Cell culture and transfection
C33a, HCC94 and ECT1/E6E7 cell lines were purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences. All cells were grown in DMEM (Hyclone; Logan, UT, USA) containing 10% FBS (AusGeneX; AUS) at 37 °C cell incubator with 5% CO2. Mimics, inhibitor of miR-17-5p and small interfering RNA of TIMP2 (si-TIMP2) were bought from GenePharma (Shanghai, China). Lipofectamine 3000 (Invitrogen; Thermo Fisher Scientific, Inc.) was used to transfected mimics, inhibitor, si-TIMP2 into C33a and HCC94 cell for 24 h respectively.
CCK-8 assay
Cell viability was evaluated by CCK-8 kit (Beyotime Institute of Biotechnology) following the manufacturer’s manual. Briefly, 100 µl of cell suspension was added in 96-well plate and added sterile PBS to the wells around the wells where the cells are added. The culture plate was incubated in an incubator for 0, 24, 48, 72 h. Then 10 µl CCK-8 was added to each well for 4 h. The absorbance at 450nm was measuredwith a microplate.
Colony formation assay
The 6 well plate was planted with the transfected cells, and it was cultured in the incubator for 1 week. After the culture medium was discarded, each well was washed three times with PBS. The cells in each well were fixed with 4% paraformaldehyde for 20 min and then stained with Gimsa for 15 min. Finally, the cells were gently washed with running water, and the colony formation units were calculated under the microscope. The number of cells in each clone was more than 50.
Transwell assay
Matrigel (BD Biosciences, USA) was used to pre-coat 24-well Transwell chamber (Corning Inc., Corning, NY, USA) for the Transwell invasion test, and migration assay did not pre-coat up-chamber. The serum-free DMEM medium was used to dilute transfected cells. Cells were added into upper chamber and DMEM with 10% FBS was added to lower chamber. After 48 h of incubation, 4% paraformaldehyde and 0.1% crystal violet were used to treat the cells on the bottom of the upper chamber respectively. Six random fields of view were selected to count the cells under the microscope.
RT-qPCR
TRizol reagent extracted total RNA from tissues and cells. MiRNA reversal reagent ( by stem-loop method) and common RNA reversal reagent were used to obtain miR-17-5p and gene cDNA respectively. qPCR reagent (by SYBR dye method) was used for three-step amplification PCR. Finally, the relative expression level was calculated according to the 2−ΔΔCt method. The calibration internal reference of miR-17-5p was U6; GAPDH was served as internal reference to normalize the expression of TIMP2, MMP2 and MMP9 mRNA.
Western blot
The protein lysate was formulated with RIPA, PMSF and protease inhibitors, which was used to extract total protein from CC tissues and cells. The protein concentration was measured using the BCA protein quantification kit. After the protein was diluted and boiled to denature, SDS-PAGE was performed. Then cut the gel to transfer the membrane. The primary (TIMP2, MMP2, MMP9 and GAPDH) and secondary antibodies were used to incubate the membrane in turn. Finally, the protein bands were displayed under the ECL chemiluminescence solution.
Dual-luciferase reporter assay
Online predictions from the starBase and TargetScan websites found that miR-17-5p and TIMP2 had base complementary pairing regions. The wild type (WT) and mutant (Mut) type of 3′UTR of TIMP2 mRNA were amplified by PCR. The WT- and Mut- fragments and psi-CHECH2 were both digested with double restriction enzymes and inserted into the luciferase reporter plasmid with T4 ligase. Lipofectamine 3000 reagent was used to co-transfect mimics/mimics NC and WT-TIMP2/Mut-TIMP2 plasmids into HEK293T cells for 24 h. Finally, the activity of firefly luciferase and Renilla luciferase was measured with a dual luciferase detection kit.
Statistical analysis
GraphPad Prism 7.0 software was performed to statistically analyze data which were expressed as mean ± SEM. Paired Student’s t-test was hired to compare the levels of miR-17-5p and TIMP2 in CC tissues and normal tissues. Unpaired Student’s t-test and one-way ANOVA was used to compare differences between groups. P < 0.05 was recognized as significant difference.
