
Abstract
Mature microRNAs (miRNAs) are single-stranded RNA molecules of 20–23-nucleotide (nt) length that control gene expression in many cellular processes. These molecules typically reduce the translation and stability of mRNAs, including those of genes that mediate processes in tumorigenesis, such as inflammation, cell cycle regulation, stress response, differentiation, apoptosis, and invasion. miRNA targeting is initiated through specific base-pairing interactions between the 5′ end (“seed” region) of the miRNA and sites within coding and untranslated regions (UTRs) of mRNAs; target sites in the 3′ UTR lead to more effective mRNA destabilization. Since miRNAs frequently target hundreds of mRNAs, miRNA regulatory pathways are complex. To provide a critical overview of miRNA dysregulation in cancer, we first discuss the methods currently available for studying the role of miRNAs in cancer and then review miRNA genomic organization, biogenesis, and mechanism of target recognition, examining how these processes are altered in tumorigenesis. Given the critical role miRNAs play in tumorigenesis processes and their disease specific expression, they hold potential as therapeutic targets and novel biomarkers.
An erratum to this chapter is available at 10.1007/978-94-007-5590-1_17
T. T. is cofounder of and scientific advisor to Alnylam Pharmaceuticals and scientific advisor to Regulus Therapeutics.
An erratum to this chapter can be found at http://dx.doi.org/10.1007/978-94-007-5590-1_17
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1.1 miRNA Overview
miRNAs were originally shown to be important in timing of larval development in C. elegans, leading to the identification of the miRNAs lin-4 and let-7 [1, 2]. Our initial understanding of miRNA-mRNA target recognition came from observations of sequence complementarity of the lin-4 RNA to multiple conserved sites within the lin-14 3′ UTR [1, 3]; molecular genetic analysis had shown that this complementarity was required for the repression of lin-14 by lin-4 [4]. Homologues of let-7 or lin-4/mir-125 were thereafter shown to have temporal expression patterns in other organisms, including mammals, and to regulate mammalian development [5–8]. Given their integral role in development, it was no surprise that miRNAs were soon found to be important in tumorigenesis, and since their discovery close to 5,000 publications associate miRNAs to cancer, including over 1,000 reviews (recent examples include [9–11]). miRNAs were initially linked to tumorigenesis due to their apparent proximity to chromosomal breakpoints [12] and their dysregulated expression levels in many malignancies [13, 14].
Given the wealth of rapidly accumulating information implicating miRNAs in cancer, to allow the reader to critically assess the reports exploring the function of miRNAs in malignancies, we first review the methods used to study the expression and role of miRNAs in tumors, and then review the evidence that relates miRNA genomic organization, biogenesis, target recognition and function to tumorigenesis. An overview of miRNA cistronic expression and sequence similarity allows a better understanding of the regulation of miRNA expression and the factors contributing to technical limitations in accuracy of miRNA detection. Understanding the regulatory potential of miRNAs based on sequence similarity families and miRNA abundance allows evaluation of which miRNAs are important regulators of tumorigenesis pathways.
1.2 Methods for Studying miRNA Genetics and Expression
1.2.1 miRNA Profiling
The main methods currently used for miRNA profiling are sequencing, microarray and real-time RT-PCR based approaches (reviewed in [15–17]). The input material initially used for these studies comprised high quality preserved fresh frozen samples, but recently it has been possible to obtain reproducible and comparable profiles using formalin-fixed paraffin-embedded tissues (FFPE), making these archived tumor collections accessible for study [18–20]. Microarrays generally provide fold-changes in miRNA expression between samples, with members of miRNA sequence families prone to cross-hybridization [21–24]. More recently, calibration cocktails of synthetic miRNAs were used in array experiments to derive absolute abundance of miRNAs [25]. RT-PCR methods are lower throughput and require normalization (i.e. candidate reference genes including other small noncoding RNAs [26, 27]). Mean expression normalization has been suggested as an alternative RT-PCR normalization method for reduction of technical variation to allow appreciation of biological changes [28]. If external miRNA standards are used for quantification (i.e. [29, 30]), the most abundant miRNA, which may vary in length due to 3′ end heterogeneity, should be used as a calibration standard. Sequencing methods, besides their obvious potential to identify new miRNAs, editing and mutation events, estimate miRNA abundance based on frequency of sequence reads (e.g. [5, 7, 8, 31–34]). Given the dramatic increase in sequencing power, bar-coding samples can allow multiple specimens to be processed at the same time, reducing the cost and effort of profiling, and paving the way for large specimen studies [34–36]. Ligation biases between miRNAs and 5′ and 3′ adapters for RT-PCR amplification exist in sequencing methods, and miRNA read frequencies may not always reflect the absolute expression levels, but these variations are irrelevant when monitoring fold-changes between samples. A study with a synthetic pool of 770 miRNA sequences showed that overall, these biases do not prevent identification of miRNAs, and allowed estimation of these biases [36]. For example, certain miRNAs could be over-represented due to higher ligation efficiency (such as miR-21, which was ∼2-fold over-represented), while other miRNAs could be under-represented (such as miR-31, which was >5-fold under-represented). However, given the increasing depth of sequencing, most under-represented miRNAs are identified with sufficient sequence reads to allow for a statistically significant comparison across parallel processed samples.
Recent studies have compared the results obtained using multiple platforms [37]. A study of miRNA expression in liposarcoma revealed excellent agreement between bar-coded next generation sequencing and microarray profiles [38], while another study of miRNA expression in breast cancer showed good agreement between bar-coded sequencing and another hybridization-based method, Northern blotting [39].
Finally, choosing the appropriate statistical analysis to evaluate the data depends on the methodology used to obtain the profiles, ranging from established SAM analysis for microarray data [40], to newly developed techniques for sequencing data [34, 41, 42]. Recent in situ hybridization (ISH) advances allowed sensitive detection of miRNAs in heterogeneous tissues, defining miRNA cellular localization [43–45]. The potential of miRNA localization to suggest function for a subpopulation of cells was demonstrated early on, as in the case of lsy-6 expressed in less than ten neurons in C. elegans controlling left/right asymmetry [46].
1.2.2 miRNA Databases and Validation
It is critical to know which miRNAs are validated and have the potential to regulate cellular functions, especially given the frequent revisions of the miRNA database, miRBase (www.mirbase.org) [47], and the dramatic increase in the number of novel and re-annotated miRNAs through the use of deep-sequencing technologies. It is extremely challenging to establish the validity of novel miRNAs, particularly when their definition is based on a handful of sequence reads. The latest release of miRBase (version 17) includes 1,424 human miRNA precursors. Compared to version 16, version 17 includes 385 novel human miRNA precursors, 45 name changes, 1 sequence revision, and the removal of 2 precursors. Given the recent explosion in acquisition of next generation sequencing profiles, miRBase has now added features to allow evaluation of microRNA annotation [48]. The database mapped reads from short RNA deep-sequencing experiments to miRNAs and developed web interfaces to view these mappings. This is an important step in characterizing the newly identified miRNAs as prototypical miRNAs (consisting of a hairpin structure and processing sites consistent with RNase III cleavage steps).
