
Abstract
The root barks of Sophora japonica L., a plant widely used in traditional Chinese medicine, were extracted with 70 % Me2CO. The antibacterial activity of the crude extracts and fractions from the subsequent purification was evaluated against two Gram-positive bacteria (Bacillus subtilis and Staphylococcus aureus) and two Gram-negative bacteria (Klebsiella pneumonia and Escherichia coli). Further purification of the EtOAc fraction, which exhibited the strongest bacterial inhibitory activity among the resultant fractions, led to the isolation of three new acylated flavonol glycosides, quercetin 3-O-(4″-(E)-caffeoyl)-α-rhamnopyranoside (1), quercetin 3-O-(4″-(Z)-caffeoyl)-α-rhamnopyranoside (2) and kaempferol 3-O-(4″-galloyl)-α-rhamnopyranoside (3), as well as a known flavonol glycoside, kaempferol 3-O-α-arabinofuranoside (4). Structures of the isolated compounds were elucidated by spectroscopic techniques such as 1D and 2D NMR, and other chemical methods. Acylated flavonol glycosides 1–3 are new natural compounds, and their structures were elucidated here for the first time. Antibacterial study indicated that compounds 1–3 showed bacteria inhibitory effects, especially against S. aureus. Compound 3 was the most potent one, with MIC values of 25, 0.78, 6.25 and 50 μg/mL against B. subtilis, S. aureus, K. pneumonia and E. coli, respectively, while compound 4 did not exhibit antibacterial capability.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Plants synthesize a diverse array of secondary metabolites, many of which have antimicrobial activities (Cowan 1999; Lima et al. 2015). In the face of the persistently rising threat of antimicrobial resistance—a major worldwide health problem (Burcu et al. 2014), there has recently been an increasing interest in the isolation of antimicrobial compounds from plants because of their structural diversity, unique bioactivity and environmental compatibility, which make them more favorable than synthetic chemicals (Harvey 1999; Zhou et al. 2007; Rashed et al. 2014).
Sophora japonica L. (Leguminosae), a deciduous tree, is cultivated throughout China, Korea and Japan (Zhang et al. 2013). In traditional Chinese medicine, seeds, fruits and buds of S. japonica have extensively been used as hemostatic agents (Wang et al. 2003). Flowers of this species are used in folk remedies to prevent paralysis on patients who have high blood pressure. S. japonica leaves have long been employed to treat various disorders, such as hemorrhoids. Stem barks of this tree have been used as anti-inflammatory and analgesic remedies (Park et al. 2002; Wang et al. 2003; Zhang et al. 2013). Recent pharmacological and clinical practices revealed that S. japonica materials possessed antifertility and anticancer activities (Panthati et al. 2012).
Previous phytochemical studies of S. japonica were mainly focused on its flowers, seeds, buds, leaves and stem barks, which resulted in the isolation of several classes of chemical constituents, including flavonoids, triterpenes, sterols, alkaloids, fatty acids, phospholipids and amino acids (Komatsu et al. 1976; Shirataki et al. 1987; Grishkovets and Gorbacheva 1995; Mukhamedova and Glushenkova 1997). However, to date, investigation on chemical composition and bioactivity of S. japonica root barks has never been carried out. With the aim to search for new antibacterial secondary metabolites, an intensive phytochemical study was initiated on the root barks of S. japonica through bioassay-guided fractionation against four Gram bacteria, which resulted in the isolation of three new acylated flavonol glycosides, quercetin 3-O-(4″-(E)-caffeoyl)-α-rhamnopyranoside (1), quercetin 3-O-(4″-(Z)-caffeoyl)-α-rhamnopyranoside (2) and kaempferol 3-O-(4″-galloyl)-α-rhamnopyranoside (3), as well as a known flavonol glycoside, kaempferol 3-O-α-arabinofuranoside (4). Herein, the isolation and structure elucidation of the three new secondary metabolites and the antibacterial activity of compositions of S. japonica root barks are described.
