
Abstract
The red flour beetle, Tribolium castaneum, is a model organism for agricultural and medical research and its complete genome is sequenced. We established a continuously replicating T. castaneum cell line to complement existing physiological, genetic, and genomic research tools. We set up trial cell cultures from egg, pupa, and adult stages as tissue sources and incubated them in six separate cell culture media to determine the optimal combination of tissue source and medium for cell replication. Our most promising culture was generated by co-culturing adult (∼75 %) and pupal tissues in EX-CELL 420 medium containing 9 % FBS. Our new cell culture is designated BCIRL-TcA-CLG1 (TcA) and it has been subcultured more than 90 times. Amplification of genomic DNA with species-specific primers yielded DNA fragments of the expected sizes and with sequences identical to those from the published Tribolium genome. Additionally, we characterized this line using DNA fingerprinting (DAF-PCR) and compared it with three other coleopteran cell lines and its conspecific pupae to confirm identity. Its doubling time is 155.2 hr. Early passages consisted of attached cells and vesicles in suspension, whereas later passages consisted primarily of attached, spherical cells. Similar to other established cell lines, the ploidy of TcA cells was variable, ranging from 20 chromosomes/cell (diploid) to above 30 chromosomes/cell. TcA cells withstood incubation at 40°C for 1 h with no decrease in viability. We recorded increased levels of one heat shock protein (43 kDa) and of the hsp68a transcript following exposure to 40°C. Taken together, this represents the first report of a continuously replicating T. castaneum cell line. We expect the BCIRL-TcA-CLG1 line will become a useful tool in Tribolium research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The red flour beetle, Tribolium castaneum (Coleoptera: Tenebrionidae), is an important pest of stored grains and has been extensively studied in field and laboratory settings. Research will expand with the recent completion of its genome (Park et al. 2008; Tribolium Genome Sequencing Consortium 2008; Kim et al. 2010). One reason for interest in T. castaneum is that Coleoptera is less evolutionarily derived than Diptera and it is possibly a better representative of the Class Insecta than Drosophila melanogaster (Schröder et al. 2008; Peel 2009). T. castaneum has become a model system for a wide variety of biological research, including anatomy, development, physiology, population genetics, and population ecology (Denell 2008). With the T. castaneum genome sequence available, investigators have pried deeper than before into numerous areas of insect biology, including immunity (Zhou et al. 2007; Knorr et al. 2009), endocrinology (Li et al. 2008), and evolutionary mechanisms of development (Denell 2008). The goal of the present work was to develop a continuous cell line from T. castaneum tissues to complement other tools for ongoing genomic and proteomic studies. Cell lines from Coleoptera are underrepresented in the cell culture arena compared to lepidopteran and dipteran cell lines (Lynn 2001; Hoshino et al. 2008); here, we provide a new line for investigations.
Materials and Methods
Cell line initiation.
Beetles were reared on whole wheat flour containing 5 % (w/w) brewer’s yeast following Haliscak and Beeman (1983). Trial cell cultures were prepared from egg, pupae, and adults. The culture producing a continuously replicating cell line was generated from 85 female beetles (Ga-1 isolates; ∼75 % Day 1 adults, ∼25 % Day 6 pupae). The beetles were surfaced sterilized as follows (10 mL/solution): 3 min wash in 0.05 % sodium hypochlorite, brief rinse in sterile water, 3 min wash in 70 % ethanol, and final rinse in sterile water. The beetles were transferred into a sterile Petri dish and minced using micro scissors in 1 mL EX-CELL 420 medium (Sigma Chem. Co., St Louis, MO) containing 200 U/mL penicillin and 0.2 mg/mL streptomycin (MP Biomedicals, Solon, OH). The resulting tissues were washed twice in 5 mL HBSS then incubated for 5 min in 1 mL trypsin–EDTA (0.05 % trypsin, 0.02 % EDTA, MP Biomedicals) while continuing to be minced and gently crushed with a pipette tip. The tissue suspension was centrifuged in two 15-mL tubes (800×g, 15 min, 4°C) and the pellets were resuspended in 13 mL EX-CELL 420 medium per tube (medium contained 9 % heat-treated FBS [Hyclone, Rockford, IL]; 100 U/mL penicillin with 0.1 mg/mL streptomycin [MP Biomedicals]; and 0.25 mg/mL amphotericin B [Sigma]). The supernatant was transferred into three T25 flasks, each containing 3–5 mL suspension, and monitored for growth. After 1 wk, the penicillin and streptomycin concentrations were reduced to half and amphotericin B was omitted. Half of the medium was replaced every 7–10 d. After approximately 6 mo, the cells from flask 1 were split 1:2 on a bi-weekly basis. Currently, the cells are maintained at 28°C and passaged on a weekly basis. The designation of this cell line is BCIRL-TcA-CLG1 (TcA).