The challenge of constantly revising and curating existing databases based on newly acquired sequencing data is illustrated in two recent studies re-evaluating mouse and human miRNAs. A recent study of 60 million small RNA sequence reads generated from a variety of adult and embryonic mouse tissues confirmed 398 annotated miRNA genes and identified 108 novel miRNA genes but was unable to find sequencing evidence for 150 previously annotated mouse miRNAs. Ectopic expression of the confirmed and newly identified miRNA hairpin sequences yielded small RNAs with the classical miRNA features but failed to support other previously annotated sequences (of the 17 tested miRNAs with no read evidence, only one yielded a single sequence read, while of 28 tested miRNAs with insufficient number of reads, only 4 were verified) [49]. A more recent study has re-annotated human miRNAs based on read evidence from over 1,000 human samples [39]. miRNAs were curated both on the basis of read counts, as well as patterns compatible with traditional miRNA processing, re-defining prototypical miRNAs (557 precursors, corresponding to 1,112 mature and star sequences (miRNA*, described in the following section), miR-451 and miR-618 being the only miRNAs without a star sequence). 269 not yet reported star sequences were added (compared to miRBase 16), putative miRNAs from miRBase, for which read evidence was not obtained, were ignored, and specific miRNAs were renamed according to the read ratio between mature and star sequences. The importance of curated miRNA databases is especially evident in assessing the statistical significance of differentially expressed miRNAs to identify potential biomarkers based on microarray studies. Including miRNAs without strong read evidence in such comparisons could skew the results.
1.3 Mechanisms of Alteration of miRNA Levels in Malignancy
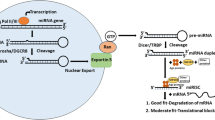
We review miRNA biogenesis (Fig. 1.1) and illustrate which steps of the biogenesis pathway are linked to malignancy, starting from miRNA genomic localization, transcriptional regulation, processing steps and post-transcriptional modification. There is evidence supporting the association of the first three processes and/or the factors that control them with tumorigenesis, whereas evidence relating post-transcriptional miRNA modifications to cancer is not clear-cut.

miRNA biogenesis pathway. miRNAs are transcribed by RNAPII to produce pri-miRNAs. Canonical miRNAs are processed by the endoribonuclease Drosha in partnership with its RBP partner DGCR8; mirtrons are instead processed by the spliceosome. The processed pre-miRNA is transported to the cytoplasm through an export complex consisting of exportin 5. The pre-miRNA is subsequently processed in the cytoplasm by another endoribonuclease Dicer in partnership with its RBP partner TRBP to form the final 21–23 nucleotide miRNA product. miR-451 is not processed by Dicer, but is rather cleaved by AGO2. Mature miRNAs (indicated in red) are then incorporated into AGO 1 through 4, forming miRNPs, also known as miRISC. miRNPs also incorporate other proteins, such as GW182. miRNPs are thought to direct miRNA mediated destabilization (i.e. through interaction with CCR4) or miRNA mediated translational repression (i.e. through interaction with ribosomes) of miRNAs without perfectly complementary mRNA targets. miRISC is thought to direct AGO2 catalyzed target mRNA cleavage of miRNA fully or nearly fully complementary mRNA targets
1.3.1 General Principles of miRNA Genomic Organization
miRNAs are frequently expressed as polycistronic transcripts. To date, 1,424 human miRNA precursor sequences have been deposited in miRBase [47]. Approximately one-third (497) of these miRNAs are located in 156 clusters, each measuring ≤51 kb in the human genome (51 kb being the longest distance between miRNAs belonging to the same cluster, Fig. 1.2). These miRNA clusters are co-expressed based on evidence from miRNA profiling data from a variety of tissues and cell lines [22, 33, 34, 49]. The genomic organization of representative oncogenic (miR-17 and miR-21) and tumor suppressor (let-7 and miR-141) sequence families (described in following section) is illustrated in Fig. 1.2. Presentation of miRNA profiles in the form of expression clusters provides a readily interpretable summary of expression data and stresses the importance of cistronic expression regulation; dysregulation of one member of the cluster should be accompanied by similar dysregulation of other cluster members [39]. Since miRNA genes are frequently multi-copy, determining the relative contribution of each genomic location to mature miRNA expression is challenging.

miRNA genomic and functional organization. The genomic and functional organization of four miRNA clusters is clarified: (a) let-7/mir-98 cluster, (b) mir-141/mir-200a cluster, (c) mir-21 cluster and (d) mir-17-92 cluster. The genomic locations for each of the miRNA members are defined. Grey lines denote intronic regions. miRNA mature sequences are color coded according to the sequence family they belong to (i.e. in the let-7/mir-98 cluster red signifies the let-7 sequence family). The star sequence is defined with a grey bar. The sequence families are depicted as sequence alignments compared to the most highly expressed miRNA family member shown on top, based on profiles of over 1,000 human specimens [39]. Shaded residues denote differences from the most highly expressed miRNA family member
1.3.2 Alterations in Genomic miRNA Copy Numbers and Location
Changes in miRNA expression between normal and tumor specimens are often attributed to the location of miRNAs in regions of chromosomal instability (amplification, translocation or deletion), or nearby chromosomal breakpoints, initially locating 52.5% of miRNA genes in cancer-associated regions or fragile sites [12]. The miRNA cluster mir-15a/16-1 is located in a frequently deleted genomic locus containing a putative tumor suppressor containing region in chronic B-cell lymphocytic leukemia (B-CLL) [50]. Other examples include deletion of let-7g/mir-135-1 in a variety of human malignancies [12], amplification of mir-17-92 cluster in lymphoma [51], translocation of mir-17-92 in T-cell acute lymphoblastic leukemia (T-ALL) [52] and amplification of mir-26a in glioblastoma [53].
1.3.3 Alterations in miRNA Transcriptional Regulation
Some autonomously expressed miRNA genes have promoter regions that allow miRNAs to be highly expressed in a cell-type-specific manner, and can even drive high levels of oncogenes in cases of chromosomal translocation. The mir-142 gene, strongly expressed in hematopoietic cells, is located on chromosome 17 and was found at the breakpoint junction of a t(8;17) translocation to MYC, which causes an aggressive B-cell leukemia [54]. The translocated MYC gene, which was also truncated at the first exon, was located only four nucleotides from the 3′ end of the mir-142 precursor, placing it under the control of the upstream mir-142 promoter. In an animal model for Hepatocellular Carcinoma (HCC), a similar event placed C-MYC downstream of the mir-122a promoter, which is active only in hepatocytes [55].
Many transcription factors regulate miRNA expression in a tissue-specific and disease state-specific fashion, and some miRNAs are regulated by well-established tumor suppressor or oncogene pathways such as TP53, MYC, and RAS (reviewed in [56]). The miRNA and its transcriptional regulators can participate in complex feedback regulation loops. Examples include the TP53 regulated mir-34a [57, 58], the RAS regulated mir-21 [33, 59, 60] and the MYC regulated mir-17-92 gene cluster [61, 62].
miRNA dysregulation has also been linked to changes in epigenetic regulation, such as the methylation status of miRNA genes, which results in alterations in their expression levels [63, 64]. Examples of methylated miRNA genes include mir-127 in bladder cancer cells [65] and mir-9-1 in breast cancer [66].
1.3.4 miRNA Biogenesis Pathway in Tumorigenesis
miRNA biogenesis has been reviewed extensively [56, 67–73] (Fig. 1.1). miRNA pathway components could either be mis-expressed in tumors or mutated (reviewed in [74, 75]). Post-transcriptional regulation of miRNAs themselves through RNA editing or terminal modifications was shown to alter miRNA targeting, processing and stability, but connection of these modifications to tumorigenesis has not yet been definitive (reviewed in [56, 75, 76]).
1.3.4.1 miRNA Biogenesis
Briefly, the mature 20–23-nt miRNA molecules are excised in a multi-step process from primary transcripts (pri-miRNAs) that contain one or more 70-nt hairpin miRNA precursors (pre-miRNA) and have their own promoters or share promoters with coding genes. These hairpin structures are recognized in the nucleus by DGCR8, a double-stranded RNA-binding protein (dsRBP), and RNASEN, also known as RNase III Drosha, and excised to yield pre-miRNAs. These molecules are subsequently transported by XPO5 (exportin 5) to the cytoplasm where they are further processed by DICER1 (Dicer) in complex with the dsRBPs TARBP2 (TRBP) and/or PRKRA to yield an RNA duplex processing intermediate composed of mature miRNA and miRNA* sequences. Some miRNAs bypass the general miRNA processing and their maturation can be independent of DGCR8 and RNASEN, such as miR-320 or miR-484 [77], or are DICER1 independent, such as erythropoiesis-related miR-451 [78, 79]. DGCR8 and RNASEN independent miRNAs include mirtrons and tailed mirtrons, which release their pre-miRNA by splicing and exonuclase trimming [80, 81]. A recent review describes alternative processing pathways and enumerates settings in which alternative miRNA pathways contribute to distinct phenotypes among miRNA biogenesis mutants [82].