Materials and methods
Plant materials
Root barks of S. japonica (5 years old) were obtained in October 2013 from the campus forest in Tianjin University of Science and Technology, China. Plant specimens were identified by Dr. D. Wang from Institute of Chemical Industry of Forest Products, Chinese Academy of Forestry, China. Voucher specimens (No. 131002) are on file in the herbarium of Tianjin Key Laboratory of Pulp and Paper, College of Materials Science and Chemical Engineering, Tianjin University of Science and Technology, China. The collected plant materials were air-dried at room temperature, ground into powder, and kept in a dark and cold place until use.
General experimental
Melting points (mp) were determined with an Electro Thermal 9100 apparatus and were uncorrected. UV spectra were recorded with a Jenway 6405 spectrophotometer. IR spectra were obtained by KBr disk method on a Perkin-Elmer BX FT-IR spectrometer. The optical rotations were measured using a JASCO DIP-1000 digital polarimeter in MeOH. NMR spectra were recorded in MeOH-d 4 with tetramethylsilane as an internal standard using a Bruker Avance DPX 400 spectrometer, with operating frequency of 400 and 100 MHz for 1H and 13C NMR, respectively. Positive fast atom bombardment mass (FAB MS) data were done with a Micromass Autospec M363 spectrometer.
Open column chromatography (OCC) was conducted with Sephadex LH-20 (Sigma) and silica gel (Merck) as packing materials. Eluents were collected with SBS-160 fraction collectors. In TLC experiments, DC-Plastikfolien Cellulose F (Merck) plates were used and t-BuOH-HOAc-H2O (3:1:1, v/v/v, solvent A) and HOAc-H2O (3:47, v/v, solvent B) were employed as developing solvents. Thin-layer chromatography (TLC) visualization and detection were carried out by exposure to UV light at 254 and 365 nm wavelengths, and then spraying with 1 % FeCl3 in EtOH solution followed by heating.
Extraction and fractionation
Root bark samples of S. japonica (4.08 kg) were extracted four times in a jar (30 L) with Me2CO-H2O (7:3, v/v) at room temperature for more than 3 days. As shown in Fig. 1, the combined extracts were filtered and concentrated with a rotary evaporator in vacuo to remove the solvent (Si et al. 2014). The resultant residue mixture was freeze-dried (221.40 g, yield 5.43 % of o.d. bark), then suspended in H2O and successively fractionated by liquid–liquid extraction in fractionators with a series of liquids with polarity gradients, including n-hexane, CHCl3, EtOAc and n-BuOH, followed by freeze-drying to give fractions soluble in n-hexane (7.61 g, yield 0.19 % of o.d. bark), CHCl3 (8.82 g, yield 0.22 % of o.d. bark), EtOAc (29.95 g, yield 0.73 % of o.d. bark), n-BuOH (23.19 g, yield 0.57 % of o.d. bark) and H2O (151.81 g, yield 3.72 % of o.d. bark).

Isolation and purification procedures
Purification and separation