Cell growth and morphological characterization.
The doubling time of the TcA line was determined after first incubating the cells for 10 min in the dyes Syto 13 (for living cells, 1.5 mM, Life Technologies-Invitrogen™, Grand Island, NY) and propidium iodide (for dead cells, 0.02 mg/mL, MP Biomedicals). Cells (2 × 105 cells/mL, 2 mL/well; passage 47 [P47]) were seeded into 12-well plates (BD Falcon, Billerica, MA) and counted periodically (three wells/time point). The cells were fed at 168 h. Cell diameters were determined using the CellometerR Vision cell counter (Nexcelom Bioscience, Lawrence, MA).
Karyotyping.
The chromosome numbers per cell were determined using a modification of Mitsuhashi (1995). Cells were grown to near confluence as described just above. Spent medium was removed and medium containing 0.5 mM colchicine was added to the culture. Cells were incubated at 28°C for 5 h, after which cells were detached by pipetting and pelletted at 1,000×g for 5 min at 4°C. The supernatant was discarded and the cells were resuspended and dispersed in 7 mL 0.58 % KCl. Cells were incubated at room temperature for 15 min and centrifuged as described. The pelletted cells were resuspended in 1 mL (1:1) 0.58 % KCl/fixative solution (3:1, methanol/glacial acetic acid) and incubated for 15 min. Cells were gently mixed with 5 mL freshly made fixative solution, incubated at room temperature for 10 min, and centrifuged as described above. Cells were then resuspended in 5 mL fixative solution, incubated at room temperature for 10 min, and centrifuged again. Lastly, cells were resuspended in 1 mL fixative solution and this suspension was added dropwise onto a glass slide. Slides were dried on a heating block, stained in Giemsa stain (diluted 1:10 with water) for 5 min, and then quickly rinsed in water twice and allowed to dry. After thoroughly drying, slides were briefly rinsed in xylene to remove excess lipids and stabilize the stain. For imaging, glycerol was placed on the slides, followed by a coverslip. Cells were imaged using an Olympus Vanox AHBT3 compound microscope attached to a Leica DFC295 color digital camera (×100 objective).
DAF-PCR.
The TcA line was characterized using DNA fingerprinting (DAF-PCR) with the primers described by McIntosh et al. (1996): mammalian aldolase, prolactin, and interluken. In addition to the TcA cell line, T. castaneum pupae and three other coleopteran cell lines were subjected to DAF-PCR for comparison: IPLB-DU from Diabrotica undecimpunctata (Lynn and Stoppleworth 1984); DSIR-HA-1179 from Heteronychus arator (Crawford 1982); and BRL-Ag3C from Anthonomous grandis (Stiles et al. 1992).
PCR characterization.
Genomic DNA was isolated from TcA cells and whole adult T. castaneum beetles using a commercial kit (Qiagen, Valencia, CA). Primer sequences for chitin synthase 1 (TcCHS1) were derived from nonspecific primers used to clone the entire gene (Arakane et al. 2004). Primers for ribrosomal protein S3 (TcrpS3) were previously published (Begum et al. 2009). Primers for tryptophan oxygenase V (Tcv) were designed to amplify a 400-bp segment of the third exon using PrimerBLAST (NCBI). Thermal cycling protocols for all primer sets included 2 min at 94°C; 40 cycles of 15 s at 94°C, 30 s at 60°C, and 30 s at 72°C. The thermal cycling protocol finished with a 5-min incubation at 72°C. PCR products were purified using a commercial kit (Qiaquick PCR Purification Kit, Qiagen) and visualized and photographed on a 2 % agarose gel impregnated with SYBRSafe gel stain (Life Technologies) using UV light. PCR products were sequenced using an Applied Biosystems 3730 DNA Analyzer (DNA Sequencing Services, Department of Plant Pathology, Kansas State University) and the resulting sequences were used as queries in BLAST searches.