While the mature miRNA is loaded into the Argonaute/EIF2C (AGO) proteins that are at the core of the miRNA-containing ribonucleoprotein complex (miRNP), sometimes also referred to as RNA-induced silencing complex (miRISC), the miRNA* is released and degraded. miR-451 is generated from an unusual hairpin structure that is processed by AGO2 instead of DICER1 [78, 79]. The miRNPs contain a member of the AGO family (1–4), which binds the miRNA and mediates target mRNA recognition. Several other RBPs have been implicated in miRNA biogenesis, including DHX9, DDX6, MOV10, DDX5, DDX17, LIN28A, HNRNPA1 and KSRP [56, 83]. Following transcription, miRNAs can be modified by several enzymes, including deaminases, resulting in miRNA editing, and terminal uridylyl transferases (TUTases), leading to pre-miRNA uridylylation, potentially affecting the amount and ratio of miRNA and miRNA* (e.g. [84]), or their sequences (e.g. [85]).
1.3.4.2 Alterations in RNASEN/DGCR8 and DICER1/TARBP2
Inhibition of the miRNA biogenesis pathway leads to severe developmental defects and is lethal in many organisms (reviewed earlier in [86], recent examples include [77, 78]), and perturbations of this pathway predispose to tumorigenesis [87]. Initial miRNA expression profiling experiments suggested that miRNAs are less abundant in tumors compared to their normal tissue counterparts [14], leading to the proposal that miRNAs are predominantly tumor suppressors rather than oncogenes. Quantification of absolute miRNA levels, not only relative abundance, in miRNA profiling methods is necessary to clarify these observations. 27% of various tumors are found to have a hemizygous deletion of the gene that encodes DICER1 [88]. Global knockdown of mature miRNAs by targeting DICER1, RNASEN and its cofactor DGCR8 increases the oncogenic potential of already transformed cancer cell lines and accelerates tumor formation [87]. Reductions in the amount of DICER1 resulting in impaired miRNA processing have also been shown to increase the rate of tumor formation in two different cancer mouse models, a K-RAS-driven lung cancer [88] and an Rb-driven retinoblastoma [89]. DICER1 is therefore considered a haploinsufficient tumor suppressor, requiring partial deletion for its associated tumorigenesis phenotype [89]. The phosphorylation of the DICER1 cofactor TARBP2 by the mitogen-activated protein kinase Erk enhances pre-miRNA processing of oncogenic miRNAs, such as miR-21, and decreases production of tumor suppressor let-7a [90]. Moreover, TARBP2 is mutated in some colon and gastric cancers with microsatellite instability, and TARBP2 frameshift mutations correlate with DICER1 destabilization; in cell lines and xenografts with TARBP2 mutations, reintroduction of wild type TARBP2/DICER1 slowed tumor growth [91, 92]. Finally, DICER1 was also recently implicated as a metastasis suppressor (reviewed in [93]).
1.3.4.3 Alterations in Other Pathway-Related RBPs
Firstly, expression of LIN28A blocks processing of tumor suppressor pri- and pre-let-7 [94–98], thus maintaining expression of genes that drive self-renewal and proliferation (reviewed in [99]); tumors that express LIN28A were indeed shown to be poorly differentiated and more aggressive than LIN28A-negative tumors. Secondly, the helicases DDX5 and DDX17 are thought to stimulate processing of one third of all murine miRNAs by acting as a scaffold and recruiting factors to the RNASEN complex and thereby promoting pri-miRNA processing [100]. Association of DDX17 and DDX5 RNA helicases through interactions mediated by the tumor suppressor TP53 with the RNASEN/DGCR8 complex facilitates the conversion of pri- to pre-miRNAs [101]. Specifically, the DDX5-mediated interaction of the RNASEN complex with the tumor suppressor TP53 was shown to have a stimulatory effect on the tumor suppressor pri-miR-16-1, pri-miR-143 and pri-miR-145 processing in response to DNA damage in cancer cells [101]. Thus, TP53 mutations, often observed in malignancies, led to a decrease in pre-miRNA production. Thirdly, oncogenic SMADs, downstream effectors of the TGF-β superfamily pathways, have been shown to control RNASEN-mediated miRNA maturation through interaction with DDX5, promoting expression of oncogenic miR-21 [102]. KSRP promotes the biogenesis of a subset of miRNAs, including let-7a, by serving as a component of both DICER1 and RNASEN complexes affecting proliferation, apoptosis and differentiation [103]. In a final example, inactivating mutations of XPO5 in tumors with microsatellite instability result in the nuclear retention of miRNAs [104]. Restoration of XPO5 function reverses the impaired export of pre-miRNAs and has tumor suppressor features.
1.4 Dysregulation of miRNA-mRNA Target Recognition
1.4.1 miRNA Function/Mechanism
As described above, miRNAs function through the AGO proteins, containing both RNA-binding domains and RNase H domains (reviewed in [105]). The four human Ago genes are co-expressed and bind to miRNAs irrespective of their sequence. AGO2, in contrast to the other members, retains an active RNase H domain and thus is able to directly cleave target RNAs with extensive complementarity to the bound miRNAs. The assembly of the miRNP complex involves multiple AGO conformational transitions captured in a series of crystal structures (reviewed in [106]). The mRNA target is recognized by pairing of the miRNA seed region (position 2–8) to complementary sequences located mainly in the target 3′ UTR, but also in the coding regions. Target mRNA recognition and regulation involves members of the GW182/TNRC6 family. TNRC6 proteins act at the effector step of silencing, downstream of AGO proteins, and play a crucial role in miRNA silencing in animals (reviewed in [107]). Proteomic approaches identified additional AGO-interacting proteins, some of which likely represent mRNA-interacting partners that co-purified with miRNA-targeted mRNPs; their function in RNA silencing processes and potentially tumorigenesis remains to be established.
In mammalian cells under steady state conditions, miRNAs have been shown to destabilize targeted transcripts [108–111] through a variety of mechanisms, including de-capping and de-adenylation; target mRNA and protein abundance changes track closely [108, 109, 112, 113]. These studies also showed that miRNAs destabilize mRNAs preferably through binding sites located in their 3′ UTRs [114–118]. Ribosome profiling studies demonstrated that the ribosome density of miRNA targets was unaltered, while changes in miRNA levels were inversely correlated to mRNA and protein abundance, emphasizing the role of miRNAs in regulation of mRNA stability but not translation [119]. Translational regulation by miRNA targeting is considered to predominantly act at the level of translation initiation. Identification of miRNA/mRNA ribonucleoprotein components in processing bodies (P-bodies) also implies their role in mRNA storage and RNA turnover. An excellent recent review describes the different mechanisms implicated in miRNA function, highlighting the different experiments supporting translational repression versus mRNA decay and the evolution in our current thinking [107].
1.4.2 Organization of miRNAs into Sequence Families
Certain miRNAs share sequence similarity in regions that are critical for mRNA target recognition, specifically the seed region, and are best viewed as a family when considering mRNA target regulation and functional consequences of altered miRNA expression. miRNAs can be grouped in sequence families, based not only on their seed sequence similarity but also overall sequence similarity given that the miRNA 3′ end also contributes to miRNA targeting, although to a lesser extent (reviewed in [68]) (Fig. 1.2). Changes in the overall abundance of miRNA sequence families relate directly to target regulation. In a Myc-driven B-cell lymphoma mouse model, a conditional knockout of the oncogenic miR-17-92 gene cluster induces apoptosis, which can be reduced by reintroduction of only one of the four sequence families produced from the cluster [120].