As demonstrated in Fig. 1, 20.82 g of the above-obtained EtOAc fraction powder was loaded to open column chromatography (OCC) (Ø 5 cm × 60 cm) packed with silica gel and a gradient of EtOAc–MeOH-H2O (38:1:1 → 28:1:1 → 18:1:1 → 8:1:1, v/v, 4200 mL) used as the solvents system to yield six fractions labeled as SJRE1 (1.12 g), SJRE2 (0.85 g), SJRE3 (6.11 g), SJRE4 (1.38 g), SJRE5 (9.26 g) and SJRE6 (1.02 g), which were guided and grouped by TLC experiments. Then, fraction SJRE3 was further chromatographed over an open column (Ø 3.0 cm × 50 cm) packed with silica gel and eluted with Me2CO-CHCl3 (23:1 → 9:1 → 4:1, v/v, 2100 mL) to forward four fractions SJRE31–SJRE34. Fraction SJRE33 (3.86 g) was also applied to OCC with Sephadex LH-20 as packing materials (Ø 2 cm × 50 cm) eluted with MeOH-H2O twice (3:1 and 2:3, v/v, 800 mL each) to afford three fractions SJRE331, SJRE332 and SJRE333. SJRE332 (1.89 g) was chromatographed over an open column (Ø 1.5 cm × 40 cm) with Sephadex LH-20 as packing materials and EtOH-n-hexane (2:1 and 1:2, v/v, 500 mL each) serving as mobile solvents to get a yellowish amorphous powder (compound 1, 45.3 mg). Fraction SJRE5 was subjected to SiO2 OCC eluted with EtOAc–MeOH-H2O (95:4:1 → 46:3:1) to give five fractions SJRE51–SJRE55. Fraction SJRE52 (6.02 g) was further loaded over a Sephadex LH-20 open column (Ø 3.0 cm × 50 cm) and washed with EtOH-n-hexane (3:1, v/v, 4300 mL) to present four fractions. The third fraction (SJRE523, 3.29 g) was also chromatographed over an open column (Ø 2.0 cm × 50 cm) packed with silica gel and eluted with 2800 mL of EtOAc-petroleum ether (1:2, v/v) to obtain three fractions SJRE5231, SJRE5232 and SJRE5233. SJRE5232 (2.35 g) was applied to Sephadex LH-20 OCC with MeOH-H2O (3:1 and 1:2, v/v, 1100 mL each) as eluting solvents to get compound 2 (41.8 mg) and four other fractions. SJRE52324 (1.31 g) was loaded to Sephadex LH-20 OCC (Ø 1.5 cm × 40 cm) using EtOH-n-hexane (2:3, v/v, 650 mL) as washing solvents to yield 35.4 mg of compound 3 as white amorphous powder. Fraction SJRE54 (2.60 g) was further chromatographed over open columns (Ø 1.5 cm × 40 cm) successively packed with SiO2 and Sephadex LH-20, with EtOAc-petroleum ether (2:3, v/v, 3200 mL) and MeOH-H2O (7:3 and 2:5, v/v, 1200 mL each) used as washing solvents, respectively, to give 50.2 mg of compound 4.
Compound 1 Yellowish amorphous powder; mp 172–174 °C; [α] 20 D −123.8° (MeOH, c 0.5); IR (KBr) ν max cm−1 3396 (OH), 1658 (conjugated ketone C=O), 1605 (aromatic C=C); UV λmax (MeOH) nm: 262, 330; R f 0.22 (solvent A) and 0.51 (solvent B); FAB MS (positive mode) m/z: [M + H]+ and [M + Na]+ at m/z 611 and 633, respectively, corresponding to molecular mass 610 and calculated for C30H26O14; 1H (400 MHz, δ, MeOH-d 4) and 13C (100 MHz, δ, MeOH-d 4) NMR data are shown in Table 1.
Compound 2 Yellowish amorphous powder; mp 165–167 °C; [α] 20 D −97.6° (MeOH, c 0.5); IR (KBr) ν max cm−1 3390 (OH), 1660 (conjugated ketone C=O), 1600 (aromatic C=C); UV λmax (MeOH) nm: 268, 336; R f 0.54 (solvent A) and 0.18 (solvent B); FAB MS (positive mode) m/z: [M + H]+ and [M + Na]+ at m/z 611 and 633, respectively, corresponding to molecular mass 610 and calculated for C30H26O14; 1H (400 MHz, δ, MeOH-d 4) and 13C (100 MHz, δ, MeOH-d 4) NMR data are shown in Table 1.
Compound 3 White amorphous powder; mp 191–193 °C; [α] 20 D −108.1° (MeOH, c 0.5); IR (KBr) ν max cm−1 3402 (OH), 1672 (conjugated ketone C=O), 1610 (aromatic C=C); UV λmax (MeOH) nm: 260, 325; R f 0.73 (solvent A) and 0.49 (solvent B); FAB MS (positive mode) m/z: [M + H]+ and [M + Na]+ at m/z 585 and 607, respectively, corresponding to molecular mass 584 and calculated for C28H24O14; 1H (400 MHz, δ, MeOH-d 4) and 13C (100 MHz, δ, MeOH-d 4) NMR data are shown in Table 1.
