
Abstract
Because altered microRNAs (miRNAs) expression patterns have been observed in a variety of diseased tissues, miRNA expression was compared in human cervical cancer tissues relative to adjacent normal cervical tissues in the present study. Microarray chips with 924 probes were used to detect the expression of miRNAs in cervical cancer tissue and adjacent normal cervical tissue of 13 patients with cervical cancer (11 squamous cervical cancers, one cervical adenocarcinoma, and one cervical sarcoma), all of whom were infected with human papilloma virus (HPV) 16 and/or HPV18. Compared to the expression levels in normal cervical tissues, 18 miRNAs (1.9%) were significantly upregulated (increase of ≥2×), and 19 miRNAs (2.1%) were significantly downregulated (decrease of ≤0.5×) in cervical cancer tissues. miRNA expression was independent of lymph node involvement, vascular invasion, and pathological differentiation. Taken together, cervical cancer tissues have altered expression of miRNAs relative to adjacent normal tissues. Further studies are necessary to determine whether aberrant miRNA expression is related to the pathogenesis of cervical cancer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNAs) are small endogenous noncoding RNAs that can specifically silence gene expression, and thereby alter cell and organism phenotype. Previous studies have indicated that miRNAs are involved in developmental timing, cell proliferation, apoptosis, morphogenesis [1], antiviral defense [2], and tumorigenesis [3]. At present, the physiological function and identity of target genes are only known for few miRNAs. Analysis of the entire miRNAome has become possible by the use of microarrays that contain all known human miRNAs [4, 5]. Screening for miRNAs that are expressed differently in normal and cancerous tissues may help to identify miRNAs involved in cancer pathogenesis.
Accumulating evidence indicates that various miRNAs are aberrantly expressed or mutated in diverse viral diseases and cancers [6–9], suggesting a role for miRNAs in the pathogenesis of these diseases. In fact, more than 50% of miRNA genes are located at chromosomal regions that are genetically altered in human cancers, such as fragile chromosome sites, or chromosomal regions where there is extensive deletion or amplification [9]. All of these suggest an important role of miRNAs in cancer pathogenesis.
Cervical cancer is the second most common cancer in women, and the human papilloma virus (HPV) infection is the primary cause of cervical cancer [10, 11]. miRNA genes are commonly observed at HPV integration sites many of which are associated with cancers [12]. Furthermore, the involvement of the particular miRNAs in the pathogenesis of cervical cancer has been suggested [13–15]. Thus, systematically screening the miRNA expression profiles in between cancerous and normal tissues might facilitate the identification of more candidate miRNAs that might play roles in the pathogenesis of cervical cancer. To pursue the above objective, 13 patients diagnosed with HPV16- or HPV18-positive cervical cancer were included in the present study. The differential miRNA expression profiles in between cervical cancer tissues and adjacent normal tissues were compared using miRNA chip technology.
Methods
Patients and materials
Cervical cancer tissues and adjacent normal cervical tissues were obtained from 13 patients treated at the Second Affiliated Hospital of Sun Yat-sen University (Guangzhou, China) from June to December of 2006 (Table 1). Eleven patients had squamous cervical cancer, one had cervical adenocarcinoma, and one had cervical sarcoma. Approval from the institutional ethics committee of the Second Affiliated Hospital of Sun Yat-sen University and the patients’ written informed consent were obtained before surgery. None of the patients received any prior treatment intervention, such as radiotherapy, chemotherapy, immunotherapy, or recent partial physiotherapy.