1.4.3 miRNA-mRNA Stoichiometry
The majority of miRNA profiling studies do not provide an estimate of miRNA abundance, which is critical in our understanding of the role of miRNA-mRNA mediated regulation in tumorigenesis. Only the most abundantly expressed miRNAs occupy a substantial fraction of their available mRNA target sites and affect target mRNA stability [118]. Abundant miRNAs that behave as “switches”, turned on or off during the tumorigenesis process, as shown in developmental processes, have the most significant regulatory potential, given that miRNAs usually only lead to modest 1.5- to 4-fold regulation of their target expression [112, 113, 115]. However, given that specific mRNAs are subject to regulation by multiple miRNAs of unrelated families, cumulative effects of lower expressed miRNAs may be relevant [67, 121, 122]. Furthermore, in the rare circumstance that miRNAs share near perfect complementarity to mRNAs, they may act in a siRNA-like catalytic mode, cleaving mRNA targets even at low miRNA abundance. To conclude, the interplay between miRNAs expressed in particular tissues, the levels of their respective expressed targets, as well as other post-transcriptional gene regulatory mechanisms (such as regulation by RBPs or other competing interactions – see below) is likely responsible for balancing miRNA conferred regulation.
1.4.4 Changes in the miRNA Targets
The binding sites of miRNAs in mRNAs can be altered through a variety of mechanisms, such as point mutations, translocations, shortening of the 3′ UTR, competition with other RBPs or decoy molecules for mRNA binding. Point mutations in miRNA targets can both create or destroy a miRNA binding site [123–125]. Chromosomal translocations can remove miRNA binding sites from their regulated oncogenes, such as in the case of let-7 targeting of the 3′ UTR of the Hmga2 gene [126]. Shortening of the 3′ UTR through alternative polyadenylation can relax miRNA mediated regulation of known oncogenes, such as IGF2BP1/IMP1, and lead to oncogenic transformation [127], as does use of decoy pseudogenes, as in the case of PTEN, by saturating miRNA binding sites [128]. Finally, cooperativity or competition of miRNAs for mRNA target site binding with other RBPs, such as ELAVL1 (HuR), DND1 and PUM1, can also de-repress target expression [129–132]. This topic is discussed in a recent review [83].
1.5 Cancer Tissues Have Distinct miRNA Profiles
We will first discuss the state of current miRNA profile databases, and then explore the issue of tissue heterogeneity in the tissue profiles before summarizing the role of miRNA dysregulation in malignancies.
1.5.1 miRNA Cancer Database
The development of miRNA microarrays, RT-PCR platforms and deep sequencing methodologies has resulted in an exponential acquisition of miRNA profiles. Some of the published miRNA profiles are available in the NCBI Gene Expression Omnibus, similarly to mRNA profiles (other resources include www.microrna.org, http://www.mirz.unibas.ch). Larger cancer and blood-borne disease collections have recently been published using various platforms [133–135]. However, there is no database or viewer that allows for cross-platform comparison of existing data.
1.5.2 Tissue Heterogeneity
Tissues are generally composed of multiple cell types, each with their distinct gene expression program. Disease not only alters the expression programs of the affected cell type, but often also its cell type composition. To best separate these effects in the profiling of heterogeneous tumor samples, it may be useful to profile tumor cell lines and individual cell types that may be present in a tumor sample, or define miRNA cellular localization by performing RNA ISH. Figure 1.3 compares miRNA abundance profiles of normal breast, an estrogen receptor positive invasive ductal breast carcinoma, the estrogen receptor positive ductal cell line MCF7, human fat and blood [38, 39]. Strikingly, we can model the profile of a human cancer by simply combining tumor cell line and human fat profiles at equal ratio. This demonstrates that the MCF7 tumor cell line may be a good disease model for deciphering miRNA regulatory networks, as it expresses many of the miRNAs present in the predominant tumor derived cell type and highlights the need for individual cell type miRNA profiles.

miRNA breast tumor and cell line profiles. Comparison of abundance profiles of the top expressed miRNA sequence families of normal breast, an estrogen receptor positive invasive ductal carcinoma breast tumor (ER+), the MCF7 ductal derived cell line, human subcutaneous adipose tissue and red blood cells
1.5.3 miRNAs as Tumor Suppressors and Oncogenes
miRNA dysregulation could be used as a diagnostic tool even if the particular miRNAs do not serve any regulatory function. Alternatively, miRNA dysregulation could drive tumorigenesis through the roles miRNAs can adopt as tumor suppressors or oncogenes. miRNAs that are up- or down-regulated in malignancies are respectively referred to as oncogenic or tumor-suppressor miRNAs, sometimes even if there is no evidence for their causative role in tumorigenesis. Some of the most commonly dysregulated miRNAs are summarized in Table 1.1 (reviewed in [11]).
Functional studies performed in cancer cell lines or mouse models of various malignancies through over-expression or knockdown of miRNAs have supported a role for some of these miRNAs in tumorigenesis. Over-expression of tumor suppressor miRNAs, such as let-7g, reduced tumor burden in a K-RAS murine lung cancer model [172]. Over-expression of the oncogenic mir-17-92 gene cluster led to a lymphoproliferative disorder, and higher level expression of the cluster in MYC-driven B-cell lymphomas dramatically increased tumorigenicity [62, 173]. Over-expression of another oncogene, miR-21, frequently highly expressed in solid and hematologic malignancies, resulted in a pre-B malignant lymphoid like phenotype whereas subsequent miR-21 inactivation in the same model led to apoptosis and tumor regression [174]. Transgenic mice models with loss and gain of function of miR-21 combined with a model of lung cancer confirmed the role of miR-21 as an enhancer of tumorigenesis when over-expressed, or a partial protector when genetically deleted [59]. Ectopic expression of miR-155 in bone marrow induced polyclonal pre-B cell proliferation progressing to B-cell leukemia or myeloproliferation in mice [175, 176].
Metastasis-related miRNAs have been identified in various malignancies mainly from cell line and xenograft experiments (reviewed in [177]). Examples include breast cancer-related miR-10b, miR-9, miR-31 and miR-335 among others. The interesting regulatory roles of these miRNAs cannot easily be validated in large clinical studies. Two clinical studies with long-term follow-up data instead identified miR-210 to be associated with tumor aggressiveness [178, 179], pointing to difficulties reconciling cell line, xenograft model and patient materials, due to tissue heterogeneity discussed earlier, the heterogeneous nature of the malignancy and timing of clinical specimen acquisition. Tumor miRNA profiles cannot dissect contributions from sub-populations of cells that may be important for tumor characteristics such as metastasis, while cell line miRNA profiles cannot capture the cellular interactions from supporting cell types in the tumor microenvironment. Patient samples are often collected at time of diagnosis, by which time a tumor is already well established and cannot unravel early changes that may be critical in tumor initiation or later changes important in metastasis.
1.5.4 miRNA-Regulated Pathways
The observed effects of miRNA mis-expression on tumor initiation, maintenance or metastasis can be explained by the mRNA targets and pathways they regulate, which include known tumor suppressors and oncogenes (reviewed in [11]). miRNAs regulate a large number of genes, some estimates reporting miRNA regulation of up to 60% of the human genome, making it challenging to attribute a phenotype after mis-expression of a particular miRNA through its action on only a subset of targets [111, 180]. If a few of these targets control rate-limiting steps in the studied tumorigenesis processes within the specified tissues and cell types, such as metastasis, then miRNA regulation of a handful of targets could potentially explain the phenotype resulting from miRNA mis-expression [181].