Mycoplasma testing.
The TcA line was tested for the presence of Mycoplasma using the MycoAlert™ Kit (Lonza, Allendale, NJ).
Monitoring heat shock protein levels and gene expression.
Heat shock protein and mRNA levels were monitored using Western blotting and qPCR. Prior to heat shock treatments, cells (P90) were set up in T25 flasks (at ∼50 % confluency) and incubated for 48 h until ∼90 % confluency (three flasks/treatment/time point). Flasks were incubated in water baths at either 28°C (control) or 40°C (heat shock) for 20, 40, or 60 min. After the incubations, cells were resuspended in the spent medium (5 mL) and 1 mL of the suspension was transferred into a 1.5-mL tube for qPCR and 4 mL transferred into a 15-mL tube for SDS-PAGE. The cells were centrifuged at 1,000×g for 5 min at 4°C and the pellet was processed as indicated below. The impact of heat treatment on cell viability was tested after 60 min at 40°C. Cell counts and percent cell viability were determined using the Cellometer as just described.
For Western blots, 40 μL of 10 mM Tris–HCl, pH 7 (containing protease inhibitor cocktail, 1:75; Sigma, #P8340) was added to the cell pellets. Cells were sonicated on ice (3×; 35 W, 10 sec; Sonifier Cell Disruptor, Model W185, Heat Systems Ultrasonics, Planview, NY), followed by centrifugation (16,000×g, 10 min, 4°C). The supernatants were used as protein samples. Before storing at −80°C, a 5-μL aliquot was used to determine protein concentrations with a BCA protein assay (Thermo Science Pierce, Rockford, IL). Samples were diluted with sample buffer (0.0625 M Tris–HCl, pH 6.8, 25 % glycerol, 2 % SDS, 5 % mercaptoethanol, and 0.01 % bromophenol blue) for a final concentration of 20 μg/25 μL and heated at 95°C for 5 min. Samples (25 μL/lane) were applied to SDS-PAGE gels (4 % acrylamide loading gel and 10 % acrylamide resolving gel) and run at 125 V for ∼2.5 h using a Mini Protean cell (Bio-Rad, Hercules, CA). Proteins were transferred onto nitrocellulose membranes in Towbin’s buffer (25 mM Tris, 192 mM glycine, 20 % methanol, pH 8.3) using a Mini Trans-Blot Cell (Bio-Rad) at 30 V overnight. The following standards were applied to monitor protein transfer and estimate molecular weights: prestained standards (Bio-Rad, #161-0309; 48 to 204 kDa) and unstained standards (Bio-Rad, #161-0363, 10 to 250 kDa: this lane was stained separately with 0.1 % amido black in 10 % acetic acid, 25 % isopropanol). Blots were washed 3× rapidly in 25-mL Tris-buffered saline (TBS, 20 mM Tris, pH 7.6, 0.14 M NaCl), then blocked for 1 h in 25 mL SuperBlock blocking buffer (Thermo Sci. Pierce, #37545), and rocked (speed 3, Rocker Platform, Bellco Biotech., Vineland, NJ). The blots were washed 3× in 25-mL TBS with 0.05 % Tween 20 (TTBS) for 5 min/wash, rocked (speed 3), then incubated overnight at 4°C in the presence of the primary antibody (0.1 μg/mL anti-HSP70 [ab47455, Abcam, Cambridge, MA] or 0.02 μg/mL anti-HSP70/HSC70 [sc-59570, Santa Cruz Biotech, Inc., Santa Cruz, CA]) in antibody buffer (10 % SuperBlock buffer + 0.05 % Tween 20). The next day, the blots were washed 3× in 25-mL TTBS (5 min/wash; rocked, speed 3) and incubated in 25 mL secondary antibody (goat anti-mouse alkaline phosphatase conjugate, dilution, Bio-Rad, #170-6520) in antibody buffer for 1 h (rocked, speed 3). Finally, blots were washed 3× in 25-mL TTBS (5 min/wash; rocked, speed 3) and incubated in a nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate substrate solution (Bio-Rad, #170-6432) until bands were visualized (rocked, speed 3). Blots were imaged using a G:Box Chemi-XT4 imager (Syngene, Frederick, MA) and the protein band volumes were determined using GeneTools protein analysis software (Syngene).
