
Abstract
The DSIR-HA-1179 coleopteran cell line is a susceptible and permissive host to the Oryctes rhinoceros nudivirus (OrNV), which has been used as a biocontrol agent against the coconut rhinoceros beetle (Oryctes rhinoceros); a pest of palms in the Asia-Pacific region. However, little is known about growth and metabolism of this cell line, knowledge of which is necessary to develop an in vitro large-scale OrNV production process. The strong anchorage-dependent characteristics of the cell line, its particular fragility and its tendency to form dense clumps when manipulated, are the most likely reasons that have precluded further development of the cell line. In order to characterize DSIR-HA-1179 cells, there was first a need for a reliable technique to count the cells. A homogenous cell suspension suitable for enumeration could be produced by treatment with TrypLE Express™ with optimum mean time for cell release calculated as 30 min. The cell line was adapted to grow in four serum-supplemented culture media namely TC-100, IPL-41, Sf-900 II and Sf-900 III and cell growth, glucose consumption, lactate and ammonia production were assessed from static-batch cultures. The maximum viable cell density was reached in Sf-900 II (17.9 × 105 cells/ml), with the maximum specific growth rate observed in this culture medium as well (0.0074 h−1). Higher production of OrNV was observed in IPL-41 and TC-100 (4.1 × 107 TCID50/ml) than in cultures infected in Sf-900 III (2.0 × 107 TCID50/ml) and Sf-900 II (1.4 × 107 TCID50/ml). At the end of the growth period, glucose was completely consumed in cultures grown in TC-100, while remained in excess in the other three culture media. The cell line produced lactate and ammonia to very low levels in the TC-100 culture medium which is a promising aspect for its cultivation at large-scale.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The coconut rhinoceros beetle, Oryctes rhinoceros, L. (Coleoptera: Dynastinae) is a major pest of coconut and oil palms found throughout Southeast Asia, the Indian Subcontinent and the Pacific Islands (Bedford 1980), that causes economic losses that reach millions of dollars annually (Jackson et al. 2005; Moore 2009). Control of the beetle using chemical pesticides has had limited success owing to the life cycle of the beetle with damage to the tops of palms caused by feeding adults. The use of the Oryctes rhinoceros nudivirus (OrNV), a natural pathogen of the beetle, has been an effective method for controlling the coconut rhinoceros beetle (Young 1986; Huger 2005).
The traditional in vivo method of production of the virus relies on its propagation in infected beetle larvae (Huger 2005). However, this process has several disadvantages, including the requirement for a facility for growing and maintaining the insects, the need for numerous personnel and a final product that suffers from inconsistencies in virus concentration and purity. In vitro large-scale production of OrNV in insect cell cultures would be an important step towards the feasible and robust production of this virus, provided that engineering and economical constrains are solved (Visnovsky et al. 2003; Gioria et al. 2006). The profitable production of viral biopesticides in vitro requires the efficient scale up of a host cell line, the use of low-cost chemically defined and preferably animal-component-free cell culture media, and the maintenance of high viral specific productivity (Claus et al. 2012). The characterization of growth and metabolism of the host cell line will play a key role in understanding both cell growth kinetics and nutrient requirements. Cell growth kinetics will be very relevant to the management and prediction of the cell and virus production process, and an understanding of nutrient requirements, important to tailor a specific culture medium that will support both high cell and virus yield.
To date, the only cell line that has proven a susceptible and permissive host to infection with OrNV is the DSIR-HA-1179 cell line, established in 1979 by Crawford from sterilized eggs of the black beetle Heteronychus arator (Crawford 1981). There have been studies on the molecular and structural biology of OrNV (Payne 1974; Richards et al. 1999; Mohan and Gopinathan 1989; Crawford and Zelazny 1990), its viral pathogenesis in DSIR-HA-1179 cells (Crawford and Sheehan 1985), and more recent studies on nudivirus genomics (Wang et al. 2011). However, no literature currently exists on the characterization of this cell line from a technological perspective, with the exception of an initial estimation of the population doubling time of 6 days shortly after establishment of the DSIR-HA-1179 cell line (Crawford 1982). The strong anchorage-dependent characteristics of this cell line, the fact that it grows forming clumped clusters that become even larger when the cells are lifted, its particular fragility, and the lack of a method to dissociate and reliably count individual cells, are reasons that have hampered any further work on understanding the growth and metabolism of the cell line over the past 30 years. Additionally, of the 500 reported insect cell lines, only 22 are coleopteran derived and these, as a group, have not been fully understood in terms of their long term cultivation, manipulation, and the possibility of using them as hosts for mass production of viruses (Hoshino et al. 2009; Goodman et al. 2012).
In animal cell cultures, rapid, accurate determination of cell concentration and viability are essential in monitoring cell growth and the effects that the culture environment has on the viability of cells, including their interaction with biological, chemical, and mechanical agents that are part of the same environment. Attachment-dependent cell lines can suffer a loss in their viability if they are not handled properly when sampled. In the case of DSIR-HA-1179 cells, the situation is even more complex, since the cells tend to aggregate when they are removed from the growth surface. Thus, there is an added requirement to dissociate cell clumps into a homogenous single cell suspension to allow individual cells to be scored for viability. The simplest methods for dissociating an adherent cell monolayer into a single cell suspension involve mechanical detachment, such as tapping the flask to dislodge cells, repeated aspiration through a pipette, and cell scraping. The addition of chemicals such as sulfated polyanions (e.g. heparin and dextran sulfate) to the culture medium may reduce aggregation in cell clumps, while the addition of chelators, like EDTA, sequester divalent cations such as calcium and magnesium, which play an important role in cell adhesion (Freshney 1987). Alternatively, an adherent cell monolayer maybe dissociated using an enzymatic method which involves treating the monolayer with a proteolytic enzyme for a short duration (Freshney 1987). Of these methods, enzymatic dissociation has been most widely used because of its ability to release a large number of cells whilst preserving cellular integrity and viability (Cunningham 1999). Fragile cell lines that suffer damage upon treatment with strong enzymes such as trypsin and collagenase may instead be treated with TrypLE™ Express, a recombinant fungal trypsin-like protease that is gentler and less toxic to cells. TrypLE™ Express is a serine protease which cleaves peptides at the C-terminal of arginine and lysine amino acids much like trypsin. Its stability at room temperature and the fact that it does not require inactivation with serum or other protease inhibitors make it an attractive alternative to using trypsin (Nestler et al. 2004).