Examples of miRNA regulated cancer pathways include differentiation, apoptosis, proliferation and stem cell maintenance, a process important for disease relapse and/or metastasis. The skeletal muscle-specific miR-206 blocks human rhabdomyosarcoma growth in mouse xenograft models by inducing myogenic differentiation [30], while the mir-141/200a cluster is critical in the epithelial to mesenchymal transition (EMT) in various malignancies (reviewed in [182]). Sustained expression of endogenous mir-17-92 cluster is required to suppress apoptosis in Myc-driven B-cell lymphomas in a conditional knockout allele of mir-17-92 cluster [120]. TP53-regulated, ectopically expressed miR-34 induced cell cycle arrest in both primary and tumor derived cell lines, downregulating genes promoting cell cycle progression (reviewed in [58]). In a final example of miRNA regulated cancer pathways, isolation of a subset of highly tumorigenic breast cancer cells that were thought to have stemness properties showed that these cells do not express let-7 family members and that expression of let-7 or its known target RAS leads to loss of self renewal [183].
1.6 Alterations of miRNA Sequence
miRNA dysregulation could be a result of mutations in miRNA genes in well-conserved regions in their mature sequence affecting mRNA targeting, or the remainder of the miRNA precursor potentially affecting processing and stability of the mature miRNA (reviewed in [75]). For example, a mutation in the seed region of mir-96 was shown to lead to hearing loss in a mouse model [184] and was identified in families with non-syndromic progressive sensorineural hearing loss [185], while a point mutation in the viral mir-K5 precursor stem loop was shown to interfere with its processing and reduce mature miR-K5 accumulation [186]. Germline deletion of the mir-17-92 gene cluster was another recent example causing skeletal growth defects in humans [187]. If miRNAs are drivers of oncogenic and tumor suppressor pathways we would expect to find miRNA mutations that can also be causative of the disease. So far the only mutation identified in a miRNA that could lead to malignancy is miR-16, where a germline mutation potentially affects miR-16 biogenesis and abundance in a kindred with familial CLL [188] and New Zealand black mice that naturally develop CLL-like disease [189]. Single nucleotide polymorphisms (SNPs), located both in precursor and mature miRNA sequences, have been examined in the context of disease risk for various malignancies but have not been validated as causative (reviewed in [75]).
1.7 miRNA Target Identification
The currently available target prediction databases (reviewed in [68]) do not easily allow prioritizing the involvement of reported targets in certain phenotypes, thus necessitating the selection of a few targets from a list of hundreds for further study and validation, based on a priori knowledge of potentially involved biological pathways. Since the prediction algorithms do not always produce identical target lists, use of multiple algorithms and comparison or intersection of their results narrows the list to higher confidence targets. Targets are only relevant to a specific phenotype if they are expressed in the studied tissue, an issue not addressed by most computational prediction algorithms. Recently, new algorithms are trying to prioritize computationally predicted targets using integrated miRNA and mRNA profiles [134]. Biochemical identification methods in cell lines and tissues are being established and further refine our understanding of miRNA-mRNA target binding recognition. These methods involve two approaches: over-expression or down-regulation of studied miRNAs followed by assessment of transcriptome-wide mRNA levels by mRNA microarray analysis (e.g. [118]) or deep sequencing technology after immunoprecipitation of miRNAs and mRNAs complexed with Ago, the main component of the miRNA effector complex, to not only identify mRNA targets, but also localize their precise binding sites [190, 191].
1.8 miRNAs as Diagnostics
miRNAs demonstrated their potential as diagnostic tumor markers early on when their profiles were shown to correlate with the tumor embryonic origin, thus defining tumors of unknown origin indistinguishable by histology and assigned based on clinical information [14]. miRNA expression patterns have been linked to clinical outcomes given that miRNAs modulate tumor behavior such as tumor progression and metastasis. Expression of let-7 is downregulated in non-small cell lung cancer patients [192] and is associated with poor prognosis [125, 193], whereas a miRNA signature was identified to be associated with prognosis in CLL [188]. Advances in miRNA detection, such as ISH or RT-PCR, may allow miRNAs to be used as diagnostic and prognostic markers in the clinic.
1.9 miRNAs as Therapeutics
Because miRNAs affect the expression of multiple genes and thereby tune multiple points in disease pathways, miRNAs and their regulated genes represent interesting drug targets. Antisense oligonucleotide targeting experiments in human cell lines, mice [117, 194–197] and non-human primates [198] have demonstrated the feasibility of manipulating miRNA levels. miR-143 was initially shown to promote adipocyte differentiation and could be a target for therapies in obesity and metabolic diseases [194]. Alternatively, “miRNA sponges” have been exploited to reduce miRNA expression in mammalian cells and mouse models by using RNA transcripts expressed from strong promoters containing miRNA-complementary binding sites (reviewed in [199]). Systemic administration of antisense oligonucleotide therapeutics to miR-122, a liver-enriched miRNA, in mice and primates was shown to alter lipid metabolism and hepatitis C viral load, resulting in reduced liver damage [117, 195–197, 200, 201]. At the same time, systemic delivery of a miRNA mimic for miR-26a in a murine model of HCC reduced tumor size [148]. The new and exciting advances in delivery of miRNA inhibitors and mimics hold the promise of quickly translating our knowledge of miRNAs into treating disease.
References
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854
Reinhart BJ, Slack FJ, Basson M et al (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403:901–906
Wightman B, Ha I, Ruvkun G (1993) Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75:855–862
Wightman B, Burglin TR, Gatto J et al (1991) Negative regulatory sequences in the lin-14 3′-untranslated region are necessary to generate a temporal switch during Caenorhabditis elegans development. Genes Dev 5:1813–1824
Lagos-Quintana M, Rauhut R, Lendeckel W et al (2001) Identification of novel genes coding for small expressed RNAs. Science 294:853–858
Lagos-Quintana M, Rauhut R, Yalcin A et al (2002) Identification of tissue-specific microRNAs from mouse. Curr Biol 12:735–739
Lau NC, Lim LP, Weinstein EG et al (2001) An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294:858–862
Lee RC, Ambros V (2001) An extensive class of small RNAs in Caenorhabditis elegans. Science 294:862–864
Garofalo M, Croce CM (2010) microRNAs: master regulators as potential therapeutics in cancer. Annu Rev Pharmacol Toxicol 51:25–43
Medina PP, Slack FJ (2008) microRNAs and cancer: an overview. Cell Cycle 7:2485–2492
Ventura A, Jacks T (2009) MicroRNAs and cancer: short RNAs go a long way. Cell 136:586–591
Calin GA, Sevignani C, Dumitru CD et al (2004) Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 101:2999–3004
Calin GA, Liu CG, Sevignani C et al (2004) MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci U S A 101:11755–11760
Lu J, Getz G, Miska EA et al (2005) MicroRNA expression profiles classify human cancers. Nature 435:834–838
Aravin A, Tuschl T (2005) Identification and characterization of small RNAs involved in RNA silencing. FEBS Lett 579:5830–5840
Creighton CJ, Reid JG, Gunaratne PH (2009) Expression profiling of microRNAs by deep sequencing. Brief Bioinform 10:490–497
