

The regio- and stereoselectivity of [3+2] cycloaddition reactions of (Z)-1-(anthracen-9-yl)-N-methyl nitrone with analogs of trans-β-nitrostyrene were studied within the molecular electron density theory at the B3LYP/6-31G(d) and MPWB95/6-311G(d,p) theory levels. Analysis of the reactivity indices for presented reactions suggests that nitrone participates as nucleophile, while studied nitroalkenes play a role of electrophiles. According to electron localization function and conceptual density functional theory, kinetic and thermodynamic aspects of processes as well as analysis of all critical structures, the most favored reaction path is the formation of (3RS,4RS,5SR)-3-(anthracen-9-yl)-5-aryl-2-methyl-4-nitroisoxazolidine, independently of simulated solvent.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Isoxazolidines are a group of five-membered, saturated heterocyclic compounds containing both nitrogen and oxygen in the skeleton.1 This class of compounds, due to their biological activity, found use in many aspects of life.2 Isoxazolidines are widely used in medicine as antibacterial and antiviral drugs.3,4 An example to be mentioned is cycloserine which is a common structural motif in antibiotics5 (Fig. 1). Besides, isoxazolidines are used in industry as a part of scrubbers to remove sulfur-containing compounds6 or in polymer chemistry as a part of copolymers.7

Structure of cycloserine (4-amino-3-isoxazolidinone).
The most universal and common protocols for the synthesis of isoxazolidines and their derivatives, comprise [3+2] cycloaddition (32CA) reactions of nitrones with olefins,8–10 proceeding under mild conditions in absence of catalyst and giving high yields.11,12 This method also allows to obtain products with high or complete selectivity.13,14 Moreover, 32CA reactions are characterized by complete atom economy. The full atom economy of the above processes lies in accordance with green chemistry rules.15,16
In our research, the study on possibility of synthesis of sterically crowded N-methylisoxazolidines was carried out (Scheme 1). In order to achieve assumed purpose, (Z)-1-(anthracen-9-yl)-N-methyl nitrone (1) was applied as a three-atom component (TAC).17 Nitrone 1 has already been tested in 32CA reaction giving high yields of products.3 Simultaneously, the protocol of synthesis of nitrone 1 is not complicated and according to the literature, the molecule possesses biological activity.18 In turn, the series of trans-β-nitrostyrene analogs 2a–c were used as alkenes. It is well known, that conjugated nitroalkenes are commonly used in 32CA reactions.19,20 Moreover, as in vitro tests show, trans-β-nitrostyrene-containing analogs can be successfully used as antimicrobial agents.21

Scheme 1
For such defined substrates, four competitive 32CA reaction paths are possible. They are composed of two regioisomeric pathways, of which each one has two, likely, stereoisomeric pathways.
Complexity of the described process led us to use the computational methods based on molecular electron density theory (MEDT)22 in order to understand the reaction mechanism and, as a result, make an attempt of synthetic study in a future.
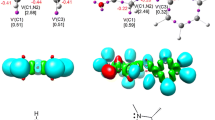
This theoretical research was divided into four parts. First, a topological analysis of electron localization function (ELF)23 and natural population analysis (NPA)24,25 for substrates 1 and 2a–c at the ground state were done. These analyses were presented in order to characterize the electronic structure of the substrates and to predict their reactivity in 32CA reaction based on electron density distribution in molecules.17 ELF localization domains, ELF basin attractor positions, together with the valence basin populations are shown in Figure 2. The analysis was performed using B3LYP/6-31G(d) theory level in gas phase.