For qPCR, RNA from TcA cell pellets was extracted using the TRIzol Reagent (Life Technologies-Invitrogen, Carlsbad, CA) following the manufacturer’s protocol and RNA concentrations determined on a NanoDrop 2000 Spectrometer at 260 nm (Thermo Scientific, Wilmington, DE). The first-strand cDNA was synthesized from ∼3 μg total RNA using superscript II reverse transcriptase (Life Technologies-Invitrogen) and oligo(dT) primers according to the manufacturer’s protocol.
Real-time PCR amplification and analysis was performed on an Eppendorf Master Cycle Rep Realplex 4S and analyzed with the Realplex software (Eppendorf, Hauppauge, NY). The qPCR reactions were carried out using QuantiFast SYBR Green PCR Kit (Qiagen) according to the manufacturer’s procedure in 20 μl reaction volumes. The qPCR cycling program used was: hold at 95°C for 5 min followed by 50 cycles at 95°C for 15 s, 58°C for 30 s, then 60°C for 30 s. The specificity of the SYBR Green PCR signal was further confirmed by melting curve analysis. The mRNA expression was quantified using the comparative CT (cross threshold, the PCR cycle number that crosses the signal threshold) method (Livak and Schmittgen 2001). The cDNA amplification was performed using gene-specific primers. The primers for T. castaneum hsp68a were designed based on sequence from http://www.ncbi.nlm.nih.gov/gene/ (Gene ID, 100142317, hsp68a): 5′-TGG TGG CAC TTT CGA CGT GTC TAT-′3 and 5′-AAG CCC TCG GAT TGC TTC TCA GAT-′3. The normalizing housekeeping gene used was rsp3 (5-CAG GGC TTG CTA TGG TGT ATC-′3 and 5′-ACG CAG TGT TCC AGA AAC TAC-′3) (Mahroof et al. 2005a).
Data from the Western blot and qPCR studies were analyzed statistically using ANOVA in combination with Fisher’s LSD test, p < 0.05 (NCSS 2007, Kaysville, UT).
Baculovirus replication.
Cells (1 × 105 cells/mL, 2 mL/well) were seeded into a 12-well plate and allowed to attach. Cells were inoculated with 10 multiplicity of infection (MOI) of one of the following viruses (in triplicate): a recombinant Autographa californica multiple nucleopolyhedrovirus expressing a red fluorescent protein under the Drosophila hsp70 promoter (rAcMNPV-hsp70Red), the Galleria mellonella multiple nucleopolyhedrovirus (GmMNPV), and Helicoverpa zea single nucleopolyhedrovirus (HzSNPV). Cells were rocked (speed 3) for 2 h and incubated at 28°C for 7 d. Wells containing no virus were used as controls.
Results
We initially worked with eggs, pupae, and adults but found that the co-culturing of adult and pupal tissues in EX-Cell 420 medium lead to the only long-term culture (BCIRL-TcA-CLG1), which has since been passaged over 90 times. Other media did not result in viable long-term cultures. These included: Shields & Sang, L-15, IPL-41, TNM-FH, and Schneider’s medium.
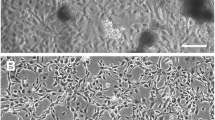
The cell line consists of two morphologically distinct cell types: a spherical and an elongated cell type (Fig. 1). In early passages, the cells periodically adhered to each other and formed ball-shaped vesicles that detached from the substrate and remained in suspension (Fig. 1B ). Later passage cells slowly lost the ability to form these vesicles and eventually primarily spherical, attached cells remained (Fig. 1D ).

Photomicrographs of the BCIRL-TcA-CLG1 cell line (×200): (A) attached cells, P4 (elongated and spherical cells); (B) cells in suspension, P4 (cells forming a ball-like vesicle); (C) typical cells (primarily attached cells; both elongated and spherical), P28; (D) typical cells (attached cells only; primarily spherical), P86.
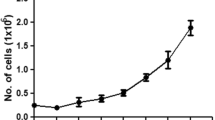
The doubling time of the TcA line is 155.2 h (Fig. 2) and its mean cell diameter is 8.5 ± 0.5 μm (±SEM). The chromosome count of this cell line ranged from approximately 20 to ∼32 chromosomes per cell (Fig. 3).