The quantification of nutrients consumed and metabolites produced by insect cell lines is an important step towards the characterization of these cell lines. Nutritional limitations and by-products accumulation can affect cell multiplication and viability. This in turn, can have an effect on the synthesis of products produced by cells such as recombinant proteins or viruses. Among the metabolites involved in insect cell cultivation, glucose, lactate and ammonia emerge as important ones. Glucose has been indicated as one of the main energy sources for insect cells (Drugmand et al. 2012), while lactate and ammonia have been demonstrated to adversely affect them because of the toxic effects they produce when they accumulate in the culture medium (Cruz et al. 2000). Metabolic profiles vary among insect cells lines. Glucose has been found to be the major carbohydrate source in a range of insect cell lines including Sf-9 (Reuveny et al. 1992), High-Five (Rhiel et al. 1997), saUFL-Ag-286 (Gioria et al. 2006) and BCIRL-HZ-AM1 (Lua and Reid 2003). BTI-Tn-5B1-4 and BCIRl-HZ-AM1 cells display an inherent tendency to produce lactate and ammonia even when provided with sufficient glucose to grow in batch culture (Rhiel et al. 1997; Lua and Reid 2003), while Sf-9 cells will produce them only under glucose deprivation (Ohman et al. 1995; Drews et al. 2000). On the other hand, the metabolism of the saUFL-AG-286 cell line is characterized by the production of ammonia but not of lactate (Gioria et al. 2006). To date, no studies have been conducted on nutrient consumption or metabolite production by DSIR-HA-1179 cells. Understanding the metabolic behaviour of this cell line is both a prerequisite for tailoring an appropriate low-cost serum-free culture medium which can meet the specific nutritional requirements of the cell as well as to design a cultivation strategy in order to achieve high cell and virus yields in a large-scale production process.
This paper describes first time attempts to understand, accurately manipulate and characterize the DSIR-HA-1179 coleopteran cell line in order to develop a feasible process for the in vitro large-scale production of OrNV. The cell line was adapted to grow in a range of commercially available insect cell culture media and screened for their ability to support cell growth and OrNV production. Since the understanding of the nature of this very adherent cell line is at a nascent stage, the difficulties faced while culturing it and techniques developed in overcoming these difficulties are described. The kinetic parameters of growth in the different culture media as well as glucose consumption, lactate and ammonia production were also evaluated to allow for future development of a tailored chemically-defined serum-free culture medium that suits the specific requirements of the DSIR-HA-1179 cell line.
Materials and methods
Cell cultures and culture media
DSIR-HA-1179 cells were obtained from the AgResearch (Lincoln, New Zealand) cell culture collection and were maintained as adherent cultures in 25 cm2 tissue culture flasks (Corning®, Kennebunk, MN, USA) at 27 °C in 5 ml of 10 % foetal bovine serum (FBS, Life Technologies, Auckland, New Zealand) supplemented PS-100 culture medium, which was prepared in-house. PS-100 is a modified preparation of Grace’s insect medium made to mimic TC-100. It was prepared by supplementing Grace’s medium (Sigma, Auckland, New Zealand) with tryptose phosphate broth (Sigma) at 2.95 g/l and TC-100 vitamins (Sigma) at 1 ml/l, and then adjusting the pH of the medium to 6.2 using 10 M potassium hydroxide (Sigma).
A 5-months sequential process was used in adapting DSIR-HA-1179 cultures to grow from PS-100 to four commercial culture media: TC-100 (Sigma), IPL-41 (Life Technologies, Auckland, New Zealand), Sf-900 II (Life Technologies) and Sf-900 III (Life Technologies), that were supplemented with 10 % FBS. Once adapted, the cells were routinely passaged in the appropriate culture medium when the cell monolayer reached 80 % confluence.
Cell dissociation and cell counting methods
For nuclei counting, a citric acid lysis buffer was made by dissolving 0.9 g of citric acid powder (Sigma) in 100 ml of distilled water to which 0.01 g crystal violet nuclear stain (Sigma) was added and homogenously dispersed. A 1 ml sample of cell suspension was centrifuged at 12,000g for 5 min and the supernatant discarded. The cell pellet was re-suspended in 1 ml of the lysis buffer, vortexed in a whirl mixer for 1 min, and then incubated for 1 h at 37 °C. The individually stained nuclei were counted in duplicate using a Neubauer hemocytometer.
The role of heparin in eliminating cell aggregation was investigated by cultivating duplicate 25 cm2 attached DSIR-HA-1179 cultures (seeded at approximately 2 × 105 cells/ml) in 5 ml of 10 % FBS-supplemented TC-100 containing heparin (Beparine®, 5,000 IU/ml, Biological E, Hyderabad, India) at concentrations of either 100, 250 or 667 μg/ml. Cultures were visually inspected over a 14 day growth period for dissociated single cells. Dissociated cells were collected and the trypan blue dye exclusion assay was used to assess culture viability. Briefly, cell suspensions were stained with 0.4 % trypan blue (Sigma) and then both the total and viable cells were counted in duplicate using a Neubauer haemocytometer (Phillips 1973).
TrypLE™ Express was used to dissociate cells. Prior to adding TrypLE™ Express (Life Technologies), 2 ml of Dulbecco’s phosphate buffered saline (D-PBS) free of calcium and magnesium (Sigma) was used to wash the DSIR-HA-1179 cell monolayer. This washing step with buffer was essential in order to remove any calcium and magnesium ions present on the monolayer as well as residues of the spent cell culture medium. 1 ml of TrypLE™ Express, pre-warmed to 27 °C was added per 25 cm2 of a confluent cell monolayer in the tissue culture flask. The flask was rocked gently to evenly coat the monolayer and then incubated at 27 °C until cells had visibly detached. An appropriate volume of pre-warmed cell culture medium containing 10 % FBS was added and the cell suspension was gently aspirated with a 10 ml pipette to break up any remaining cell aggregates. Cell counts were made in a Neubauer haemocytometer and trypan blue dye exclusion assay was used to assess cell counts and culture viability.