Antibacterial activity
In this work, the in vitro antibacterial activities of the 70 % Me2CO crude extracts, fractions and pure compounds from root barks of S. japonica were evaluated against two Gram-positive bacteria (Bacillus subtilis ATCC 9372 and Staphylococcus aureus ATCC25923) and two Gram-negative bacteria (Klebsiella pneumonia ATCC 700603 and Escherichia coli ATCC 11775), which were obtained from the College of Food Science and Biotechnology, Tianjin University of Science and Technology, P.R. China. The in vitro antibacterial activity evaluation of the 70 % Me2CO crude extracts and fractions were carried out by the disk diffusion method using Mueller–Hinton agar for bacteria (Baron and Finegold 1990; Shafaghat et al. 2014). Disks containing 30 μL of the samples (100 μg/mL) were used, and bacterial growth inhibitory zones were determined after 24 h of incubation at 37 °C. The minimum inhibitory concentrations (MICs) of the isolated compounds were conducted with procedures as described by Rabe and van Staden (2000) with slight modification. 96-well microtiter plates were used for the microdilution method (Eloff 1998). The test samples were diluted with H2O in plate wells. Then, an equal volume of a 1:100, diluted overnight, culture of organisms was put to the microtiter wells to give the final volume of 0.2 mL. The plate was covered and cultured at 37 °C overnight. For the indication of bacterial growth, 50 μL p-iodonitrotetrazolium violet (Sigma) solution at 200 μg/mL was added to each well and incubated for further 30 min at 37 °C. Then, the inhibition of bacterial growth was visible as a clear well and the presence of growth was implied by the purple color. Meanwhile, the commercial antibiotic neomycin (Sigma) was used as positive control. The results were calculated by taking the mean of all triplicated values.
Results and discussion
S. japonica has long been used in traditional Chinese medicine. However, phytochemical investigation on root barks of the species has never been carried out. In this study, the 70 % Me2CO crude extracts of S. japonica root barks and following partitions, including n-hexane, CHCl3, EtOAc, n-BuOH and H2O fractions, were evaluated for their antibacterial activities against Gram-positive and Gram-negative bacteria. Results of antibacterial potency, as listed in Table 2, demonstrated that the EtOAc fraction was the most active among the five fractions.
Successive silica gel and Sephadex LH-20 chromatography of the EtOAc fraction, as demonstrated in Fig. 1, led to the isolation of four chemical constituents, including three hitherto new acylated flavonol glycosides (1–3) and a known flavonol glycoside (4), as shown in Fig. 2. On the basis of a careful comparison of their spectroscopic data with those published in the literature, compound 4 was identified as kaempferol 3-O-α-arabinofuranoside (Kim et al. 1994; Jung et al. 2002). To the best of the authors` knowledge, compound 4 has not previously been found in genus Sophora, although it is a known component of other plant species (Geibel et al. 1990; Kim et al. 1994), while 1–3 are three new natural compounds and structurally elucidated here for the first time.

Structures of the isolated compounds 1–4
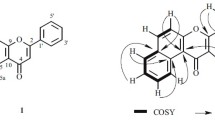
Compound 1, with mp 172–174 °C and optical rotation [α] 20 D −123.8° (MeOH, c 0.5), was isolated as a yellowish amorphous powder. The IR spectrum of 1 suggested the presence of the aromatic double bond (1605 cm−1), conjugated carbonyl (1658 cm−1) and hydroxyl group (3396 cm−1) (Akhavan et al. 2015). Its UV spectrum exhibited absorption maxima at 262 and 330 nm. Phenolic hydroxyl group in 1 was observed through TLC chromogenic reaction by the gray-green color when sprayed with ethanolic FeCl3 (R f values 0.22 and 0.51 in solvents A and B, respectively) (Imakura et al. 1985; Si et al. 2009a, 2011). The positive FAB MS spectrum of 1 presented [M + H]+ ion peak at m/z 611 and [M + Na]+ at m/z 633, respectively, corresponding to the molecular mass 610 and formula C30H26O14. In compound 1, the 1H NMR spectrum gave a pair of doublets of meta coupling (J = 2.1 Hz) at δ H 6.20 (1H) and δ H 6.37 (1H) attributable to H-6 and H-8 (Si et al. 2009b). A set of ABX type proton signals arising at δ H 7.62 (1H, d, J = 1.8 Hz, H-2′), δ H 6.88 (1H, d, J = 7.9 Hz, H-5′) and δ H 7.55 (1H, dd, J = 7.9 and 1.8 Hz, H-6′) were assigned to the protons of a 1,3,4-trisubstituted benzene ring (Moharram et al. 2006). In 13C NMR spectrum, the typical flavonol skeleton of 1 was confirmed by signals resonating at δ C 159.2 (C-2), 134.8 (C-3) and 179.5 (C-4) (Semmar et al. 2002). Thus, the aglycone was identified as quercetin. In 1H NMR spectrum of 1, the rhamnosyl sugar of α-configuration was characterized for its anomeric proton (H-1″) resonating at δ H 5.31 as a broad singlet, and its secondary methyl protons distinctively appeared at δ H 0.97 (3H, J = 6.3 Hz, H-6″) as a doublet (Si et al. 2013). In further 1H–1H correlation spectroscopy (COSY) experiments, other proton peaks, including δ H 4.14 (1H, br s, H-2″), 3.51 (1H, m, H-3″), 5.04 (1H, dd, J = 9.2 and 9.5 Hz, H-4″) and δ H 4.45 (1H, m, H-5″), were also assigned by their inner cross correlations.