Growth curve of BCIRL-TcA-CLG1 cells (P56). Each point represents the mean (±SEM) number of cells/well at the indicated time after seeding. At time 0, 4 × 105 cells were seeded into 12-well culture plates. At the indicated times after seeding, cell counts in three wells/time point were determined using the Cellometer.

TcA cells (P80) were fixed and stained to show the chromosome groupings associated with each cell nucleus: (A, B) chromosomes from a single cell; (C) chromosome groupings from five cells. Chromosome counts revealed the polyploidy of the established cell line.
We characterized the cell line using DAF-PCR with three primers (Fig. 4A ). PCR analysis produced cell line DNA patterns similar to those of their host tissues (lanes 4 and 5) but distinctly different from other coleopteran cell lines (lanes 1–3). These primer-specific patterns were consistent between early and late passages (Fig. 4B ). The TcA line was Mycoplasma-free (data not shown).

Characterization of the TcA cell line by DAF-PCR using the indicated primers. (A) Analysis of the TcA cell line, other selected coleopteran cell lines, and T. castaneum pupae. Lanes: (1) IPLB-DU (D. undecimpunctata, P737); (2) DSIR-HA-1179 (H. arator, P47); (3) BRL-Ag3C (A. grandis, P18); (4) BCIRL-TcA-CLG1 (T. castaneum, P3); (5) T. castaneum pupae; (6) DNA Ladder (1 kb); (7) water control. (B) Analysis of the TcA cell line at different passages. Lanes: (1) DNA ladder (1 kb); (2) TcA cells, P9; (3) TcA cells, P21; (4) TcA cells, P79; (5) water control.
We also characterized the TcA cell line using three species-specific sets of primers (Fig. 5). PCR of genomic DNA from either whole beetles or the TcA cell line using these primer sets resulted in amplification of a single DNA product of the predicted size as visualized on an agarose gel. The PCR products for the TcA cell line were sequenced, and BLAST searches using the resulting sequences as queries revealed 100 % identity for all PCR products with published Tribolium sequences for these genes. Additionally, we tested the TcA cell line for Wolbachia contamination by using primers specific to the Wolbachia 16S gene. No PCR products were detected following amplification (data not shown).

Additional PCR characterization of the TcA cell line. Primer sequences and predicted product sizes are shown for each gene along with photos of the agarose gels used to visualize the PCR products. In each gel photo, four lanes are shown: top lane—100 bp ladder, second lane—PCR product amplified from whole beetle genomic DNA, third lane—PCR product amplified from TcA cell line genomic DNA, bottom lane—no template negative control.
We determined the response of the cells to heat shock conditions. Cell viability (as proportions of total cells) did not change after 60 min at 40°C compared with the 28°C control: 92.1 ± 1.0 % (28°C), 92.9 ± 1.7 % (40°C) (mean ± SEM, n = 3). We determined heat shock protein levels by Western blot (Fig. 6A ). The protein specifically recognized by both antibodies had a molecular weight of 43 kDa. This protein significantly increased after 40 min of incubation at 40°C, but returned to control levels after 60 min of incubation at 40°C. Expression of hsp68a mRNA increased significantly compared to the control cells at 20 and 40 min, but decreased significantly below control levels by 60 min.

Heat shock (40°C) influences expression of heat shock-specific mRNA and protein. (A) Western blot analysis using two monoclonal antibodies (anti-HSP70 and anti-HSP70/HSC70) revealed the presence of a 43-kDa protein at the indicated times after treatment. Histogram bars represent the mean (±SEM) ratios of experimental/control values. Bars annotated with different letters for the same antibodies are significantly different. (B) qPCR analysis of hsp68a expression at the indicated times after treatment. Histogram bars represent the mean (±SEM) ratios of experimental/control expression values. Bars annotated with different letters indicate significant differences.
We inoculated TcA cells separately with three baculoviruses (rAcMNPV-hsp70-Red, GmMNPV, or HzSNPV) at a high MOI (10) to determine the presence of infection. We recorded no OBs and all cells grew similarly to the controls (data not shown), indicating that this cell line is refractory to infection by these baculoviruses.