The presence of HPV16 and/or HPV18 in all cancerous and adjacent normal tissues was confirmed using the human papillomavirus types 16 and 18 (HPV16, HPV18) Fluorescent Polymerase Chain Reaction Diagnostic Kit (Da An Gene, Guangzhou, Guangdong, China) (data not shown). All patients had clinical stages IB1, IB2, IIA, or IIB, based on the International Federation of Gynecology and Obstetrics (FIGO) staging system (Table 1). All cancerous and adjacent normal tissue samples (2–4 cm) collected from each patient were pathologically confirmed to be cancerous or normal by both hematoxylin and eosin staining and immunohistochemistry, which were also allowed us to distinguish different cell types, such as stroma cells, fibroblasts, and infiltrating cells. The adjacent normal tissue was 2 cm away from the cancerous tissue. Therefore, the ratios of epithelial to stromal cells of cancerous and normal tissues were likely to be similar. All patients received radical hysterectomy and pelvic lymphadenectomy. Samples were collected immediately after surgery and preserved in liquid nitrogen before analysis.
Preparation of miRNA samples and miRNA array
The miRNA expression profiles of tissue samples were determined by the CaptialBio mammalian miRNA array V 3.0 (CapitalBio, Beijing, China), according to the manufacturer’s instructions. This miRNA array contains 924 probes that recognize 802 known miRNAs (from human, mouse, and rat) and 122 predicted miRNA sequences. All reagents were prepared in DEPC-treated water to inactivate RNases, and total RNA was extracted from tissue samples using the Trizol reagent (Invitrogen, Carsibad, CA, USA). For each miRNA array experiment, 25 μg of total RNA was subjected to polyethylene glycol (PEG) precipitation to enrich the low-molecular-weight RNA fraction. Isolated miRNAs were labeled with 5’-phosphate-cytidyl-uridyl-cy3-3’ (CU-Cy3; Dharmacon, Lafayette, CO, USA) by T4 RNA ligase (NEB, Ipswich, MA, USA). After precipitation, the CU-Cy3-labeled miRNA was resuspended in 15 μL of hybridization buffer containing 3 × sodium chloride-sodium citrate Buffer (SSC), 0.2% sodium dodecyl sulfate (SDS), 15% formaldehyde, and 5 × Denhard’s solution. Hybridization was performed under a LifterSlipTM (Electron Microscopy Sciences, Hatfield, PA, USA) at 42°C overnight. The miRNA array was washed with 0.2% SDS and 2 × SSC solution at 42°C for 4 min, then with 0.2 × SSC for 4 min at room temperature. After air-drying, the array was scanned by a Confocal bio-chip scanner (LuxScanTM 10 K/A, CaptialBio).
Statistical and bioinformatic analysis of microarray data
Array data were normalized by simple median centering of the arrays (making the median equal to zero using Significance Analysis of Microarrays (SAM) v2.1 software) and analyzed using GenePix Pro 4.0 software (Molecular Devices, Silicon Valley, CA, USA). Statistical comparisons of the human miRNAs (excluding four rno-miRNAs from rat) were performed by SAM v2.1 software (www-stat.stanford.edu/~ tibs/SAM/), licensed by the Stanford University Office of Licensing (otl.stanford.edu). miRNAs with a q-value (false discovery rate [FDR]) of <5%, and at least a 2-fold increase or a 0.5-fold decrease in miRNA expression were considered to have significant changes in their expression between the samples. For hierarchical analysis, Cluster version 3.0 software (bonsai.ims.u-tokyo.ac.jp/~ mdehoon/software/cluster) was used to analyze average linkage clustering of the miRNAs identified by SAM between human cancer and adjacent normal tissues. Java Treeview (jtreeview.sourceforge.net/) was used for tree visualization.
Results
Microarray analysis of 924 miRNA probes of cervical cancer tissues and adjacent normal cervical tissues from 13 patients was performed. In all chip images, the signal values of positive controls (HEX, U6, U2, TRNA, ZIP25, ZIP23, ZIP21, ZIP15, ZIP13, ZIP5, Y3, has-let-7a, has-let-7b, has-let-7c) were all above 5,000, and the signal values of the negative controls (DMSO and Y2) were under 1,000, indicating that the chips were reliable.