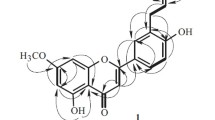
In 1H → 13C couplings of heteronuclear multiple bond correlation (HMBC) of 1, long-range correlations were observed between anomeric proton δ H 5.31 (br s, H-1″) and C-3 (δ C 134.8) of the quercetin, as shown in Fig. 3, deducing that the rhamnosyl moiety was attached to C-3 of the aglycone. The proton signals at δ H 7.48 (H-α) and 6.20 (H-β) with coupling constant of J = 16.1 Hz as doublets indicated the presence of E-configuration olefinic protons of AB type on the caffeoyl group (Zhao et al. 2008; Yahagi et al. 2012). An ortho-coupled doublet at δ H 6.71 (1H, d, J = 8.2 Hz, H-5‴), double doublets at δ H 6.90 (1H, dd, J = 8.2 Hz and J = 2.3 Hz, H-6‴) and a meta-coupled doublet at δ H 6.99 (1H, d, J = 2.3 Hz, H-2‴) suggested the typical ABX proton system. Moreover, interactions between δ H 7.48 (H-α) and δ C 168.4 (C-γ), δ H 7.48 (H-α) and δ C 127.2 (C-1‴), δ H 6.20 (H-β) and δ C 168.4 (C-γ), δ 6.20H (H-β) and δ C 127.2 (C-1‴), δ H 6.99 (H-2‴) and δ C 146.6 (C-α), δ H 6.90 (H-6‴) and δ C 146.6 (C-α) on HMBC spectrum of 1 also confirmed the presence of a (E)-caffeoyl moiety. The connection of (E)-caffeoyl moiety to rhamnosyl C-4″ was evidenced by significant downfield shift of H-4″ to δ H 5.04 (approximately Δ + 1.92, from δ H 3.12) and C-4″ to δ C 75.3 (approximately Δ + 4.1, from δ C 71.2) relative to that of quercetin 3-O-α-rhamnopyranoside (Nicoluer and Thompson 1983; Zhang et al. 2014). The appearance of HMBC correlations between δ H 5.04 (H-4″) and δ C 168.4 (C-γ) additionally implied that the (E)-caffeoyl moiety was located at C-4″ (δ C 75.3) of the rhamnosyl moiety. As shown in Table 1, the distortionless enhancement by polarization transfer (DEPT) spectrum of 1 classified 30 carbons into 1 methyl, 15 methine and 14 quaternary carbon signals. Therefore, compound 1 was established as quercetin 3-O-(4″-(E)-caffeoyl)-α-rhamnopyranoside.