Discussion
We report the establishment of a cell line from T. castaneum. The BCIRL-TcA-CLG1 line is the sole cell line from this species and the first from any insect species within the Tenebrionidae. Compared to the hundreds of cell lines established from lepidopteran and dipteran insects, fewer than 30 cell lines have been established from Coleoptera (Lynn 2001). Hoshino et al. (2008) described 22 coleopteran cell lines, representing eight species and four families, in addition to the ones they established. Emphasis on the establishment of cell lines from lepidopteran and dipteran insects has been primarily due to the need to generate lines that could propagate viruses (e.g., baculoviruses for biological control uses or plant/mammalian viruses for studying their interactions with insect vectors; Lynn 2007; Smagghe et al. 2009) or cell cultures that could be used as models for development (Schneider 1972). Until recently, few viruses were known to infect beetles, with the notable exception of five viruses discussed in Crawford (1982). Over the past few decades, this has changed and a variety of insect viruses (representing at least four virus families) have been found to infect beetles (Asgari and Johnson 2010). The number of cell lines established from coleopterans is expanding. Many of the established coleopteran cell lines now available were derived from embryonic tissues, although three originated from other tissues, hemocytes (Charpentier et al. 2002), larval fat body (Iwabuchi 1999), and whole pupae (Long et al. 2002). Similarly, our cell line was established from non-embryonic tissues using whole insects (primarily adults).
The TcA cell line has characteristics that differ from other insect cell lines. The doubling time is longer than many insect cell lines, such as 22.6 h for Sf9 from Spodoptera frugiperda (in TNM-FH medium; Hashimoto et al. 2010) and 20–22 h for four cell lines from the coleopteran A. grandis (Stiles et al. 1992). Long doubling times have been recorded for two coleopteran cell lines: 5 d for IPLB-DU (Lynn and Stoppleworth 1984) and 6 d for DSIR-HA-1179 (Crawford 1982). It is not clear whether slow replication rates of these cell lines and the TcA line are due to nutritional factors or inherent cellular properties. The mean diameter of the TcA cells, 8.5 μm, is smaller than many insect cell lines, especially lepidopteran cell lines (e.g., 17.7 μm for BCIRL-HvAM1 cells and 18.9 μm for BCIRL-Hz-AM1 cells [Ringbauer, unpublished results]). The diameter of our cell line is within the reported range of 5–11 μm for D. melanogaster S2 cells, which is much smaller than other D. melanogaster lines produced at the same time (Schneider 1972).
Continuously replicating cell lines typically undergo increases in chromosome counts as they adapt to their culture environment (Mitsuhashi 2002), as is also true for our cell line. Standard in vivo haploid T. castaneum cells contain 10 chromosomes (20 for diploid) (Beeman and Stuart 1990; Stuart and Mocelin 1994). The TcA line contains 20 to 32 chromosomes/cell, indicating an increase in chromosome count which took place during the adaptation process. This is similar to other beetle cell lines, such as the longicorn beetle cell line which has 19 to 36 chromosomes per cell (standard diploid value = 20; Hoshino et al. 2008), and the Xylotrechus pyrrhoderus cell line with 18 to 42 chromosomes/cell (Iwabuchi 1999). Eighty percent of the cells from two D. undecimpunctata beetle cell lines had 18–21 chromosomes/cell (Lynn and Stoppleworth 1984).
The TcA cell line generated DNA band patterns distinctly different from other coleopteran cell lines, but similar to conspecific pupae. This confirms the specific identity of the TcA cell line as originating from T. castaneum. The TcA DNA profiles were stable over numerous passages, similar to other insect cell lines (McIntosh et al. 1996). Additional characterization with three genes, TcCHS1, Tcv, and TcrpS3, indicate all three genes are present in the established TcA line, all with 100 % identity to the published sequences. TcA is certainly derived from T. castaneum and free of Mycoplasma and Wolbachia contamination. This detailed characterization firmly validates TcA as a reliable tool for research into the biology of T. castaneum and possibly other representatives of the Coleoptera.
The TcA cell line responded to heat shock with increases in a heat shock protein and an hsp transcript. There was no difference in viability between cells incubated at 28°C and 40°C after 1 h. This observation documents an important point for this insect cell line: it is competent for stress-related experiments in insect biology. Heat shock at 40°C has been used in previous studies with intact T. castaneum (Mahroof et al. 2005a). We determined protein and gene expression levels at 28°C and 40°C over three time points (20, 40, 60 min). The two HSP70 antibodies we used recognized a 43-kDa protein that significantly increased in expression over the control after 40 min. This protein is the same size as the 43-kDa heat shock protein reported in the nematode Caenorhabditis elegans (Ding and Candido 2000), but possibly differs from an earlier study in T. castaneum which has shown increases in a ∼70-kDa protein (Mahroof et al. 2005b). We recorded significant increases in the expression of hsp68a in TcA cells at 20 and 40 min relative to the control. This work documents that the TcA line is a robust research platform for gene and protein expression studies.