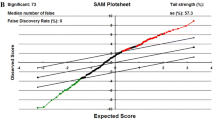
SAM analysis of cervical cancer tissues relative to adjacent normal cervical tissues indicated that the FDR was 0.0159 (Fig. 1a); 18 miRNAs were significantly upregulated (Table 2), and 19 miRNAs were significantly downregulated (Table 3). As shown in Fig. 1b, cluster analysis of the human miRNAs was performed (excluding the four rno-miRNAs from rat, which might non-specifically bound to human samples). The top row of this figure shows that the clustering algorithm yielded two main clusters: cancerous tissues (labels beginning “1R”) and normal tissues (labels beginning “2R”), with the exception of cancerous tissue sample 1R283, which appeared in the normal tissue cluster.

Differentially expressed miRNAs in human cervical cancer. a Significance Analysis of Microarrays (SAM) of differentially expressed miRNAs in human cervical cancer tissues and adjacent normal tissues. Red dots represent the 18 significantly upregulated miRNAs (increase of 2 × or more); green dots represent the 19 significantly downregulated miRNAs (decrease of 0.5 × or less); black dots represent miRNAs with no significant difference in expression. b Cluster analysis of miRNA expression in adjacent normal (I) and human cervical cancer tissues (II). Yellow represents increased expression and blue represents decreased expression. Top row : tissue samples. Labels beginning “1R” are cancerous tissues (n = 13), labels beginning “2R” are normal tissues (n = 13). Two samples, 1R20 (cancerous) and 2R14 (normal), were assayed twice. Right column : miRNAs that were significantly upregulated (III) or downregulated (IV) in cancerous cervical tissue relative to adjacent normal cervical tissue. Four rno-miRNAs (from rat) were not included in the cluster analysis; therefore, only the 33 human miRNAs with altered expression patterns were included in the analysis (Color figure online)
SAM analysis of the miRNA data for all cervical cancer patients analyzed in this study indicated that miRNA expression was not dependent on lymph node metastasis (FDR = 0.529, Fig. 2a), vascular invasion (FDR = 0.371, Fig. 2b), or pathological differentiation (FDR = 0.163, Fig. 2c).

Significance Analysis of Microarrays (SAM) of differentially expressed miRNAs in cervical cancer tissues with different pathologies. a lymph node metastasis, b vascular invasion, and c pathological differentiation
Discussion
Microarray analysis of 924 miRNAs in cervical cancer tissues and adjacent normal cervical tissues of 13 patients who were infected with HPV16 and/or HPV18 was performed in the present study. Based on statistical comparisons performed by SAM, significant upregulation of 18 miRNAs (1.9%) and downregulation of 19 miRNAs (2.1%) in cervical cancer tissues relative to normal cervical tissues were observed. The mean expression of the 18 upregulated miRNAs was at least double that of normal cervical tissues; the mean expression of the 19 downregulated miRNAs was at least half that of normal cervical tissues. These results are in agreement with previous studies of HPV-positive human cervical cancer tissues in which aberrant expression of selected miRNAs was reported to be involved in the pathogenesis of human cervical cancer [13–15].
HPV infection is the principle cause of cervical cancer [10, 11], and all of the patients analyzed in the present study were HPV-positive. miRNA genes are commonly observed at HPV integration sites many of which are associated with cancers [12]. In squamous cell carcinoma of the head and neck (SCCHN) cell lines, altered miRNA expression was observed in HPV16-positive cells when compared to HPV16-negative cells [16]. Specifically, HPV16 reduced miR-218 expression [15], and the miR-218 single-nucleotide polymorphism, rs11134527, may contribute to cervical cancer susceptibility in Chinese women [17]. Further studies are necessary to determine the role of these miRNAs in cervical cancer progression.