Key HMBC correlations of compounds 1–3
Compound 2 was obtained as a yellowish amorphous powder with optical rotation [α] 20 D -97.6° (MeOH, c 0.5) and mp 165–167 °C. Its R f values were 0.54 and 0.18 in solvents A and B, respectively. The molecular formula was the same as 1 by its positive FAB MS spectrometric analysis. The 1H, 13C and 2D NMR spectra of 2 showed signals closely resembling those observed for 1 (see Table 1). The only significant difference was the coupling constants of olefin protons at δ H 6.77 (H-α) and δ H 5.78 (H-β) decreased to J = 12.8 Hz, indicative of the presence of the (Z)-caffeoyl moiety (Zhao et al. 2008; Yahagi et al. 2012). The linkage location of the quercetin aglycone, rhamnosyl and (Z)-caffeoyl moieties was supported by its key HMBC correlations, as demonstrated in Fig. 3. Thus, the structure of 2 was elucidated to be the Z-type isomer of 1. Thus, compound 2 was determined as quercetin 3-O-(4″-(Z)-caffeoyl)-α-rhamnopyranoside.
Compound 3 was isolated as a white amorphous powder having a negative optical rotation ([α] 20 D −108.1° (MeOH, c 0.5)) and mp 191–193 °C. Its molecular formula was determined to be C28H24O14 by FAB MS (positive mode) m/z [M + H]+ and [M + Na]+ at m/z 585 and 607, respectively, corresponding to its molecular mass 584. The 1H and 13C NMR data of 3 were similar to those of 1 and 2, except for two differences: 1) The presence of one AA’BB’ style proton signal (δ H 8.01 and 6.90 as two doublets with coupling constant of J = 8.5 Hz assignable to H-2′, 6′ and H-3′, 5′) in 3 due to the para-substituted B-ring in the kaempferol aglycone (Kanchanapoom 2007); 2) Two singlet protons observed at δ H 7.09 ascribed to H-2‴ and 6‴, along with 13C NMR signals (δ C 121.9 (C-1‴), 110.1 (C-2‴, 6‴), 146.4 (C-3‴, 5‴) and 139.9 (C-4‴)) indicated that the galloyl instead of caffeoyl group was linked to C-4″ of the rhamnosyl moiety in 3 (Samy et al. 2014). The DEPT (Table 1), COSY and HMBC (Fig. 3) spectroscopic data corroborated the assumption above. Consequently, compound 3 was elucidated as kaempferol 3-O-(4″-galloyl)-α-rhamnopyranoside.
Isolated compounds 1–4 were assessed for their individual antibacterial activities against Gram-positive and Gram-negative bacteria B. subtilis, S. aureus, K. pneumonia and E. coli. The results, as summarized in Table 3, revealed that the three new acylated flavonol glycosides 1–3 exhibited antibacterial potency with their MICs ranging from 0.78 to 100 μg/mL, comparing with MICs 0.20–1.56 μg/mL for positive control neomycin. Compound 3 was found to be the most active with MIC values of 25, 0.78, 6.25 and 50 μg/mL against B. subtilis, S. aureus, K. pneumonia and E. coli, respectively. Among the four tested organisms, S. aureus was the most sensitive with MICs 0.78–3.13 μg/mL for compounds 1–3. However, compound 4 showed no antibacterial activities against the four tested Gram bacteria. Previous phytochemical investigation on Tagetes minuta led to the identification of several acylated flavonol glycosides, and these glycosides showed significant antibacterial activity against Gram-positive bacteria, especially S. aureus (Shahzadi and Shah 2015), which agreed well with the current investigations. Thus, the three new acylated flavonol glycosides (1–3), especially compound 3, might serve as hit compounds for the development of new antibacterial agent.
Conclusion
The objectives of this investigation were to isolate secondary metabolites from root barks of S. japonica, and to further determine the antibacterial activity through bioassay-guided fractionation against two Gram-positive bacteria and two Gram-negative bacteria. The 70 % Me2CO crude extracts of root barks of S. japonica were fractionated sequentially using solvents of increasing polarity. The EtOAc fraction, which exhibited most significant antibacterial activities among all the solvent fractions, was further purified by silica gel and Sephadex LH-20 OCC, yielding three new acylated flavonol glycosides, quercetin 3-O-(4″-(E)-caffeoyl)-α-rhamnopyranoside (1), quercetin 3-O-(4″-(Z)-caffeoyl)-α-rhamnopyranoside (2) and kaempferol 3-O-(4″-galloyl)-α-rhamnopyranoside (3), together with a known flavonol glycoside, kaempferol 3-O-α-arabinofuranoside (4). Acylated flavonol glycosides 1–3 are new natural compounds isolated and structurally elucidated in the present study for the first time, and compound 4 has not previously been reported in Sophora. Compounds 1–3 exhibited inhibitory activities against the tested bacterial species, especially against S. aureus, while compound 4 was not active on the tested bacteria. This study indicated that S. japonica root barks could be a source of antibacterial natural compounds.