Results
miR-17-5p was increased, while TIMP2 was reduced in CC tissues and cells
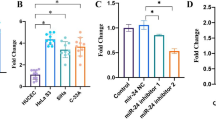
To investigate miR-17-5p and TIMP2 mRNA levels of in CC tissues and cells, RT-qPCR was performed to detect. miR-17-5p in CC patient tissues was much higher than adjacent tumor tissues (Fig. 1A), while miR-17-5p similarly rise in C33a and HCC94 contrasted to that of ECT1/E6E7 cells (Fig. 1B). However, TIMP2 mRNA level was greatly suppressed in CC tissues and cell lines (Fig. 1C, D). Western blot confirmed that TIMP2 protein levels in CC patient tissues were much lower than adjacent tumor tissues (Fig. 1E), and TIMP2 protein was similarly suppressed in the two CC cell lines (C33a and HCC94) (Fig. 1F).

miR-17-5p was increased, while TIMP2 was decreased in CC tissues and cell lines. A CC tissues and adjacent tissues were detected miR-17-5p expression using RT-qPCR. B RT-qPCR was performed to determine the levels of miR-17-5p in CC cell lines and human cervical immortalized squamous cell line ECT1/E6E7. C TIMP2 mRNA level in CC tissues and adjacent tissues were detected by RT-qPCR. D TIMP2 mRNA expression were examined by RT-qPCR in CC cell lines and ECT1/E6E7 cells. E, F TIMP2 protein was measured with western blot in tissues and cells. **p < 0.01
miR-17-5p promoted CC cells’ growth
Overexpression or silencing of miR-17-5p on C33a and HCC94 cells was set up to investigate the role of miR-17-5p on proliferation. The efficiency of overexpressed or inhibited miR-17-5p was determined, which confirmed that miR-17-5p mimics obviously up-regulated miR-17-5p level in C33a cells, while miR-17-5p expression was repressed on HCC94 cells transfected with inhibitor (Fig. 2A). Detection of cell viability by CCK-8 displayed that C33a cells viability was significantly increased by miR-17-5p (Fig. 2B), but it could accelerate cell growth after being inhibited on HCC94 cells (Fig. 2C). Colony formation analysis once again confirmed that miR-17-5p promoted cell proliferation (Fig. 2D).

Overexpressed miR-17-5p promoted proliferation in CC cells. A RT-qPCR was hired to measure miR-17-5p expression levels after transfected mimics/inhibitor into C33a and HCC94 cells. B The cell proliferation was evaluated using CCK-8 assay after transfected C33a cells with mimic or mimics NC. C The cell viability was assessed by CCK-8 assay after inhibition of miR-17-5p on HCC94 cells. D The colony formation ability of CC cells was detected by colony formation assay after transfection. *p < 0.05; **p < 0.01
Inhibition of miR-17-5p repressed invasion and migration in CC cells
Transwell analysis was used to assess the function of miR-17-5p on CC metastasis. As displayed in Fig. 3A, cell migration and invasion capabilities in mimics group was significantly enhanced compared to the mimics NC group. miR-17-5p silencing drastically suppressed migration and invasion on HCC94 cells (Fig. 3B). In addition, MMP2 and MMP9 mRNA levels in mimics group was higher than those in mimics NC group in C33a cells (Fig. 3C). However, the mRNA levels of MMP2 and MMP9 were strongly hindered on HCC9 cells transfected with inhibitor (Fig. 3D). The same result occurred on the expression of MMP2 and MMP9 proteins (Fig. 3E, F).