Cell growth kinetics
DSIR-HA-1179 cell growth kinetics were conducted on attached cultures in 25 cm2 tissue culture flasks with a culture volume of 5 ml for each of the four commercial culture media. For each culture medium, TrypLE™ Express treatment (as described above) of a confluent cell monolayer in a 75 cm2 tissue culture flask, provided sufficient number of cells to seed fourteen replicate 25 cm2 flasks, each at an initial cell density of 2 × 105 viable cells/ml (Day 0) in a culture volume of 5 ml. Duplicate cultures were randomly selected and harvested every 48 h and total and viable cell counts determined until day 14, when cultures in TC-100, Sf-900 II and Sf-900 III reached 100 % confluence. At this stage, cultures in IPL-41 were 80 % confluent.
Nutrient analyses
Glucose concentrations were determined using a YSI 2700 biochemistry analyzer (YSI, Yellow Springs, OH, USA). Lactate concentrations were determined using a reflectometric test kit (Merck, Darmstadt, Germany). Ammonia concentration was determined using a commercial kit based on the Berthelot reaction (Wiener Labs, Rosario, Argentina) (Patton and Crouch 1977).
Rates
The DSIR-HA-1179 cell specific growth rate (μ) was calculated by identifying the linear region from the semilog plot of viable cell density versus time, followed by linear regression of these data (Gioria et al. 2006). Population doubling time (td) was calculated by Eq. 1:
Over the duration of the exponential growth phase, the mean specific consumption rate of glucose was calculated by determining cell yield (given by the slope of the plot of mean glucose concentration vs. viable cell density) and then multiplying this value by the specific growth rate (Gioria et al. 2006).
Virus and virus quantification
OrNV stock (strain X2B) at a concentration of 1 × 107 TCID50/ml was obtained from AgResearch. X2B is a highly virulent strain originally isolated in 1983 from a field population of infected coconut rhinoceros beetles on Bugsuk Island, Palawan, Philippines (Crawford et al. 1986; Zelazny et al. 1990).
The virus stock used in these experiments was prepared by infection of attached cultures during the early exponential phase of growth (approximately 4 × 105 viable cells/ml) at a MOI of 0.1 TCID50/cell. OrNV titer was quantified by end-point dilution analysis. Briefly, to determine the TCID50, suspensions of DSIR-HA-1179 cells (2.5 × 105 cells/ml) were seeded onto 96 well plates (50 μl per well) and then an equal volume of each viral supernatant dilution was added with five replicates per supernatant. The plates were placed in a humidified, disinfected plastic container and incubated at 27 °C for 11–14 days until the cytopathic effect was well developed, when the plates were scored for infection, and TCID50 was calculated (Reed and Muench 1938).
Results and discussion
Adaptation of the cell line to four culture media
The starting point in this work was the adaptation of attached DSIR-HA-1179 cell cultures grown in the PS-100 culture medium to four commercially available insect cell culture media namely TC-100, IPL-41, Sf-900 II and Sf-900 III. This was done to test those which might better support the nutritional requirements of the cell line and thereby improve cell yield and virus production, and to select a culture medium that could be used as a model to conduct initial studies on this cell line. From the large spectrum of commercially available insect cell culture media, these were selected for this initial screening for the following reasons: TC-100 was chosen because of its similarity in composition to PS-100 (Gardiner and Stockdale 1975) in which the cell line was originally maintained. The other three culture media were selected as they are formulated with additional carbohydrates and amino acids sources. For example, IPL-41 contains sucrose and maltose in addition to glucose as a carbohydrate source (Weiss et al. 1981), while Sf-900 II and Sf-900 III culture media are very rich formulations with higher concentrations of asparagine and glutamine (Bovo et al. 2008; Lua and Reid 2003). All culture media were supplemented with 10 % FBS to provide the cells a better chance to be adapted to the new culture media.
Distinct variations in cell morphology and growth patterns were observed between the four adapted cultures, as shown in Fig. 1. In the PS-100 culture medium, cells grew in a fibroblast-like shape with clear cytoplasm and the formation of cell ‘towers’ upon reaching confluence. Cells adapted to the TC-100 culture medium exhibited a similar morphology and growth pattern, which is to be expected if one considers that PS-100 was formulated to mimic the TC-100 culture medium. From visual observation, cells adapted to IPL-41 culture medium were smaller in size, appeared slightly rounded and had clear, non-vacuolar cytoplasm. On the other hand, cells adapted to Sf-900 II and Sf-900 III culture media were larger in size (Fig. 1). Cells in Sf-900 II appeared more round in shape and contained cytoplasmic vacuoles which could be observed as clear round spots at 400× magnification. Cells in PS-100, TC-100, IPL-41 and Sf-900 III adhered strongly to the flask surface, while cells grown in Sf-900 II showed a decreased degree of adherence to the flask. This tendency for reduced adherence and rounded morphology of cells in Sf-900 II indicates potential for this culture medium to be used in the future for adapting the cell line to grow as a free suspension culture. Cells that are round in shape have been found to be less susceptible to hydrodynamic stresses in the culture environment during cultivation in agitated suspension (Chisti 2001; Papoutsakis 1991). Cultivation of DSIR-HA-1179 cells in suspension rather than attached cultures, would potentially improve the virus volumetric productivity (Micheloud et al. 2011).

Micrographs of uninfected DSIR-HA-1179 cells grown in stationary culture in four culture media supplemented with 10 % FBS. The images were obtained using ×400 magnification once cultures reached 70–80 % confluence
Development of a technique to count DSIR-HA-1179 cells
To evaluate kinetic parameters of the DSIR-HA-1179 cell line, there was a need to develop a method for reliable cell counting; a task never achieved to date because of the particular growth characteristics of this cell line. It was first necessary to produce a homogenous suspension of single cells. Several methods, including mechanical dispersion, nuclei counting, heparin and enzymatic treatments were evaluated.