B3LYP/6-31G(d) ELF localization domains of nitrone 1 and nitroalkenes 2a–c represented at an isosurface value of ELF 0.75 and ELF basin attractor positions, together with the most significant valence basin populations. ELF valence basin populations are given in average number of electrons (e). Protonated basins are shown in blue, monosynaptic basins in red, disynaptic basins in green. The ELF attractors are shown as purple spheres.
The ELF topological analysis of (Z)-1-(anthracen-9-yl)-N-methyl nitrone (1) shows the presence of two monosynaptic basins, V(O1) and V'(O1), integrating a total electron population of 5.94 e, located at the more nucleophilic O1 atom; one V(O1,N2) disynaptic basin, integrating 1.36 e, associated with an underpopulated O1–N2 single bond; and one V(N2,C3) disynaptic basin, integrating 4.04 e, associated with a N2=C3 double bond (Fig. 2). Due to the absence of pseudoradical center nor a carbenoid center as well as the absence of a double bond, nitrone 1 can be classified as zwitterionic TAC (zw-type).17
The ELF topology of trans-β-nitrostyrene (2b) presents one pair of disynaptic basins, V(C4,C5) and V'(C4,C5), integrating a total electron population of 3.52 e. A similar situation is observed both for trans-4-amino-β-nitrostyrene (2a) and trans-4-nitro-β-nitrostyrene (2c). The ELF topological analysis of nitrostyrene 2a shows one pair of disynaptic basins, V(C4,C5) and V'(C4,C5), integrating a total electron population of 3.56 e. In turn, the ELF topology of nitroalkene 2c shows one pair of disynaptic basins, V(C4,C5) and V'(C4,C5), integrating a total electron population of 3.54 e. The presence of one pair of V(C4,C5) and V'(C4,C5) disynaptic basins is associated with a C4=C5 double bond in nitroalkenes 2a–c structures (Fig. 2). The presence of V(C4,C5) and V'(C4,C5) basins of trans-β-nitrostyrene analogs 2a–c is associated with a depopulated C4=C5 double bond caused by neighbourhood of groups NO2 and Ph.
Based on ELF analysis, the proposed Lewis-like structures together with the natural atomic charges for nitrone 1 and nitroalkenes 2a–c are given in Figure 3.

ELF-based Lewis-like structures for nitrone 1 and nitroalkenes 2a–c proposed by B3LYP/6-31G(d), together with the natural atomic charges. Natural atomic charges are given in average number of electrons (e).
While the ELF topological analysis provides a bonding pattern concordant with the commonly accepted Lewis structure, the NPA analysis represents reagents electronic structure. NPA analysis of the natural atomic charges shows that in (Z)-1-(anthracen-9-yl)-N-methyl nitrone (1), the atom O1 is strongly negatively charged by –0.51 e, while the atom N is weakly positive charged by +0.09 e. In turn, C3 carbon presents a formally negligible charge of –0.04 e (Fig. 3). Therefore, NPA indicates that nitrone 1 does not possess a 1,2-zwitterionic charge distribution. Presented charge distribution is a consequence of the polarization of the O–N–C framework toward the more electronegative O1 oxygen.
NPA analysis of the natural atomic charges indicates that nitroalkenes 2a–c show a similar charge distribution. Both ethylene carbons are negatively charged. More electrophilic Cα carbon atoms possess more negative charge compared to Cβ carbon atoms. In trans-β-nitrostyrene (2b), the charge of Cα carbon is –0.16 e, while charge of Cβ carbon is –0.11 e. Introduction of electron-donating NH2 group at para position of trans-β-nitrostyrene 2a causes a slight increase of electronic charge distribution of trans-4-amino-β-nitrostyrene (2a) to –0.17 e (Cα) and –0.13 e (Cβ). In turn, in a case of electron-withdrawing NO2 group electronic charge distribution values of nitroalkene 2c are reduced to –0.12 e (Cα) and –0.09 e (Cβ).
An analysis of the electronic properties of substrates and their intermolecular interactions according to conceptual density functional theory (CDFT)26,27 reactivity indices was carried out during our theoretical research on 32CA reaction. The global reactivity indices, namely, electronic chemical potential μ,26 chemical hardness η, global electrophilicity ω, and global nucleophilicity N, for the reagents involved in these 32CA reactions were calculated at the B3LYP/6-31G(d) theory level in the gas phase (Table 1).26,27
The electronic chemical potential of nitrone 1 (μ –3.50 eV) is higher in comparison to nitroalkenes 2a–c: μ –3.96 eV (nitroalkene 2a), –4.79 eV (nitroalkene 2b) and –5.58 eV (nitroalkene 2c). It means that the flux of the electron density presumably takes place from (Z)-1-(anthracen-9-yl)- N-methyl nitrone (1) to trans-β-nitrostyrene analogs 2a–c. Thus, discussed 32CA reactions can be classified as the forward electron density flux (FEDF).28
Calculated electrophilicity29 index ω of (Z)-1-(anthracen-9-yl)-N-methyl nitrone (1) is 1.90 eV and the calculated nucleophilicity30 index N for this nitrone 1 is 4.00 eV (Table 1). These values allow to conclude that nitrone 1 can be classified as strong electrophile as well as strong nucleophile in a polar reaction within the electrophilicity and nucleophilicity scale.26,31
The calculated electrophilicity index ω of nitroalkene 2b is 2.66 eV and the calculated nucleophilicity N index for this compound is 2.17 eV (Table 1). Thus, trans-β-nitrostyrene (2b) can be classified as strong electrophile and moderate nucleophile in a polar reaction based on the electrophilicity and nucleophilicity scale.26
Introduction of electron-donating group NH2 at para position has a slight impact on the electrophilicity index ω of nitroalkene 2a with a decrease to 2.17 eV and significant increase of nucleophilicity index N to 3.34 eV (Table 1). In consequence, trans-4-amino-β-nitrostyrene (2a) can be considered a strong electrophile as well as a strong nucleophile.26
In turn, introduction of electron-withdrawing group NO2 significantly increases the electrophilicity index ω of nitroalkene 2c to 3.70 eV and slightly reduces its nucleophilicity index N to 1.44 eV (Table 1). So, trans-4-nitro-β-nitrostyrene (2c) can be classified as a superelectrophile and marginal nucleophile.26,31
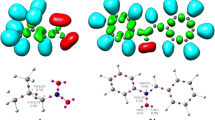
Overall, for all studied 32CA reactions, it can be assumed that (Z)-1-(anthracen-9-yl)-N-methyl nitrone (1) most evidently can participate as nucleophilic component, while nitroalkenes 2a–c remain electrophilic substrates. Thus, the regioselectivity of 32CA including the participation of nonsymmetric reagents can be defined through interaction between the most electrophilic center of the electrophile and the most nucleophilic center of the nucleophile.32 Therefore, in order to characterize the most nucleophilic and the most electrophilic centers of the species involved, the electrophilic Pk+ and nucleophilic Pk– Parr functions together with local electrophilicity ωk and local nucleophilicity Nk of substrates 1 and 2a–c were analyzed (Fig. 4).33