References
Arakane Y.; Hogenkamp D. G.; Zhu Y. C.; Kramer K. J.; Specht C. A.; Beeman R. W.; Kanost M. R.; Muthukrishnan S. Characterization of two chitin synthase genes of the red flour beetle, Tribolium castaneum, and alternate exon usage in one of the genes during development. Insect Biochem. Molec. Biol. 34: 291–304; 2004.
Asgari S.; Johnson K. N. Insect virology. Caister Academic Press, Norfolk; 2010.
Beeman R. W.; Stuart J. J. A gene for lindane + cyclodiene resistance in the red flour beetle (Coleoptera: Tenebrionidae). J. Econ. Entomol. 83(5): 1745–1751; 1990.
Begum K.; Li B.; Beeman R. W.; Park Y. Functions of ion transport peptide and ion transport peptide-like in the red flour beetle Tribolium castaneum. Insect Biochem. Molec. Biol. 39: 717–725; 2009.
Charpentier C.; Tian L.; Cossette J.; Lery X. Characterization of cell lines developed from the Colorado potato beetle, Leptinotarsa decemlineata Say (Coleoptera: Chrysomelidae). In Vitro Cell Dev Biol — Animal 38: 73–78; 2002.
Crawford A. M. A coleopteran cell line derived from Heteronychus arator (Coleoptera: Scarabaeidae). In Vitro 18(10): 813–816; 1982.
Denell R. Establishment of Tribolium as a genetic model system and its early contributions to evo-devo. Genetics 180: 1779–1786; 2008.
Ding L.; Candido E. P. M. HSP43, a small heat-shock protein localized to specific cells of the vulva and spermatheca in the nematode Caenorhabditis elegans. Biochem. J. 349: 409–412; 2000.
Haliscak J. P.; Beeman R. W. Status of malathion resistance in 5 genera of beetles infesting farm-stored corn, wheat, and oats in the United States. J. Econ. Entomol. 76(4): 717–722; 1983.
Hashimoto Y.; Zhang S.; Blissard G. W. Ao3, a new cell line from eggs of the black witch moth, Ascalapha odorata (Lepidoptera: Noctuidae), is permissive for AcMNPV infection and produces high levels of recombinant proteins. BMC Biotechnol 10(50): 1–16; 2010.
Hoshino K.; Hirose M.; Iwabuchi K. A new insect cell line from the longicorn beetle Plagionotus christophi (Coleoptera: Cerambycidae). In Vitro Cell Dev Biol — Animal 45: 19–22; 2008.
Iwabuchi K. An established cell line from the beetle, Xylotrechus pyrrhoderus (Coleoptera: Cerambycidae). In Vitro Cell Dev Biol – Animal 35: 612–615; 1999.
Kim H. S.; Murphy T.; Xia J.; Caragea D.; Park Y.; Beeman R.; Lorenzen M. D.; Butcher S.; Manak J. R.; Brown S. J. BeetleBase in 2010: revisions to provide comprehensive genomic information for Tribolium castaneum. Nucleic Acids Res. 38: D437–D442; 2010.
Knorr E.; Schmidtberg H.; Vilcinskas A.; Altincicek B. MMPs regulate both development and immunity in the Tribolium model insect. PLoS One 4(3, e4751): 1–14; 2009.
Li B.; Predel R.; Neupert S.; Hauser F.; Tanaka Y.; Cazzamali G.; Williamson M.; Arakane Y.; Verleyen P.; Schoofs L.; Schachtner J.; Grimmelikhuijzen C. J. P.; Park Y. Genomics, transcriptomics, and peptidomics of neuropeptides and protein hormones in the red flour beetle Tribolium castaneum. Genome. Res. 18: 113–122; 2008.
Livak K. J.; Schmittgen T. D. Analysis of relative gene expression data using real-time quantitative PCR and 2-∆∆CT method. Methods 25: 402–408; 2001.