Simple mechanical means such as tapping the flask surface to dislodge cells and repeated gentle aspiration of the culture did not homogenously disperse the cell monolayer. Sections of the monolayer detached, and cells aggregated in clumps of differing sizes, preventing cell counting in a haemocytometer. More vigorous pipetting partially disaggregated the clumps but resulted in cell damage (visible cell debris), thus significantly reducing culture viability. Following these trials, it was confirmed that DSIR-HA-1179 is a strongly attachment dependent cell line, and its tendency to form dense clumps during growth, present a myriad of difficulties to the cell culturist. The presence of clumps introduces diffusion limitations in nutrients, including oxygen, in the culture media from reaching the cells. Furthermore, virus infection in such aggregated cultures will be less effective and therefore unproductive, because of the physical impediment of the virus to effectively reach every single cell. The DSIR-HA-1179 cell line is highly sensitive to shear forces so disruption of the clumps by mechanical means is not a viable option.
The possibility of using nuclei counting as a method to determine total cell number was investigated. The inability to monitor culture viability using nuclei counting is a disadvantage (Van Wezel 1973) because monitoring this parameter over batch growth is important and more so in large-scale cell cultivations. Moreover, the presence of bi-nucleated cells during the exponential growth phase introduces errors in the total cell count estimated by this method (Berry et al. 1996). Despite these disadvantages, the technique was still assayed on DSIR-HA-1179 cells since it was not known at that point of the research whether it would be possible to develop a more reliable method to determine total cell counts. Assessments of total cell counts of DSIR-HA-1179 cells by nuclei counting were later compared with those made using TrypLE™ Express (a more reliable technique for counting both viable and total cells that was developed in parallel). This confirmed that nuclei counting overestimated the total cell count by 14.8 % (mean total cell count from DSIR-HA-1179 cultures on day 14 of growth that were treated with TrypLE™ Express was 17 × 105 ± 2.1 cells/ml vs. nuclei counts that estimated 19.5 × 105 ± 2.8 cells/ml), corroborating the findings of Berry et al. (1996). In the light of this result and success achieved later with TrypLE™ Express, nuclei counting was deemed an unreliable method for total cell counting.
The use of heparin as an anti-aggregation agent to inhibit cell-to-cell adhesion has been reported for some cell lines. It has been successful in facilitating the suspension adaptation process in the HEK293 mammalian cell line (Tsao et al. 2001) and the TN 5B-1-4 insect cell line (Taticek et al. 1997), both used as hosts in virus production applications. The optimal heparin concentrations that promoted the anti-aggregation effect in these two cell lines were 100 and 667 μg/ml, respectively. On the other hand, for the suspension adaptation of recombinant CHO-TS28 cells, 250 μg/ml heparin was reported to effectively induce cell dispersion (Li et al. 2011). Based on these previous findings, and aiming to reduce cell clumping, this range of heparin concentrations were tested on attached cultures of the DSIR-HA-1179 cells. Heparin treatment did not produce any disaggregation when assayed on the cells at the two lower concentrations, while the highest concentration of 667 μg/ml induced cell death possibly due to the dose of heparin being toxic to the cells. Further investigation contemplating the use of heparin in a concentration ranged between 250 and 667 μg/ml could be done to conclusively learn whether heparin can be effectively used to disaggregate this cell line.
It was then attempted to dissociate DSIR-HA-1179 cells grown in TC-100 supplemented with 10 % FBS using the TrypLE™ Express enzyme treatment, which was followed by a D-PBS wash step to ensure the removal of Ca2+ and Mg2+ ions from the cell monolayer, since these two ions have a role in increasing cell to substrate adhesion in animal cell cultures (Takeichi and Okada 1972). In preliminary experiments, cultures that were treated with TrypLE™ Express were microscopically observed at 5 min intervals to gauge the time it would take for the complete dissociation of the monolayer. It was found that the maximum number of cells released between 20 and 30 min post treatment. Due to a lack of literature on TrypLE™ Express incubation times and volumes to be used for the DSIR-HA-1179 cell line, an experiment was conducted to optimize these parameters according to the conditions shown in Table 1. Duplicate 25 cm2 T-flasks containing 5 ml cultures of DSIR-HA-1179 cells labeled 1–4 were seeded at an estimated density of 2 × 105 viable cells/ml, and incubated at 27 °C until day 10 of growth when 70 % confluence was reached. TrypLE™ Express treatment was then used to dissociate cell monolayers (as described in the “Materials and methods” section), using the treatment conditions listed in Table 1.
All four treatments were equally effective in releasing cells from the attached monolayer and in dissociating cell clumps to enable the creation of a homogenous single cell suspension. From Table 1, cell viability in cultures incubated for 30 minutes, either with 1 or 2ml of TrypLE Express, were comparable, appearing that the use of 2 ml of TrypLE™ Express instead of 1 ml had no detrimental effect on culture viability (94.0 %). However, since 1 ml of TrypLE™ Express was sufficient to coat the monolayer, this was deemed the optimal volume of TrypLE™ Express to use. Prolonged exposure of cells to TrypLE™ Express for up to 60 min followed by gentle mixing led to a slight decrease in culture viability (91.8 %). Exposure for up to 80 min combined with stronger mixing led to a further decrease in viability (89.9 %). Thus, the optimal duration of incubation of the DSIR-HA-1179 cells with TrypLE™ Express and handling approach were determined to be 30 min with gentle mixing of the cell suspension prior to counting. One explanation for the relatively long duration of TrypLE™ Express treatment could be because incubation was carried out at 27 °C, which is lower than the enzyme’s optimum operating temperature of 37 °C. Nevertheless, this duration is similar to the mean time required for cell release of the highly adherent MDCK canine kidney cell line which is reported at 28.5 min (Nestler et al. 2004).
To determine whether long term exposure to protease carryover would lead to a loss in cell growth and viability, the effect of passaging DSIR-HA-1179 cells with the inclusion of TrypLE™ Express at every passage was investigated (Fig. 2). At each passage, 1 ml of TrypLE™ Express was added to the parent flask to dissociate and count cells, which were then seeded into a new flask at a concentration of 2 × 105 viable cells/ml. The small percentage of TrypLE™ Express (4 %) carried over at each passage did not affect cell growth and viability between the first and fourth passage. However, at the fifth passage, the culture viability suffered a significant drop (from 93 to 73 %) and continued to decrease over the next two passages, suggesting that the use of TrypLE™ Express in routine passages after the fourth passage would not be recommended. The enzyme however proved to be an efficient tool in achieving the objective of creating a DSIR-HA-1179 single cell suspension culture, which was a prerequisite for both making accurate total and viable cell counts (i.e. to assess cell growth kinetics) and manipulating cells (i.e. for seeding a culture at a particular cell density).