References
Akhavan M, Jahangiri S, Shafaghat A (2015) Studies on the antioxidant and antimicrobial activity and flavonoid derivatives from the fruit of Trigonosciadium brachytaenium (Boiss.) Alava. Ind Crops Prod 63:114–118
Baron EJ, Finegold SM (1990) Methods for testing antimicrobial effectiveness. In: Stephanie M (ed) Diagnostic microbiology. Mosby, Baltimore, pp 171–194
Burcu B, Aysel U, Nurdan S (2014) Antimicrobial, antioxidant, antimutagenic activities, and phenolic compounds of Iris germanica. Ind Crops Prod 61:526–530
Cowan MM (1999) Plant products as antimicrobial agents. Clin Microbiol Rev 12:564–582
Eloff JN (1998) A sensitive and quick microplate method to determine the minimal inhibitory concentration of plant extracts for bacteria. Planta Med 64:711–713
Geibel M, Geiger H, Treutter D (1990) Tectochrysin 5- and genistein 5-glucosides from the bark of Prunus cerasus. Phytochemistry 29:1351–1353
Grishkovets V, Gorbacheva L (1995) Triterpene glycosides of Sophora japonica seeds. Chem Nat Compd 31:596–599
Harvey AL (1999) Medicines from nature: are natural products still relevant to drug discovery? Trends Pharmacol Sci 20:196–198
Imakura Y, Kobayashi S, Mima A (1985) Bitter phenyl propanoid glycosides from Campsis chinensis. Phytochemistry 24:139–146
Jung HA, Kim AR, Chung HY, Choi JS (2002) In vitro antioxidant activity of some selected Prunus species in Korea. Arch Pharm Res 25:865–872
Kanchanapoom T (2007) Aromatic diglycosides from Cladogynos orientalis. Phytochemistry 68:692–696
Kim HJ, Woo ER, Park H (1994) A novel lignan and flavonoids from Polygonum aviculare. J Nat Prod 57:58–586
Komatsu M, Yokoe I, Shirataki Y (1976) Studies on the constituents of Sophora specie. X. Constituents of the root of Sophora japonica L. Yakugaku Zasshi 96:254–257
Lima B, Sanchez M, Agüero MB, Tapia A, Palermo JA, Feresin GE (2015) Antibacterial activity of extracts and compounds isolated from the Andean medicinal plant Azorella cryptantha (Clos) Reiche, Apiaceae. Ind Crops Prod 64:152–157
Moharram FA, Marzouk MS, Ibrahim MT, Mabry TJ (2006) Antioxidant galloylated flavonol glycosides from Calliandra haematocephala. Nat Prod Res 20:927–934
Mukhamedova KS, Glushenkova A (1997) Phospholipids of ripe Sophora japonica seeds. Chem Nat Compd 33:445–448
Nicoluer G, Thompson AC (1983) Flavonoids of Desmanthus illioensis. J Nat Prod 46:112–114
Panthati MK, Rao KNV, Sandhya S, David B (2012) A review on phytochemical, ethnomedical and pharmacological studies on genus Sophora, Fabaceae. Rev Bras Farmacogn 22:1145–1154
Park YK, Lee HJ, Choi DH, Kwon YH, Oh JS (2002) Extractives from the bark of Sophora japonica L. Mokchae Konghak 30:42–47
Rabe T, van Staden J (2000) Isolation of an antibacterial sesquiterpenoid from Warburgia salutaris. J Ethnopharmacol 73:171–174
Rashed K, Ćirić A, Glamočlija J, Soković M (2014) Antibacterial and antifungal activities of methanol extract and phenolic compounds from Diospyros virginiana L. Ind Crops Prod 59:210–215
Samy MN, Sugimoto S, Matsunami K, Otsuka H, Kamel MS (2014) Bioactive compounds from the leaves of Eugenia uniflora. J Nat Prod 7:37–47
Semmar N, Fenet B, Gluchoff-Fiasson K, Comte G, Jay M (2002) New flavonol tetraglycosides from Astragalus caprinus. Chem Pharm Bull 50:981–984