Long S. H.; McIntosh A. H.; Grasela J. J.; Goodman C. L. The establishment of a Colorado potato beetle (Coleoptera: Chrysomelidae) pupal cell line. App. Entomol. Zool. 37: 447–450; 2002.
Lynn D. E. Novel techniques to establish new insect cell lines. In Vitro Cell Dev Biol – Animal 37: 319–321; 2001.
Lynn D. E. Available lepidopteran insect cell lines. In: Murhammer D. W. (ed) Methods in molecular biology series. Baculovirus and insect cell expression protocols. Springer, New York, pp 117–144; 2007.
Lynn D. E.; Stoppleworth A. Established cell lines from the beetle, Diabrotica undecimpunctata (Coleoptera: Chrysomelidae). In Vitro 20(5): 365–368; 1984.
Mahroof R.; Zhu K. Y.; Neven L.; Subramanyam B.; Bai J. Expression patterns of three heat shock protein 70 genes among developmental stages of the red four beetle, Tribolium castaneum (Coleoptera: Tenebrionidae). Comp. Biochem. Physiol. Part A 141: 247–256; 2005a.
Mahroof R.; Zhu K. Y.; Subramanyam B. Changes in expression of heat shock proteins in Tribolium castaneum (Coleoptera: Tenebrionidae) in relation to developmental stage, exposure time, and temperature. Ann. Entomol. Soc. Amer. 98: 100–107; 2005b.
McIntosh A.; Grasela J. J.; Matteria R. L. Identification of insect cell lines by DNA amplification fingerprinting (DAF). Insect Molec. Biol. 5(3): 187–195; 1996.
Mitsuhashi J. Invertebrate tissue culture. Springer, Tokyo; 2002.
Mitsuhashi J. A. Continuous cell line from pupal ovaries of the common cutworm, Spodoptera litura (Lepidoptera: Noctuidae). Appl. Entomol. Zool. 30(1): 75–82; 1995.
Park Y.; Aikins J.; Wang L. J.; Beeman R. W.; Oppert B.; Lord J. C.; Brown S. J.; Lorenzen M. D.; Richards S.; Weinstock G. M.; Gibbs R. A. Analysis of transcriptome data in the red flour beetle, Tribolium castaneum. Insect Biochem Molec Biol 38: 380–386; 2008.
Peel A. D. Forward genetics in Tribolium castaneum: opening new avenues of research in arthropod biology. J Biology 8(106): 1–4; 2009.
Schneider I. Cell lines derived from late embryonic stages of Drosophila melanogaster. Embryo. ex. Morph. 27(2): 353–36; 1972.
Schröder R.; Beermann A.; Wittkopp N.; Lutz R. From development to biodiversity—Tribolium castaneum, an insect model organism for short germband development. Dev. Genes Evol. 218: 119–126; 2008.
Smagghe G.; Goodman C. L.; Stanley D. Insect cell culture and applications to research and pest management. In Vitro Cell Dev Biol - Animal 45: 93–105; 2009.
Stiles B.; McDonald I. C.; Gerst J. W.; Adams T. S.; Newman S. M. Initiation and characterization of five embryonic cell lines from the cotton boll weevil Anthonomus grandis in a commercial serum-free medium. In Vitro Cell Dev Biol — Animal 28: 355–363; 1992.
Stuart J. J.; Mocelin G. Cytogenetics of chromosome rearrangements in Tribolium castaneum. Genome 38: 673–680; 1994.
Tribolium Genome Sequencing Consortium. The genome of the model beetle andn pest Tribolium castaneum. Nature 452: 949–955; 2008.
Zhou Z.; Evans J. D.; Lu Z.; Zhao P.; Williams M.; Sumathipala N.; Hetru C.; Hultmark D.; Jiang H. Comparative genomic analysis of the Tribolium immune system. Genome Biol. 8(R177): 1–16; 2007.
Acknowledgments
Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. All programs and services of the U.S. Department of Agriculture are offered on a nondiscriminatory basis without regard to race, color, national origin, religion, sex, age, marital status, or handicap.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: T. Okamoto
Rights and permissions
About this article
Cite this article
Goodman, C.L., Stanley, D., Ringbauer, J.A. et al. A cell line derived from the red flour beetle Tribolium castaneum (Coleoptera: Tenebrionidae). In Vitro Cell.Dev.Biol.-Animal 48, 426–433 (2012). https://doi.org/10.1007/s11626-012-9524-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-012-9524-x