Effect of prolonged use of TrypLE™ Express on viable cell concentration and viability of DSIR-HA-1179 cultures in TC-100 supplemented with 10 % FBS
Cell growth kinetics
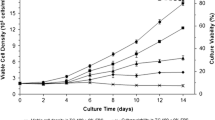
To begin characterizing DSIR-HA-1179 cell growth kinetics in the four culture media selected for this study, cell density and culture viability assays were assessed. As shown in Fig. 3, cells went through an initial lag phase which lasted for approximately 48 h after which the exponential phase of growth commenced. All cultures were allowed to grow until they reached confluence, as explained in the “Materials and methods” section. Of the four culture media, Sf-900 II gave the highest maximum viable cell density (17.9 × 105 viable cells/ml) and the mean highest specific growth rate over the batch culture (0.0074 h−1, PDT: 3.9 days). Additionally, high culture viability was observed in Sf-900 II (99.2 %), which is possibly because cells grew with a rounded shape in this culture medium and were therefore able to tolerate hydrodynamic forces of shear during cell dissociation and pipetting better. Kinetic parameters in Sf-900 III and TC-100 culture media were broadly comparable to Sf-900 II. Mean maximum viable cell densities of 16.9 × 105 and 16.8 × 105 viable cells/ml were reached, respectively, in Sf-900 III and TC-100 culture media, with mean specific growth rates of 0.0072 h−1 in Sf-900 III (PDT: 4.0 days) and 0.0069 h−1 in TC-100 culture media (PDT: 4.2 days). Mean culture viabilities in both of these culture media remained high, at 92.3 % (Sf-900 III) and 92.9 % (TC-100), over the batch growth period measured. In comparison, cultures in IPL-41 grew relatively slower with a mean specific growth rate of 0.0057 h−1 (PDT: 5.1 days). The mean maximum viable cell density reached in IPL-41 was 12.5 × 105 viable cells/ml, which was appreciably lower than in the other three culture media though mean culture viability was 93 %.

Mean growth and culture viability profiles of the DSIR-HA-1179 cell line in four culture media. Error bars represent standard deviations from the mean of duplicate cell counts
Typical specific growth rates for insect cell lines range between 0.01 and 0.03 h−1 (Schmid 1996). In comparison, the specific growth rate of the DSIR-HA-1179 cell line is very slow. In nature, the development of the parent beetle, Heteronychus arator takes approximately 6–8 months. The very slow growth rate of the DSIR-HA-1179 cell line in vitro is likely an inherent characteristic of the embryonic tissue from which the cell line derives (Crawford 1982). Across the four culture media tested, population doubling times varied between 3.9 and 5.1 days which is still an improvement from the population doubling time of 6 days estimated at the time of establishment of the cell line (Crawford 1982). The growth characteristics of continuous cell lines are known to evolve over time along with putative changes at the genome level. Such evolution maybe induced by selection pressures resulting from modifications made to the culture environment. The improvement of growth rate could have been a result of adaptation of the cells to the new culture media—especially because the highest growth rate was observed in Sf-900 II, which is one of the richest existing insect cell culture media (Bovo et al. 2008). Over the last 30 years the cell line has been through over 400 passages. The growth rate improvement could also be attributed to an effect of this long-term passaging of the cell line (Donaldson and Shuler 1998).
Metabolites analyses
Glucose has been identified as a preferred energy and carbon source for insect cells and is considered the most important carbohydrate to monitor during batch culture (Drews et al. 1995). As Fig. 4 shows, the glucose consumption profile of DSIR-HA-1179 cells in TC-100 indicates that this was the only culture medium in which glucose was exhausted at the end of the 14 days of batch growth. The mean overall specific rate of glucose consumption in TC-100 was determined to be 4.1 × 10−12 g cell−1 h−1 while a higher value of 5.0 × 10−12 g cell−1 h−1 was observed in IPL-41. Mean specific consumption rates of glucose in Sf-900 II and Sf-900 III were 4.3 × 10−12 and 3.1 × 10−12 g cell−1 h−1, respectively. Although specific rates of glucose consumption reported for insect cell lines vary under differing culture medium and cultivation conditions, one study has estimated the specific glucose consumption rates in suspension cultures of lepidopteran cell lines Sf-9, Sf-21 and BTI-Tn-5B1-4 at 3.7 × 10−12, 5.1 × 10−12 and 14.5 × 10−12 g cell−1 h−1, respectively (Kwon et al. 2003). BTI-Tn-5B1-4 is one of the highest yielding cell lines (up to 8 × 106 cells/ml in fed-batch culture) and correspondingly has a high specific rate of glucose consumption (Schmid 1996). The DSIR-HA-1179 cell line has a slower specific growth rate but not a slower specific rate of glucose consumption compared to Sf-9 and Sf-21 cell lines. The specific rate of glucose consumption ranged between 3 and 5 × 10−12 g cell−1 h−1 across the four culture media and these differences could be attributed to the varying concentrations of glucose present in the culture media (Micheloud et al. 2011). However, the similarity in the specific rates of glucose consumption between cultures grown in TC-100 and Sf-900 II, despite the glucose concentration of Sf-900 II being almost 9 times higher than in TC-100 (1 g/l), suggests that the cells are able to metabolize glucose very efficiently in the TC-100 culture medium.