Expression of miRNAs was independent of tumor stage, lymph node involvement, vascular invasion, and pathological differentiation, suggesting that although dysregulation of miRNA expression may be related to the occurrence of cervical cancer, it is unrelated to cancer invasion and metastasis. These results conflict with those of Hu et al. [18], who reported that the expression levels of miR-200a and miR-9 were useful in cervical cancer prognosis and proposed that they may have important roles in cervical cancer pathogenesis. These conflicting results may be due in part to the ethnicities of the patients analyzed; Hu et al. [18] was carried out in the United States whereas the present study analyzed Chinese patients.
miRNA genes are frequently located in chromosomal regions characterized as “fragile”, with high rates of aberrations evident in diverse human cancers [9], suggesting that miRNA expression might be altered by these cancer-related chromosomal abnormalities. In the present study, miR-100, located at chromosome 11q24.1, was downregulated in cervical cancers (Table 3). Zhang et al. [19] reported that the 11q23.3-11q24.1a region is associated with the loss of heterozygosity (LOH) in cervical cancer. Therefore, the downregulation of miR-100 observed in this study may have a role in the LOH that is observed in cervical cancer. Further studies are necessary to determine the exact role of miR-100 in cervical cancer pathogenesis.
Several of the miRNAs whose expression was altered in the present study were also altered in a variety of other cancerous tissues. Specifically, miR-218, which was significantly downregulated in the present study, was also downregulated in Helicobacter pylori-associated gastric cancer [20]. Infection by H. pylori significantly decreased miR-218 expression, which may be related to upregulation of NF-κB [20]. In addition, miR-429 expression was upregulated in the present study, which is in agreement with Wu et al. [21], who reported its upregulation in endometrioid adenocarcinoma. Furthermore, miR-7, which was upregulated in the present study, is considered an oncomiRNA, mediating epidermal growth factor receptor (EGFR) signaling in lung cancer; ectopic expression increased lung cancer cell proliferation and tumor formation [22]. miR-7, along with miR-214, was also expressed during neuroblastoma differentiation; however, reduced miR-214 was observed in the present study [23].
The altered miRNA expression during carcinogenesis implies that miRNAs may be of use as cancer biomarkers or therapeutic targets [24]. For example, Blenkiron et al. [25] reported that analysis of miRNA expression could distinguish between basal and luminal breast tumors. In lung squamous cell carcinoma, miR-155, let-7, and miR-146a are of prognostic use [26]. In addition, altering miRNA expression, both knockdown and re-expression, can induce sensitivity of cancer cells to anti-cancer drugs [24]. Further studies are necessary to determine which miRNA, if any, can serve as prognostic biomarkers or therapeutic targets in cervical cancer.
There are several limitations to the present study that warrant discussion. First, the difficulty in obtaining normal cervical tissue from healthy individuals necessitated using adjacent normal cervical tissue from cervical cancer patients as a control. However, use of this tissue was important in ruling out interindividual differences that may be present; highly variable miRNA expression has been reported in normal cervical tissues [27]. Furthermore, while the present study assessed the expression levels of miRNAs in cervical cancer, their role in its pathogenesis remains to be elucidated. Further studies assessing their expression in premalignant (CIN 1, CIN 2, CIN 3) lesions will help clarify the impact of miRNAs on cervical cancer carcinogenesis.
Conclusion
In summary, the present study observed dysregulation of numerous miRNA genes in human cervical cancer. Further studies that employ larger samples are necessary to determine the role of miRNA dysregulation in the pathogenesis of cervical cancer. This line of research will help to identify miRNAs that are related to HPV-positive cervical cancer and may lead to new therapeutic approaches for this disease.
Abbreviations
- CU-Cy3:
-
5′-phosphate-cytidyl-uridyl-cy3-3′
- FBS:
-
Fetal bovine serum
- FDR:
-
False discovery rate
- FIGO:
-
International federation of gynecology and Obstetrics
- HMEC:
-
Human mammary epithelial cell line
- HPV:
-
Human papilloma virus
- LOH:
-
Loss of heterozygosity (LOH)
- miRNAs:
-
MicroRNAs
- Q-PCR:
-
Quantitative PCR
- SAM:
-
Significance analysis of microarrays
References
Kim VN. Small RNAs: classification, biogenesis, and function. Mol Cells. 2005;19:1–15.
Lecellier CH, et al. A cellular microRNA mediates antiviral defense in human cells. Sci. 2005;308:557–60.