Meyer SU, Pfaffl MW, Ulbrich SE (2010) Normalization strategies for microRNA profiling experiments: a ‘normal’ way to a hidden layer of complexity? Biotechnol Lett 32(12):1777–1788
Lawrie CH, Soneji S, Marafioti T et al (2007) MicroRNA expression distinguishes between germinal center B cell-like and activated B cell-like subtypes of diffuse large B cell lymphoma. Int J Cancer 121:1156–1161
Weng L, Wu X, Gao H et al (2010) MicroRNA profiling of clear cell renal cell carcinoma by whole-genome small RNA deep sequencing of paired frozen and formalin-fixed, paraffin-embedded tissue specimens. J Pathol 222:41–51
Xi Y, Nakajima G, Gavin E et al (2007) Systematic analysis of microRNA expression of RNA extracted from fresh frozen and formalin-fixed paraffin-embedded samples. RNA 13:1668–1674
Barad O, Meiri E, Avniel A et al (2004) MicroRNA expression detected by oligonucleotide microarrays: system establishment and expression profiling in human tissues. Genome Res 14:2486–2494
Baskerville S, Bartel DP (2005) Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11:241–247
Thomson JM, Parker JS, Hammond SM (2007) Microarray analysis of miRNA gene expression. Methods Enzymol 427:107–122
Nelson PT, Baldwin DA, Scearce LM et al (2004) Microarray-based, high-throughput gene expression profiling of microRNAs. Nat Methods 1:155–161
Bissels U, Wild S, Tomiuk S et al (2009) Absolute quantification of microRNAs by using a universal reference. RNA 15:2375–2384
Peltier HJ, Latham GJ (2008) Normalization of microRNA expression levels in quantitative RT-PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA 14:844–852
Fiedler SD, Carletti MZ, Christenson LK (2010) Quantitative RT-PCR methods for mature microRNA expression analysis. Methods Mol Biol 630:49–64
Mestdagh P, Van Vlierberghe P, De Weer A et al (2009) A novel and universal method for microRNA RT-qPCR data normalization. Genome Biol 10:R64
Smith RD, Brown B, Ikonomi P et al (2003) Exogenous reference RNA for normalization of real-time quantitative PCR. Biotechniques 34:88–91
Taulli R, Bersani F, Foglizzo V et al (2009) The muscle-specific microRNA miR-206 blocks human rhabdomyosarcoma growth in xenotransplanted mice by promoting myogenic differentiation. J Clin Invest 119:2366–2378
Berezikov E, Thuemmler F, van Laake LW et al (2006) Diversity of microRNAs in human and chimpanzee brain. Nat Genet 38:1375–1377
Houbaviy HB, Murray MF, Sharp PA (2003) Embryonic stem cell-specific microRNAs. Dev Cell 5:351–358
Landgraf P, Rusu M, Sheridan R et al (2007) A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129:1401–1414
Witten D, Tibshirani R, Gu SG et al (2010) Ultra-high throughput sequencing-based small RNA discovery and discrete statistical biomarker analysis in a collection of cervical tumours and matched controls. BMC Biol 8:58
Vigneault F, Sismour AM, Church GM (2008) Efficient microRNA capture and bar-coding via enzymatic oligonucleotide adenylation. Nat Methods 5:777–779
Hafner M, Renwick N, Brown M et al (2011) RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA 17:1697–1712
Git A, Dvinge H, Salmon-Divon M et al (2010) Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA 16:991–1006
Ugras S, Brill E, Jacobsen A et al (2011) Small RNA sequencing and functional characterization reveals microRNA-143 tumor suppressor activity in liposarcoma. Cancer Res 71:5659–5669
Farazi TA, Horlings HM, Ten Hoeve JJ et al (2011) MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer Res 71:4443–4453
Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 98:5116–5121
Berninger P, Gaidatzis D, van Nimwegen E et al (2008) Computational analysis of small RNA cloning data. Methods 44:13–21
Robinson MD, McCarthy DJ, Smyth GK (2010) EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140
Nelson PT, Baldwin DA, Kloosterman WP et al (2006) RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain. RNA 12:187–191
Pena JT, Sohn-Lee C, Rouhanifard SH et al (2009) miRNA in situ hybridization in formaldehyde and EDC-fixed tissues. Nat Methods 6:139–141
Sempere LF, Christensen M, Silahtaroglu A et al (2007) Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res 67:11612–11620
Johnston RJ, Hobert O (2003) A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature 426:845–849
Griffiths-Jones S, Saini HK, van Dongen S et al (2008) miRBase: tools for microRNA genomics. Nucleic Acids Res 36:D154–D158
Kozomara A, Griffiths-Jones S (2010) miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39:D152–D157
Chiang HR, Schoenfeld LW, Ruby JG et al (2010) Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev 24:992–1009
Calin GA, Dumitru CD, Shimizu M et al (2002) Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 99:15524–15529
Tagawa H, Seto M (2005) A microRNA cluster as a target of genomic amplification in malignant lymphoma. Leukemia 19:2013–2016
Mavrakis KJ, Wolfe AL, Oricchio E et al (2010) Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol 12:372–379
Huse JT, Brennan C, Hambardzumyan D et al (2009) The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev 23:1327–1337
Gauwerky CE, Huebner K, Isobe M et al (1989) Activation of MYC in a masked t(8;17) translocation results in an aggressive B-cell leukemia. Proc Natl Acad Sci U S A 86:8867–8871
Etiemble J, Moroy T, Jacquemin E et al (1989) Fused transcripts of c-myc and a new cellular locus, hcr in a primary liver tumor. Oncogene 4:51–57
Krol J, Loedige I, Filipowicz W (2010) The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 11:597–610
Chang TC, Wentzel EA, Kent OA et al (2007) Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 26:745–752
He L, He X, Lowe SW et al (2007) microRNAs join the p53 network – another piece in the tumour-suppression puzzle. Nat Rev Cancer 7:819–822
Hatley ME, Patrick DM, Garcia MR et al (2010) Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell 18:282–293
Huang TH, Wu F, Loeb GB et al (2009) Up-regulation of miR-21 by HER2/neu signaling promotes cell invasion. J Biol Chem 284:18515–18524
O’Donnell KA, Wentzel EA, Zeller KI et al (2005) c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435:839–843
He L, Thomson JM, Hemann MT et al (2005) A microRNA polycistron as a potential human oncogene. Nature 435:828–833
Han L, Witmer PD, Casey E et al (2007) DNA methylation regulates MicroRNA expression. Cancer Biol Ther 6:1284–1288
Saito Y, Jones PA (2006) Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle 5:2220–2222
Saito Y, Liang G, Egger G et al (2006) Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 9:435–443
Lehmann U, Hasemeier B, Christgen M et al (2008) Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J Pathol 214:17–24
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215–233
Brodersen P, Voinnet O (2009) Revisiting the principles of microRNA target recognition and mode of action. Nat Rev Mol Cell Biol 10:141–148
Carthew RW, Sontheimer EJ (2009) Origins and mechanisms of miRNAs and siRNAs. Cell 136:642–655
Ghildiyal M, Zamore PD (2009) Small silencing RNAs: an expanding universe. Nat Rev Genet 10:94–108
Kim VN, Han J, Siomi MC (2009) Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10:126–139
Winter J, Jung S, Keller S et al (2009) Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 11:228–234
Kwak PB, Iwasaki S, Tomari Y (2010) The microRNA pathway and cancer. Cancer Sci 101(11):2309–2315
Ryan BM, Robles AI, Harris CC (2010) Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer 10:389–402
Nishikura K (2010) Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79:321–349
Yi R, Pasolli HA, Landthaler M et al (2009) DGCR8-dependent microRNA biogenesis is essential for skin development. Proc Natl Acad Sci U S A 106:498–502
Cheloufi S, Dos Santos CO, Chong MM et al (2010) A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465:584–589
Yang JS, Maurin T, Robine N et al (2010) Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc Natl Acad Sci U S A 107:15163–15168
Babiarz JE, Ruby JG, Wang Y et al (2008) Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev 22:2773–2785
Berezikov E, Chung WJ, Willis J et al (2007) Mammalian mirtron genes. Mol Cell 28:328–336