Mean glucose consumption profiles for DSIR-HA-1179 cultures in four culture media. Error bars represent the standard deviations from the mean of duplicate samples. Error bars are difficult to see because standard deviations were very small
The buildup of waste metabolites such as lactate and ammonia to high concentrations in batch culture could be potentially toxic to animal cells. Insect cell lines differ in their ability to produce lactate and ammonia. For example, Sf-9 cells under well oxygenated culture conditions do not produce lactate and ammonia, even in cultures supplemented with glucose concentrations higher than 40 mM (Rhiel et al. 1997). In contrast, BTI-Tn-5B1-4 cells accumulate relatively high amounts of lactate, between 10 and 20 mM (Sugiura and Amann 1996; Yang et al. 1996). The saUFL-AG-286 cell line, for example, is characterized by the production of ammonia but not of lactate (Gioria et al. 2006). This reinforces the idea that each specific cell line must be tested for production of these metabolites to shed light on its individual metabolic characteristics. It was found that lactate was produced to very low levels in DSIR-HA-1179 cells cultivated in TC-100 supplemented with 10 % FBS with a mean value of 0.1 g/l, while the ammonia production was almost negligible at 0.02 g/l. The very low level of lactate production indicates that the cell line is able to metabolize glucose from the culture medium efficiently. In this respect, DSIR-HA-1179 cells share a similarity with the metabolic nature of the Sf-9 cell line. It would be well worth investigating the production of ethanol and glycerol which have been identified as by-products of glucose metabolism in the Sf-9 cell line (Drews et al. 2000). In addition, in order to gain a complete understanding into the metabolism of the cell line, detailed amino acid analyses must be performed. Nevertheless, this initial finding that DSIR-HA-1179 cells produce very low levels of lactate and ammonia is a promising prospect for the large-scale cultivation of the cell line to high densities for the production of OrNV in future.
Infection of DSIR-HA-1179 cell cultures with OrNV
Cultures of DSIR-HA-1179 cells adapted to the four culture media were infected with OrNV at a MOI of 0.1 TCID50/cell at the 5th day post-seeding (early exponential growth phase). By visual observation, as the infection progressed the rate of growth slowed. The typical cytopathic effect in OrNV infected cultures at the 14th day post-infection (d.p.i) is shown in Fig. 5. (refer to Fig. 1 for comparison for morphology of uninfected cells). Infected cells rounded-up and small vesicles were observed. DSIR-HA-1179 cells infected in Sf-900 II and Sf-900 III were noticeably larger than cells infected in TC-100 and IPL-41 culture media.

Micrographs of DSIR-HA-1179 cells infected with Oryctes rhinoceros nudivirus in stationary cultures in four culture media. Cells were infected during the early-exponential growth phase at a MOI of 0.1. The images were obtained using ×200 magnification, at 14 days post infection
Before the inclusion of TrypLE™ Express could be recommended in a process for producing OrNV in DSIR-HA-1179 cell cultures, it was necessary to ascertain that the enzyme would not affect replication of the virus. Infected cultures that were inoculated on cells either with or without TrypLE™ Express™ treatment, were harvested at the 14th day post infection, and the virus yields were determined (Fig. 6). In all four culture media, OrNV production was consistently improved in the experimental condition where cells had been inoculated following treatment with TrypLE™ Express. A possible explanation for this could be that TrypLE™ Express effectively breaks down the extracellular matrix of the treated culture and creates a homogenous single cell suspension free of cell aggregates. Such an evenly dispersed cell inoculum resulted in the formation of uniform, clump free cell monolayers that facilitated virus attachment and binding to a greater extent than in the absence of TrypLE™ Express, where cells growing in clumps caused a physical barrier to virus attachment and where mass transfer limitations of nutrients in the culture media could have occurred.

Comparison of Oryctes rhinoceros nudivirus production with and without the use of TrypLE™ Express in four culture media. Error bars represent standard deviations from the mean of duplicate samples, except for IPL-41 with TrypLE™ Express where the same virus titer was recorded for both samples
From Fig. 6, it is interesting to note that despite Sf-900 II producing the highest cell yield, OrNV production in this culture medium was the lowest (1.4 × 107 TCID50/ml). Virus production was only marginally improved in Sf-900 III (2.0 × 107 TCID50/ml) despite the high specific rate of growth in this culture media. This could be due to some components of the culture media inhibiting virus binding. The formulations of Sf-900 II and III are proprietary and detailed testing of the culture media is required in order to elucidate this information.
It is interesting to note that despite having the lowest specific growth rate, the highest virus production was obtained in IPL-41 culture medium (4.1 × 107 TCID50/ml). TC-100 culture medium followed closely, yielding 3.8 × 107 TCID50/ml. The complete exhaustion of glucose from the culture medium in TC-100 cultures on day 14 of batch growth (Fig. 4) could explain why the virus titer achieved in TC-100 was slightly lower than in IPL-41. For virus production, the infection of the cultures lasted for 14 days post time of infection (TOI), which was 5 days after the cells were seeded. Hence, the infection phase lasted through days 5–20 of batch culture. In asynchronous infections at low MOI, only a small proportion of cells become initially infected. Uninfected cells continue to grow and become targets during the subsequent round of ‘secondary’ infection. During this period, both infected and uninfected cells require glucose to support their basal metabolic needs, and the premature depletion of glucose might result in inhibition of viral replication in cells infected secondarily (Visnovsky and Claus 1994). In IPL-41, an excess of glucose remaining in the culture medium on day 14 suggests the possibility that the cells were provided sufficient glucose for their metabolic activities during infection between days 14 and 20; which was not the case for TC-100 cultures. The consumption of glucose in cultures in IPL-41 during infected growth will be investigated in future experiments. Nonetheless, the virus titers obtained in TC-100 (3.8 × 107 TCID50/ml) were comparable with those obtained in IPL-41 (4.1 × 107 TCID50/ml), indicating that these two culture media are potential candidates for further studies on OrNV production in DSIR-HA-1179 cell cultures.
Conclusions
This work describes for the first time, attempts to manipulate and characterize the unique DSIR-HA-1179 coleopteran cell line, the only cell line susceptible and permissive to infection by the Oryctes rhinoceros nudivirus known so far. The cell line was successfully adapted to grow as attached cultures in a range of culture media supplemented with 10 % FBS, including TC-100, IPL-41, Sf-900 II and Sf-900 III. It was found that the cells underwent morphological changes when adapted to new growth conditions. Simultaneously, a method for dissociating and accurately assessing DSIR-HA-1179 cell numbers and viability using the recombinant serine protease TrypLE™ Express was developed and optimized. The use of 1 ml of pre-warmed TrypLE™ Express per 25 cm2 of flask surface area and incubation for 30 min at 27 °C enabled the creation of a homogenous single cell suspension of DSIR-HA-1179 cells while preserving cell viability (94 %). TrypLE™ Express did not affect cell viability when it was used in routine cell passages until the fourth passage. In addition, it was verified that the inclusion of this enzyme in the cell culture process did not have any detrimental effect on the in vitro infection and production of OrNV.