Shafaghat A, Pirfarshi F, Shafaghatlonbar M (2014) Luteolin derivatives and antimicrobial activity of Achillea tenuifolia Lam. methanol extract. Ind Crops Prod 62:533–536
Shahzadi I, Shah MM (2015) Acylated flavonol glycosides from Tagetes minuta with antibacterial activity. Front Pharmacol 6:195
Shirataki Y, Tagaya Y, Yokoe I, Komatsu M (1987) Sophoraside A, a new aromatic glycoside from the roots of Sophora japonica. Chem Pharm Bull 35:1637–1640
Si CL, Kim JK, Bae YS, Li SM (2009a) Phenolic compounds in the leaves of Populus ussuriensis and their antioxidant activities. Planta Med 75:1165–1167
Si CL, Wu L, Zhu ZY, Kim JK, Kwon DJ, Bae YS (2009b) Apigenin derivatives from Paulownia tomentosa Steud. var. tomentosa stem barks. Holzforschung 63:440–442
Si CL, Xu J, Kim JK, Bae YS, Liu PT, Liu Z (2011) Antioxidant properties and structural analysis of phenolic glucosides from bark of Populus ussuriensis Kom. Wood Sci Technol 45:5–13
Si CL, Jiang JZ, Liu SC, Hu HY, Ren XD, Yu GJ, Xu GH (2013) A new lignan glycoside and phenolics from the branch wood of Pinus banksiana Lambert. Holzforschung 67:357–363
Si CL, Wu L, Shen T, Huang XF, Du ZG, Ren XD, Luo XG, Hu WC (2014) Recovery of low-molecular mass galloyltannins from agricultural residue of Juglans sigillata dode seed husks and their tyrosinase inhibitory effect. Bioresources 9:2226–2236
Wang JH, Lou FC, Wang YL, Tang YP (2003) A flavonol tetraglycoside from Sophora japonica seeds. Phytochemistry 63:463–465
Yahagi T, Daikonya A, Kitanaka S (2012) Flavonol acylglycosides from flower of Albizia julibrissin and their inhibitory effects on lipid accumulation in 3T3-L1 cells. Chem Pharm Bull 60:129–136
Zhang LB, Lv JL, Chen HL (2013) Japonicasins A and B, two new isoprenylated flavanones from Sophora japonica. Fitoterapia 87:89–92
Zhang W, Li C, You LJ, Fu X, Chen YS, Luo YQ (2014) Structural identification of compounds from Toona sinensis leaves with antioxidant and anticancer activities. J Funct Foods 10:427–435
Zhao P, Tanaka T, Hirabayashi K, Zhang YJ, Yang CR, Kouno I (2008) Caffeoyl arbutin and related compounds from the buds of Vaccinium dunalianum. Phytochemistry 69:3087–3094
Zhou LG, Li D, Wang JG, Liu YS, Wu JY (2007) Antibacterial phenolic compounds from the spines of Gleditsia sinensis Lam. Nat Prod Res 21:283–291
Acknowledgments
This project was financially supported by the State Key Laboratory of Tree Genetics and Breeding (Northeast Forestry University) (K2013101), Natural Science Foundation of Tianjin City (13JCZDJC29400, 13JCZDJC33700), State Key Laboratory of Pulp & Paper Engineering (201503, 201359), Innovation Foundation for Young Teachers in Tianjin University of Science & Technology (2014CXLG14) and Foundation (201405) of Tianjin Key Laboratory of Marine Resources & Chemistry (Tianjin University of Science & Technology), P.R. China.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Si, CL., An, LL., Xie, DN. et al. New acylated flavonol glycosides with antibacterial activity from root barks of Sophora japonica . Wood Sci Technol 50, 645–659 (2016). https://doi.org/10.1007/s00226-016-0809-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00226-016-0809-1