Yang JS, Lai EC (2011) Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants. Mol Cell 43:892–903
van Kouwenhove M, Kedde M, Agami R (2011) MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer 11:644–656
Hagan JP, Piskounova E, Gregory RI (2009) Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nat Struct Mol Biol 16:1021–1025
Kawahara Y, Zinshteyn B, Chendrimada TP et al (2007) RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep 8:763–769
Stefani G, Slack FJ (2008) Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol 9:219–230
Kumar MS, Lu J, Mercer KL et al (2007) Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet 39:673–677
Kumar MS, Pester RE, Chen CY et al (2009) Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev 23:2700–2704
Lambertz I, Nittner D, Mestdagh P et al (2010) Monoallelic but not biallelic loss of Dicer1 promotes tumorigenesis in vivo. Cell Death Differ 17:633–641
Paroo Z, Ye X, Chen S et al (2009) Phosphorylation of the human microRNA-generating complex mediates MAPK/Erk signaling. Cell 139:112–122
Melo SA, Ropero S, Moutinho C et al (2009) A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat Genet 41:365–370
Garre P, Perez-Segura P, Diaz-Rubio E et al (2010) Reassessing the TARBP2 mutation rate in hereditary nonpolyposis colorectal cancer. Nat Genet 42:817–818; author reply 818
Valastyan S, Weinberg RA (2010) Metastasis suppression: a role of the Dice(r). Genome Biol 11:141
Newman MA, Thomson JM, Hammond SM (2008) Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA 14:1539–1549
Piskounova E, Viswanathan SR, Janas M et al (2008) Determinants of microRNA processing inhibition by the developmentally regulated RNA-binding protein Lin28. J Biol Chem 283:21310–21314
Rybak A, Fuchs H, Smirnova L et al (2008) A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat Cell Biol 10:987–993
Viswanathan SR, Daley GQ, Gregory RI (2008) Selective blockade of microRNA processing by Lin28. Science 320:97–100
Viswanathan SR, Powers JT, Einhorn W et al (2009) Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet 41:843–848
Viswanathan SR, Daley GQ (2010) Lin28: a microRNA regulator with a macro role. Cell 140:445–449
Fukuda T, Yamagata K, Fujiyama S et al (2007) DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat Cell Biol 9:604–611
Suzuki HI, Yamagata K, Sugimoto K et al (2009) Modulation of microRNA processing by p53. Nature 460:529–533
Davis BN, Hilyard AC, Lagna G et al (2008) SMAD proteins control DROSHA-mediated microRNA maturation. Nature 454(7200):56–61
Trabucchi M, Briata P, Garcia-Mayoral M et al (2009) The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 459:1010–1014
Melo SA, Moutinho C, Ropero S et al (2010) A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell 18:303–315
Ender C, Meister G (2010) Argonaute proteins at a glance. J Cell Sci 123:1819–1823
Parker JS (2010) How to slice: snapshots of Argonaute in action. Silence 1:3
Huntzinger E, Izaurralde E (2011) Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 12:99–110
Linsley PS, Schelter J, Burchard J et al (2007) Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol Cell Biol 27:2240–2252
Zhao Y, Ransom JF, Li A et al (2007) Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 129:303–317
Bagga S, Bracht J, Hunter S et al (2005) Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 122:553–563
Lim LP, Lau NC, Garrett-Engele P et al (2005) Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433:769–773
Baek D, Villen J, Shin C et al (2008) The impact of microRNAs on protein output. Nature 455(7209):64–71
Selbach M, Schwanhausser B, Thierfelder N et al (2008) Widespread changes in protein synthesis induced by microRNAs. Nature 455(7209):58–63
Grimson A, Farh KK, Johnston WK et al (2007) MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27:91–105
Hausser J, Landthaler M, Jaskiewicz L et al (2009) Relative contribution of sequence and structure features to the mRNA binding of Argonaute/EIF2C-miRNA complexes and the degradation of miRNA targets. Genome Res 19:2009–2020
Karginov FV, Conaco C, Xuan Z et al (2007) A biochemical approach to identifying microRNA targets. Proc Natl Acad Sci U S A 104:19291–19296
Krützfeldt J, Rajewsky N, Braich R et al (2005) Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438:685–689
Landthaler M, Gaidatzis D, Rothballer A et al (2008) Molecular characterization of human Argonaute-containing ribonucleoprotein complexes and their bound target mRNAs. RNA 14:2580–2596
Guo H, Ingolia NT, Weissman JS et al (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466:835–840
Mu P, Han YC, Betel D et al (2009) Genetic dissection of the miR-17 92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev 23:2806–2811
Wu S, Huang S, Ding J et al (2010) Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3′ untranslated region. Oncogene 29:2302–2308
Krek A, Grun D, Poy MN et al (2005) Combinatorial microRNA target predictions. Nat Genet 37:495–500
Chin LJ, Ratner E, Leng S et al (2008) A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res 68:8535–8540
Jiang S, Zhang HW, Lu MH et al (2010) MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res 70:3119–3127
Takamizawa J, Konishi H, Yanagisawa K et al (2004) Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 64:3753–3756
Mayr C, Hemann MT, Bartel DP (2007) Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 315:1576–1579
Mayr C, Bartel DP (2009) Widespread shortening of 3′ UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138:673–684
Poliseno L, Salmena L, Zhang J et al (2010) A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465:1033–1038
Bhattacharyya SN, Habermacher R, Martine U et al (2006) Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125:1111–1124
Kim HH, Kuwano Y, Srikantan S et al (2009) HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev 23:1743–1748
Kedde M, Strasser MJ, Boldajipour B et al (2007) RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell 131:1273–1286
Kedde M, van Kouwenhove M, Zwart W et al (2010) A Pumilio-induced RNA structure switch in p27-3′ UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol 12:1014–1020
Volinia S, Galasso M, Costinean S et al (2010) Reprogramming of miRNA networks in cancer and leukemia. Genome Res 20:589–599
Mestdagh P, Lefever S, Pattyn F et al (2011) The microRNA body map: dissecting microRNA function through integrative genomics. Nucleic Acids Res 39(20):e136
Keller A, Leidinger P, Bauer A et al (2011) Toward the blood-borne miRNome of human diseases. Nat Methods 8(10):841–843
Calin GA, Cimmino A, Fabbri M et al (2008) MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A 105:5166–5171
Roush S, Slack FJ (2008) The let-7 family of microRNAs. Trends Cell Biol 18:505–516
Peter ME (2009) Let-7 and miR-200 microRNAs: guardians against pluripotency and cancer progression. Cell Cycle 8:843–852
Osada H, Takahashi T (2011) let-7 and miR-17-92: small-sized major players in lung cancer development. Cancer Sci 102:9–17
O’Day E, Lal A (2010) MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res 12:201
Aqeilan RI, Calin GA, Croce CM (2010) miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ 17:215–220
Finnerty JR, Wang WX, Hebert SS et al (2010) The miR-15/107 group of microRNA genes: evolutionary biology, cellular functions, and roles in human diseases. J Mol Biol 402:491–509
Mendell JT (2008) miRiad roles for the miR-17-92 cluster in development and disease. Cell 133:217–222
Uziel T, Karginov FV, Xie S et al (2009) The miR-17 92 cluster collaborates with the Sonic Hedgehog pathway in medulloblastoma. Proc Natl Acad Sci U S A 106:2812–2817
Poliseno L, Salmena L, Riccardi L et al (2010) Identification of the miR-106b∼25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci Signal 3:ra29
Jazbutyte V, Thum T (2010) MicroRNA-21: from cancer to cardiovascular disease. Curr Drug Targets 11:926–935
Sander S, Bullinger L, Klapproth K et al (2008) MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood 112:4202–4212
Kota J, Chivukula RR, O’Donnell KA et al (2009) Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 137:1005–1017