Inhibition of miR-17-5p suppressed migration and invasion of CC cells. A The capabilities of cell migration and invasion was examine by Transwell assay after overexpression of miR-17-5p on C33a cells, and inhibition of miR-17-5p on HCC94 cells (B). C, D RT-qPCR was hired to measure the expression levels of MMP2 and MMP9 mRNA on C33a and HCC94 cells. E, F Western blot was hired to measure MMP2 and MMP9 protein expression levels. **p < 0.01
miR-17-5p directly targeted TIMP2
To further explore the molecular mechanism of miR-17-5p on CC cells, the downstream target genes of miR-17-5p was predicted by bioinformatics software. As illustrated in Fig. 4A, miR-17-5p bind to TIMP2 mRNA. To verify whether miR-17-5p targeted a TIMP2, the luciferase assay was hired to prove, whose results suggested that miR-17-5p could obviously reduce luciferase activity of WT-TIMP2 plasmid (Fig. 4B). Moreover, RT-qPCR results displayed that miR-17-5p decreased TIMP2 mRNA level in C33a cells, but the TIMP2 mRNA level was greatly improved after inhibition of miR-17-5p on HCC94 cells (Fig. 4C). TIMP2 protein expression was also downregulated by miR-17-5p using western blot (Fig. 4D).

miR-17-5p directly targeted TIMP2. A Complementary pairing site in the seed sequence of miR-17-5p with the 3′-UTR of TIMP2 transcript. B The luciferase assay was employed to verify the targeting relationship between miR-17-5p and TIMP2. C TIMP2 mRNA levels in C33a and HCC94 cells were examined by RT-qPCR. D TIMP2 protein was determined by Western blot on C33a and HCC94 cells. **p < 0.01
Knockdown of TIMP2 reversed the effect of miR-17-5p on CC cells
Knock-down of TIMP2 on HHC94 cell using si-RNA to verify whether miR-17-5p affected biological characteristics of CC cells. The knockdown efficiency of si-TIMP2 was determined by western blot. And results demonstrated that si-TIMP2 could dramatically decreased TIMP2 protein compared with si-NC in HCC94 cells (Fig. 5A). It was discovered that miR-17-5p inhibitor repressed growth and colony formation, but knockdown of TIMP2 significantly reversed the inhibitory effects by CCK-8 and colony formation assays (Fig. 5B, C). The migration and invasion ability in the inhibitor + si-TIMP2 group was dramatically higher than the inhibitor + si-NC group (Fig. 5D, E). Meanwhile, knock-down of TIMP2 increased MMP2 and MMP9 mRNA and protein, which were decreased by miR-17-5p inhibitor alone (Fig. 5F, G).

Knockdown of TIMP2 reversed miR-17-5p function in CC cells. A TIMP2 protein in HCC94 cells knocked down TIMP2 was determined by western blot. (B, C) Cell proliferation ability of HCC94 cells was detect by CCK-8 (B) and colony formation assay (C). D Cell migration and invasion was determined with Transwell assay. E RT-qPCR was hired to analyze MMP2 and MMP9 mRNA in HCC94 cells. F TIMP2, MMP2 and MMP9 protein in HCC94 cells by Western blot. **p < 0.01
Discussion
CC is a female-specific gynecological tumor with fatality rate, especially women in developing countries (Banno et al. 2014; Reshmi and Pillai 2008). The regulatory mechanism exerted by miRNA involves proliferation, differentiation, immune response, pathogenicity and tumorigenesis (Zhou and Zheng 2019). MiRNAs may become potential marker for early diagnosis and prognostic evaluation of CC. Moreover, miRNAs participate in cellular processes included cell polarity, tumor suppression and apoptosis (Rao et al. 2012; Satapathy et al. 2017). In present work, miR-17-5p obviously increased in CC tissues and cells, and it may serve as a marker for the diagnosis of CC.