Growth kinetics in TC-100, Sf-900 II and III culture media were comparable, with the highest mean specific growth rate of 0.0074 h−1 obtained in Sf-900 II. This value is an improvement over the specific growth rate estimated during establishment of the cell line, suggesting a possible benefit of adaptation of the cell line to more nutritionally suitable culture media. Glucose was determined to be a useful carbohydrate source in all four culture media, and it was totally depleted at the end of batch growth when its initial concentration was 1.0 g/l. Lactate and ammonia did not accumulate during batch growth of this cell line in TC-100, indicating an efficient use of nutrients in the culture medium. OrNV production in IPL-41 and TC-100 culture media supported the highest production of the virus (4.1 × 107 and 3.8 × 107 TCID50/ml).
Future development of the use of DSIR-HA-1179 as a host cell line in a cost-effective process for producing OrNV at large-scale would benefit from a detailed understanding of key aspects, including the adaptation of the cell line to grow in free suspension culture and in-depth metabolic studies of the cell line. In addition, the development of a low-cost chemically defined serum-free culture medium will be a key feature for the use of this cell line for industrial purposes. Future work will include the optimization of parameters such as MOI, TOI and time of harvesting of OrNV, as well as bioreactor selection and the development of production process strategies.
Abbreviations
- OrNV:
-
Oryctes rhinoceros nudivirus
- MOI:
-
Multiplicity of infection
- TOI:
-
Time of infection
- TCID:
-
Tissue culture infectious dose
- PDT:
-
Population doubling time
References
Bedford GO (1980) Biology, ecology and control of palm rhinoceros beetles. Annu Rev Entomol 25:309–339
Berry JM, Huebner E, Butler M (1996) The crystal violet nuclei staining technique leads to anomalous results in monitoring mammalian cell cultures. Cytotechnology 21:73–80
Bovo R, Galesi AL, Jorge SA, Piccoli RA, Moraes AM, Pereira CA, Augusto EF (2008) Kinetic response of a Drosophila melanogaster cell line to different medium formulations and culture conditions. Cytotechnology 57:23–35
Chisti Y (2001) Hydrodynamic damage to animal cells. Crit Rev Biotechnol 21:67–110
Claus JD, Gioria VV, Micheloud GA, Visnovsky G (2012) Production of insecticidal baculoviruses in insect cell cultures: potential and limitations. In: Soloneski S, Larramendy L (eds) Insecticides—basic and other applications. InTech, pp 127–152
Crawford AM (1981) Attempts to obtain Oryctes baculovirus replication in three insect cell cultures. Virology 112:625–633
Crawford AM (1982) A coleopteran cell line derived from Heteronychus arator (Coleoptera: Scarabaidae). In Vitro 18:813–816
Crawford AM, Sheehan C (1985) Replication of Oryctes baculovirus in cell culture: viral morphogenesis, infectivity and protein synthesis. J Gen Virol 66:529–539
Crawford AM, Zelazny B (1990) Evolution in Oryctes baculovirus: rates and types of genomic change. Virology 174:294–298
Crawford AM, Zelazny B, Alfiler RA (1986) Genotypic variation in geographical isolates of Oryctes baculovirus. J Gen Virol 67:949–952
Cruz HJ, Freitas CM, Alves PM, Moreira JL, Carrondo MJT (2000) Effects of ammonia and lactate on growth, metabolism and productivity of BHK cells. Enzyme Microb Technol 27:43–52
Cunningham RR (1999) Tissue disaggregation. In: Jarvois LC (ed) Methods in molecular biology, immunocytochemical methods and protocols. Humana Press, Totowa, New Jersey, pp 257–260
Donaldson MS, Shuler ML (1998) Effects of long-term passaging of BTI-Tn5B1-4 insect cells on growth and recombinant protein production. Biotechnol Prog 14:543–548
Drews M, Paalme T, Vilu R (1995) The growth and nutrient utilization of insect cell line Spodoptera frugiperda Sf9 in batch and continuous culture. J Biotechnol 40:187–198
Drews M, Doverskog M, Öhman L, Chapman BE, Jacobsson U, Kuchel PW, Häggström L (2000) Pathways of glutamine metabolism in Spodoptera frugiperda (Sf9) insect cells: evidence for the presence of the nitrogen assimilation system, and a metabolic switch by 1H/15N NMR. J Biotechnol 78:23–37
Drugmand J-C, Schneider Y-J, Agathos SN (2012) Insect cells as factories for biomanufacturing. Biotechnol Adv 30:1140–1157
Freshney R (1987) Culture of animal cells: a manual of basic technique. Alan R. Liss, Inc., New York
Gardiner GR, Stockdale H (1975) Two tissue culture media for production of lepidopteran cells and polyhedrosis virus. J Invertebr Pathol 25:363–370
Gioria VV, Jager V, Claus JD (2006) Growth, metabolism and baculovirus production in suspension cultures of an Anticarsia gemmatalis cell line. Cytotechnology 52:113–124
Goodman CL, Stanley D, Ringbauer JA, Beeman RW, Silver K, Park Y (2012) A cell line derived from the red flour beetle Tribolium castaneum (Coleoptera: Tenebrionidae). In Vitro Cell Dev Biol Animal 48:426–433
Hoshino K, Hirose M, Iwabuchi M (2009) A new insect cell line from the longicorn beetle Plagionotus christophi (Coleoptera: Cerambycidae). In Vitro Cell Dev Biol Animal 45:19–22
Huger AM (2005) The Oryctes virus: its detection, identification, and implementation in biological control of the coconut palm rhinoceros beetle, Oryctes rhinoceros (Coleoptera: Scarabaedaie). J Invertebr Pathol 89:78–84
Jackson TA, Crawford AM, Glare TR (2005) Oryctes virus—time for a new look at a useful biocontrol agent. J Invertebr Pathol 89:91–94
Kwon MS, Dojima T, Park EY (2003) Comparative characterization of growth and recombinant protein production among three insect cell lines with four kinds of serum free media. Biotechnol Bioprocess Eng 8:142–146
Li L, Qin J, Feng Q, Liu R, Xu L, Chen Z (2011) Heparin promotes suspension adaptation process of CHO-TS28 cells by eliminating cell aggregation. Mol Biotechnol 47:9–17