Visone R, Pallante P, Vecchione A et al (2007) Specific microRNAs are downregulated in human thyroid anaplastic carcinomas. Oncogene 26:7590–7595
Kim H, Huang W, Jiang X et al (2010) Integrative genome analysis reveals an oncomir/oncogene cluster regulating glioblastoma survivorship. Proc Natl Acad Sci U S A 107:2183–2188
Cole KA, Attiyeh EF, Mosse YP et al (2008) A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol Cancer Res 6:735–742
Li N, Fu H, Tie Y et al (2009) miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett 275:44–53
Gregory PA, Bracken CP, Bert AG et al (2008) MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 7:3112–3118
Nakada C, Matsuura K, Tsukamoto Y et al (2008) Genome-wide microRNA expression profiling in renal cell carcinoma: significant down-regulation of miR-141 and miR-200c. J Pathol 216:418–427
Du Y, Xu Y, Ding L et al (2009) Down-regulation of miR-141 in gastric cancer and its involvement in cell growth. J Gastroenterol 44:556–561
Adam L, Zhong M, Choi W et al (2009) miR-200 expression regulates epithelial-to-mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy. Clin Cancer Res 15:5060–5072
Bendoraite A, Knouf EC, Garg KS et al (2009) Regulation of miR-200 family microRNAs and ZEB transcription factors in ovarian cancer: evidence supporting a mesothelial-to-epithelial transition. Gynecol Oncol 116:117–125
Park SM, Gaur AB, Lengyel E et al (2008) The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 22:894–907
Hu X, Macdonald DM, Huettner PC et al (2009) A miR-200 microRNA cluster as prognostic marker in advanced ovarian cancer. Gynecol Oncol 114:457–464
Gandellini P, Folini M, Longoni N (2009) miR-205 Exerts tumor-suppressive functions in human prostate through down-regulation of protein kinase Cepsilon. Cancer Res 69:2287–2295
Schaefer A, Jung M, Mollenkopf HJ et al (2010) Diagnostic and prognostic implications of microRNA profiling in prostate carcinoma. Int J Cancer 126:1166–1176
Wiklund ED, Bramsen JB, Hulf T et al (2011) Coordinated epigenetic repression of the miR-200 family and miR-205 in invasive bladder cancer. Int J Cancer 128:1327–1334
Iorio MV, Ferracin M, Liu CG et al (2005) MicroRNA gene expression deregulation in human breast cancer. Cancer Res 65:7065–7070
Wu H, Zhu S, Mo YY (2009) Suppression of cell growth and invasion by miR-205 in breast cancer. Cell Res 19:439–448
Feber A, Xi L, Luketich JD et al (2008) MicroRNA expression profiles of esophageal cancer. J Thorac Cardiovasc Surg 135:255–260; discussion 260
Iorio MV, Visone R, Di Leva G et al (2007) MicroRNA signatures in human ovarian cancer. Cancer Res 67:8699–8707
Negrini M, Calin GA (2008) Breast cancer metastasis: a microRNA story. Breast Cancer Res 10:203
Ferretti E, De Smaele E, Po A et al (2009) MicroRNA profiling in human medulloblastoma. Int J Cancer 124:568–577
Laios A, O’Toole S, Flavin R et al (2008) Potential role of miR-9 and miR-223 in recurrent ovarian cancer. Mol Cancer 7:35
Ma L, Young J, Prabhala H et al (2010) miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol 12:247–256
Sun Y, Wu J, Wu SH et al (2009) Expression profile of microRNAs in c-Myc induced mouse mammary tumors. Breast Cancer Res Treat 118:185–196
Kumar MS, Erkeland SJ, Pester RE et al (2008) Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci 105:3903–3908
Xiao C, Srinivasan L, Calado DP et al (2008) Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol 9:405–414
Medina PP, Nolde M, Slack FJ (2010) OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 467:86–90
Costinean S, Zanesi N, Pekarsky Y et al (2006) Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A 103:7024–7029
O’Connell RM, Rao DS, Chaudhuri AA et al (2008) Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med 205:585–594
Hurst DR, Edmonds MD, Welch DR (2009) Metastamir: the field of metastasis-regulatory microRNA is spreading. Cancer Res 69:7495–7498
Camps C, Buffa FM, Colella S et al (2008) hsa-miR-210 is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res 14:1340–1348
Foekens JA, Sieuwerts AM, Smid M et al (2008) Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci 105:13021–13026
Friedman RC, Farh KK, Burge CB et al (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19:92–105
Valastyan S, Benaich N, Chang A et al (2009) Concomitant suppression of three target genes can explain the impact of a microRNA on metastasis. Genes Dev 23:2592–2597
Cano A, Nieto MA (2008) Non-coding RNAs take centre stage in epithelial-to-mesenchymal transition. Trends Cell Biol 18:357–359
Yu F, Yao H, Zhu P et al (2007) let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 131:1109–1123
Lewis MA, Quint E, Glazier AM et al (2009) An ENU-induced mutation of miR-96 associated with progressive hearing loss in mice. Nat Genet 41:614–618
Mencia A, Modamio-Hoybjor S, Redshaw N et al (2009) Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat Genet 41:609–613
Gottwein E, Cai X, Cullen BR (2006) Expression and function of microRNAs encoded by Kaposi’s sarcoma-associated herpesvirus. Cold Spring Harb Symp Quant Biol 71:357–364
de Pontual L, Yao E, Callier P (2011) Germline deletion of the miR-17 approximately 92 cluster causes skeletal and growth defects in humans. Nat Genet 43(10):1026–1030
Calin GA, Ferracin M, Cimmino A et al (2005) A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 353:1793–1801
Raveche ES, Salerno E, Scaglione BJ et al (2007) Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood 109:5079–5086
Chi SW, Zang JB, Mele A et al (2009) Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460:479–486
Hafner M, Landthaler M, Burger L et al (2010) Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141:129–141
Johnson CD, Esquela-Kerscher A, Stefani G et al (2007) The let-7 MicroRNA represses cell proliferation pathways in human cells. Cancer Res 67:7713–7722
Yanaihara N, Caplen N, Bowman E et al (2006) Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9:189–198
Esau C, Kang X, Peralta E et al (2004) MicroRNA-143 regulates adipocyte differentiation. J Biol Chem 279:52361–52365
Krutzfeldt J, Kuwajima S, Braich R et al (2007) Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res 35:2885–2892
Elmen J, Lindow M, Silahtaroglu A et al (2008) Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res 36:1153–1162
Esau C, Davis S, Murray SF et al (2006) miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab 3:87–98
Elmen J, Lindow M, Schutz S et al (2008) LNA-mediated microRNA silencing in non-human primates. Nature 452:896–899
Ebert MS, Sharp PA (2010) MicroRNA sponges: progress and possibilities. RNA 16:2043–2050
Lanford RE, Hildebrandt-Eriksen ES, Petri A et al (2010) Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327:198–201
Meister G, Tuschl T (2004) Mechanisms of gene silencing by double-stranded RNA. Nature 431:343–349
Acknowledgements
We thank Iddo Ben-Dov for sharing his unpublished data and Miguel Brown and Aleksandra Mihailovic for assistance with figure generation. We thank Markus Hafner, Kemal Akat, and Neil Renwick for their help with editing the manuscript. T.F. is supported by Grant #UL1 TR000043 from the National Center for Research Resources and the National Center for Advancing Translational Sciences (NCATS), NIH. J.I.H. is supported by the Deutsche Forschungsgemeinschaft. T.T. is an HHMI investigator, and work in his laboratory was supported by NIH grant MH08442, RC1CA145442 and the Starr Cancer Foundation. We apologize to those investigators whose work we could not cite due to space constraints.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Farazi, T.A., Hoell, J.I., Morozov, P., Tuschl, T. (2013). MicroRNAs in Human Cancer. In: Schmitz, U., Wolkenhauer, O., Vera, J. (eds) MicroRNA Cancer Regulation. Advances in Experimental Medicine and Biology, vol 774. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-5590-1_1
Download citation
DOI: https://doi.org/10.1007/978-94-007-5590-1_1
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-5589-5
Online ISBN: 978-94-007-5590-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)