To further explore the mechanism of miR-17-5p on CC cells growth, invasion and migration were examined. It has been proved that miR-17-5p accelerate cells proliferation and invasion in cancer (Chen et al. 2020; Xie et al. 2020). Our work displayed that miR-17-5p promoted proliferation and metastasis on C33a cells, but inhibition of miR-17-5p with the opposite result. The luciferase analysis verified that miR-17-5p targeted TIMP2. TIMP2, as a tumor suppressor gene, inhibited cell growth and invasion by blocking MMPs expression (Kurzawski et al. 2017; Tjomsland et al. 2016). TIMP2 binds to MMPs to inactivate MMPs, and protect the extracellular matrix from degradation and remodeling by MMPs, thereby inhibiting tumor cell invasion (Kiani et al. 2020; Vira et al. 2020). In this study, miR-17-5p overexpression reduced the expression of TIMP2. Meanwhile, the levels of MMP2 and MMP9 were also suppressed. Knockdown of TIMP2 by si-RNA overturned the effect of miR-17-5p inhibitor on CC cell growth and invasion.
Conclusions
In summary, all data indicated that miR-17-5p enhanced cell proliferation, invasion and migration of CC cells by targeting TIMP2. CC progression may regulate through miR-17-5p/TIMP2/MMP axis, and it may become latent target for CC therapy.
Data availability
All the involved data had been included in the manuscript
References
Agrawal S, Tapmeier T, Rahmioglu N, Kirtley S, Zondervan K, Becker C (2018) The miRNA mirage: how close are we to finding a non-invasive diagnostic biomarker in endometriosis? A systematic review. Int J Mol Sci 19(2)
Asgary R, Staderini N, Mthethwa-Hleta S, Lopez Saavedra PA, Garcia Abrego L, Rusch B, Marie Luce T, Rusike Pasipamire L, Ndlangamandla M, Beideck E, Kerschberger B (2020) Evaluating smartphone strategies for reliability, reproducibility, and quality of VIA for cervical cancer screening in the Shiselweni region of Eswatini: a cohort study. PLoS Med 17(11):e1003378
Banno K, Iida M, Yanokura M, Kisu I, Iwata T, Tominaga E, Tanaka K, Aoki D (2014) MicroRNA in cervical cancer: oncomiRs and tumor suppressor miRs in diagnosis and treatment. Sci World J 178075
Cai N, Hu L, Xie Y, Gao JH, Zhai W, Wang L, Jin QJ, Qin CY, Qiang R (2018) miR-17-5p promotes cervical cancer cell proliferation and metastasis by targeting transforming growth factor-β receptor 2. Eur Rev Med Pharmacol Sci 22(7):1899–1906
Chen Y, Shen T, Ding X, Ma C, Cheng L, Sheng L, Du X (2020) lncRNA MRUL suppressed non-small cell lung cancer cells proliferation and invasion by targeting miR-17-5p/SRSF2 Axis. 9567846
Farazi TA, Hoell JI, Morozov P, Tuschl T (2013) MicroRNAs in human cancer. Adv Exp Med Biol 774:1–20
Farchoukh LF, Onisko A, Austin RM (2020) Individualized Bayesian risk assessment for cervical squamous neoplasia. J Pathol Inf 11:9
Hasanzadeh M, Movahedi M, Rejali M, Maleki F, Moetamani-Ahmadi M, Seifi S, Hosseini Z, Khazaei MA-O, Amerizadeh F, Ferns GA, Rezayi M, Avan AA-O (2019) The potential prognostic and therapeutic application of tissue and circulating microRNAs in cervical cancer. J Cell Physiol 234(2):1289–1294
He S, Yu G, Peng K, Liu S (2020) MicroRNA–145–5p suppresses fascin to inhibit the invasion and migration of cervical carcinoma cells. Mol Med Rep 22(6):5282–5292
Kiani A, Kamankesh M, Vaisi-Raygani A (2020) Activities and polymorphisms of MMP-2 and MMP-9, smoking, diabetes and risk of prostate cancer