Lua LHL, Reid S (2003) Growth, viral production and metabolism of a Helicoverpa zea cell line in serum-free culture. Cytotechnology 42:109–120
Micheloud GA, Gioria VV, Eberhardt I, Visnovsky G (2011) Production of the Anticarsia gemmatalis multiple nucleopolyhedrosis in serum-free suspension cultures of the saUFL-AG-286 cell line in stirred reactor and airlift reactor. J Virol Methods 178:106–116
Mohan KS, Gopinathan KP (1989) Quantitation of serological cross-reactivity between two geographical isolates of Oryctes baculovirus by a modified ELISA. J Virol Methods 24:203–214
Moore AM (2009) Guam Coconut Rhinoceros Beetle (CRB) Eradication Program Semi-annual Progress report. University of Guam Cooperative Extension Service, pp 9
Nestler L, Evege E, McLaughlin J, Munroe D, Tan T, Wagner K, Stiles B (2004) TrypLE™ Express: a temperature stable replacement for animal trypsin in cell dissociation applications. Quest 1:42–47
Ohman L, Ljunggren J, Haggstron L (1995) Induction of a metabolic switch in insect cells by substrate limited fed batch cultures. Appl Microbiol Biotechnol 43:1006–1013
Papoutsakis ET (1991) Fluid-mechanical damage of animal cells in bioreactors. Trends Biotechnol 9:427–437
Patton CJ, Crouch SR (1977) Spectrophotometric and kinetics investigation of the Berthelot reaction for the determination of ammonia. Anal Chem 49:464–469
Payne CC (1974) The isolation and characterization of a virus from Oryctes rhinoceros. J Gen Virol 25:105–116
Phillips HJ (1973) Dye exclusion tests for cell viability. In: Kruse PF, Patterson MK (eds) Tissue culture. Academic Press, New York, pp 406–408
Reed LJ, Muench H (1938) A simple method of estimating 50% endpoints. Am J Epidemiol 27:493–497
Reuveny S, Kemp CW, Eppstein L, Shiloach J (1992) Carbohydrate metabolism in insect cell cultures during cell growth and recombinant protein production. Ann NY Acad Sci 665:230–237
Rhiel M, Mitchell-Logan CM, Murhammer DW (1997) Comparison of Trichoplusia ni BTI-Tn-5B1-4 (High Five) and Spodoptera frugiperda Sf-9 insect cell line metabolism in suspension cultures. Biotechnol Bioeng 55:909–920
Richards NK, Glare TR, Aloali’l I, Jackson TA (1999) Primers for the detection of Oryctes virus from Scarabaeidae (Coleoptera). Mol Ecol 8:1552–1553
Schmid G (1996) Insect cell cultivation: growth and kinetics. Cytotechnology 2:43–56
Sugiura T, Amann E (1996) Properties of two insect cell lines useful for the baculovirus—expression system in serum-free culture. Biotechnol Bioeng 51:494–499
Takeichi M, Okada TS (1972) Roles of magnesium and calcium ions in cell-to-substrate adhesion. Exp Cell Res 74:51–60
Taticek RA, McKenna KA, Granados RR, Shuler ML (1997) Rapid initiation of suspension cultures of Trichoplusia ni insect cells (TN 5B-1-4) using heparin. Biotechnol Techniques 11:237–240
Tsao YS, Condon R, Schaefer E, Lio P, Liu Z (2001) Development and improvement of a serum-free suspension process for the production of recombinant adenoviral vectors using HEK293 cells. Cytotechnology 37:189–198
Van Wezel AL (1973) Microcarrier cultures of animal cells. In: Kruse PF, Patterson MK (eds) Tissue culture: methods and applications. Academic Press, New York, pp 372–377
Visnovsky G, Claus JD (1994) Influence of time and multiplicity of infection on the batch production of Anticarsia gemmatalis nuclear polyhedrosis virus in lepidopteran insect cell cultures. In: Galindo E, Ramirez OT (eds) Advances in bioprocess engineering. Kluwer Academic Publishers, Netherlands, pp 123–128
Visnovsky G, Claus JD, Merchuk JC (2003) Cultivation of insect cells in airlift reactors: influence of reactor configuration and superficial gas velocity. Lat Am Appl Res 33:207–211
Wang Y, Binida-Emonds OR, van Oers MM, Vlak JM, Jehle JA (2011) The genome of Oryctes rhinoceros nudivirus provides novel insight into the evolution of nuclear arthropod-specific large circular double-stranded DNA viruses. Virus Genes 42:444–456
Weiss SA, Smith GC, Kalter SS, Vaughn JL (1981) Improved method for the production of insect cell cultures in large volumes. In Vitro 17:495–502
Yang JD, Gecik P, Collins A, Czarnecki S, Hsu HH, Lasdun A, Sundaram R, Muthukumar G, Silberklang M (1996) Rational scale-up of a baculovirus-insect cell batch process based on medium nutritional depth. Biotechnol Bioeng 52:696–706
Young EC (1986) The rhinoceros beetle project: history and review of the research programme. Agric Ecosyst Environ 15:149–166
Zelazny B, Lolong A, Crawford AM (1990) Introduction and field comparison of baculovirus strains against Oryctes rhinoceros (Coleoptera: Scarabaeidae) in the Maldives. Environ Entomol 19:1115–1121
Acknowledgments
We sincerely thank the authorities of the College of Engineering, University of Canterbury, New Zealand, for the understanding and help received to continue with this research during the difficult time faced as a consequence of the earthquakes that affected Christchurch in 2010 and 2011. We extend heartfelt thanks to Universidad Nacional del Litoral, Santa Fe, Argentina for making the stay of Charlotte Pushparajan at the Laboratory of Virology, Faculty of Biochemistry and Biological Sciences there possible. The technical assistance provided by Dr. Veronica Gioria is appreciated. This research was partially funded by the New Zealand Foundation for Research, Science and Technology Research Fund, contract C10X0804.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pushparajan, C., Claus, J.D., Marshall, S.D.G. et al. Characterization of growth and Oryctes rhinoceros nudivirus production in attached cultures of the DSIR-HA-1179 coleopteran insect cell line. Cytotechnology 65, 1003–1016 (2013). https://doi.org/10.1007/s10616-013-9632-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10616-013-9632-9