O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc–regulated microRNAs modulate E2F1 expression. Nat. 2005;435:839–43.
Thomson JM, Parker J, Perou CM, Dang CV, Mendell JT. A custom microarray platform for analysis of microRNA gene expression. Nat Methods. 2004;1:47–53.
Barad O, et al. MicroRNA expression detected by oligonucleotide microarrays: system establishment and expression profiling in human tissues. Genome Res. 2004;14:2486–94.
Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nat. 2005;435:682–6.
Gupta A, Gartner JJ, Sethupathy P, Hatzigeorgiou AG, Fraser NW. Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nat. 2006;442:82–5.
Burnside J, et al. Marek’s disease virus encodes microRNAs that map to meq and the latency-associated transcript. J Virol. 2006;80:8778–86.
Calin GA, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004;101:2999–3004.
Walboomers JM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–9.
Bosch FX, Lorincz A, Muñoz N, Meijer CJ, Shah KV. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol. 2002;55:244–65.
Nambaru L, Meenakumari B, Swaminathan R, Rajkumar T. Prognostic significance of HPV physical status and integration sites in cervical cancer. Asian Pac J Cancer Prev. 2009;10:355–60.
Wang X, et al. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS ONE 2008;e2557.
Pang RT, et al. MicroRNA-34a suppresses invasion through downregulation of Notch1 and Jagged1 in cervical carcinoma and choriocarcinoma cells. Carcinogenesis. 2010;31:1037–44.
Martinez I, et al. Human papillomavirus type 16 reduces the expression of microRNA-218 in cervical carcinoma cells. Oncogene. 2008;27:2575–82.
Wald AI, Hoskins EE, Wells SI, Ferris RL, Khan SA. Alteration of microRNA profiles in squamous cell carcinoma of the head and neck cell lines by human papillomavirus. Head Neck 2010 Jul 22 [Epub ahead of print].
Zhou X, et al. Polymorphisms involved in the miR-218-LAMB3 pathway and susceptibility of cervical cancer, a case-control study in Chinese women. Gynecol Oncol. 2010;117:287–90.
Hu X, et al. A microRNA expression signature for cervical cancer prognosis. Cancer Res. 2010;70:1441–8.
Zhang Y, et al. Microarray profile of micro-ribonucleic acid in tumor tissue from cervical squamous cell carcinoma without human papillomavirus. Obstet Gynaecol Res. 2009;35:842–9.
Gao C, et al. Reduced microRNA-218 expression is associated with high nuclear factor kappa B activation in gastric cancer. Cancer. 2010;116:41–9.
Wu W, Lin Z, Zhuang Z, Liang X. Expression profile of mammalian microRNAs in endometrioid adenocarcinoma. Eur J Cancer Prev. 2009;18:50–5.
Chou YT, et al. EGFR promotes lung tumorigenesis by activating miR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF. Cancer Res. 2010;70:8822–31.
Chen H, et al. miR-7 and miR-214 are specifically expressed during neuroblastoma differentiation, cortical development and embryonic stem cells differentiation, and control neurite outgrowth in vitro. Biochem Biophys Res Commun. 2010;394:921–7.
Sarkar FH, Li Y, Wang Z, Kong D, Ali S. Implication of microRNAs in drug resistance for designing novel cancer therapy. Drug Resist Updat. 2010;13:57–66.
Blenkiron C, et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007;8:R214.
Raponi M, et al. MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer Res. 2009;69:5776–83.
Pereira PM, Marques JP, Soares AR, Carreto L, Santos MA. MicroRNA expression variability in human cervical tissues. PLoS One. 2010;5:e11780.
Acknowledgments
This work was supported by National Natural Science Foundation of China No. 30872743.
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rao, Q., Zhou, H., Peng, Y. et al. Aberrant microRNA expression in human cervical carcinomas. Med Oncol 29, 1242–1248 (2012). https://doi.org/10.1007/s12032-011-9830-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12032-011-9830-2