Kumar R, Mandal S, Arora P, Mala YM, Khurana N (2020) The expression of p16 and galectin-3 in cervical intraepithelial neoplasia (CIN) and squamous cell carcinoma (SCC) uterine cervix. J Obstetr Gynaecol 1–6
Kurzawski M, Kaczmarek M, Kłysz M, Malinowski D, Kazienko A, Kurzawa R, Droździk M (2017) MMP2, MMP9 and TIMP2 polymorphisms affect sperm parameters but not fertility in Polish males. Andrologia 49(5)
Ono A, Koshiyama MA-O, Nakagawa M, Watanabe Y, Ikuta E, Seki K, Oowaki M (2020) The preventive effect of dietary antioxidants on cervical cancer development. Medicina 56(11):E604
Rao Q, Shen Q, Zhou H, Peng Y, Li J, Lin Z (2012) Aberrant microRNA expression in human cervical carcinomas. Med Oncol (Northwood Lond Engl) 29(2):1242–1248
Reshmi G, Pillai MR (2008) Beyond HPV: oncomirs as new players in cervical cancer. FEBS Lett 582(30):4113–4116
Satapathy S, Batra J, Jeet V, Thompson EW, Punyadeera C (2017) MicroRNAs in HPV associated cancers: small players with big consequences. Expert Rev Mol Diagn 17(7):711–722
Shen K, Cao Z, Zhu R, You L, Zhang T (2019) The dual functional role of microRNA-18a (miR-18a) in cancer development. Clin Transl Med 8(1):32
Shukla V, Varghese VK, Kabekkodu SP, Mallya S, Chakrabarty S, Jayaram P, Pandey D, Banerjee S, Sharan K, Satyamoorthy K (2019) Enumeration of deregulated miRNAs in liquid and tissue biopsies of cervical cancer. Gynecol Oncol 155(1):135–143
Siegel RL, Miller KD, Jemal A (2020) Cancer statistics, 2020. Cancer J Clin 70(1):7–30
Tjomsland V, Pomianowska E, Aasrum M, Sandnes D, Verbeke CS, Gladhaug IP (2016) Profile of MMP and TIMP expression in human pancreatic stellate cells: regulation by IL-1α and TGFβ and implications for migration of pancreatic cancer cells. Neoplasia (New York NY) 18(7):447–456
Tornesello ML, Faraonio R, Buonaguro L, Annunziata C, Starita N, Cerasuolo A, Pezzuto F, Tornesello AL, Buonaguro FM (2020) The role of microRNAs, long non-coding RNAs, and circular RNAs in cervical cancer. Front Oncol 10:150
Vira HJ, Pradhan VD, Umare VD, Chaudhary AK, Rajadhyksha AG, Nadkar MY, Ghosh K, Nadkarni AH (2020) Expression of the matrix metalloproteinases MMP-2 and MMP-9 and their inhibitors TIMP-1 and TIMP-2 in systemic lupus erythematosus patients. Neth J Med 78(5):261–268
Wei Q, Li YX, Liu M, Li X, Tang H (2012) miR-17-5p targets TP53INP1 and regulates cell proliferation and apoptosis of cervical cancer cells. IUBMB Life 64(8):697–704
Wong P, Iwasaki M, Somervaille TC, Ficara F, Carico C, Arnold C, Chen CZ, Cleary ML (2010) The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer Res 70(9):3833–3842
Xie W, Wang Y, Zhang Y, Xiang Y, Wu N, Wu L, Li C, Cai T, Ma X, Yu Z, Bai L, Li Y (2020) SNP rs4142441 and MYC co-modulated lncRNA OSER1-AS1 suppresses non-small cell lung cancer by sequestering ELAVL1. Cancer Sci
Zhou L, Zheng SJ (2019) The roles of MicroRNAs (miRNAs) in avian response to viral infection and pathogenesis of avian immunosuppressive diseases. Int J Mol Sci 20(21)
Acknowledgements
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zou, M., Zhang, Q. miR-17-5p accelerates cervical cancer cells migration and invasion via the TIMP2/MMPs signaling cascade. Cytotechnology 73, 619–627 (2021). https://doi.org/10.1007/s10616-021-00482-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10616-021-00482-3