The local electronic properties are presented as 3D models of Mulliken atomic spin densities for nitroalkenes 2a–c˙ˉ radical anions and nitrone 1˙+ radical cation, together with the electrophilic Pk+ Parr functions of nitroalkenes 2a–c and the nucleophilic Pk– Parr functions of nitrone 1 and indices of the local electrophilicity ωk (eV) of nitroalkenes 2a–c, given in red, and the local nucleophilicity Nk (eV) of nitrone 1, given in blue.
Four different isomeric products can be formed in 32CA reaction between (Z)-1-(anthracen-9-yl)-N-methyl nitrone (1) and trans-β-nitrostyrene analogs 2a–c (Scheme 1).
Analysis of the nucleophilic Pk– Parr functions of (Z)-1-(anthracen-9-yl)-N-methyl nitrone (1) indicates that O atom of nitrone fragment includes the most nucleophilic center of this species, presenting the maximum value PO– 0.22, and the values of the local nucleophilicity index Nk is 0.88 eV (Fig. 4).
The electrophilic Pk+ Parr functions of trans-β-nitrostyrene analogs 2a–c indicates that Cα atom is the most electrophilic, presenting the maximum value PCα+ 0.29 (nitrostyrene 2a), 0.25 (nitrostyrene 2b), and 0.12 (nitrostyrene 2c) and the values of the local electrophilicity index ωk are 0.63 (nitrostyrene 2a), 0.67 (nitrostyrene 2b), and 0.44 eV (nitrostyrene 2c, Fig. 4).
So, based on CDFT theory, the most favorable reaction paths are determined by the nucleophilic attack of O atom of (Z)-1-(anthracen-9-yl)-N-methyl nitrone (1) on electrophilic Cα atom of trans-β-nitrostyrene analogs 2a–c. Therefore, the creation of compounds 3a–c and/or 4a–c as the most favored regioisomeric adducts is equally probable (Scheme 1).
Further, the kinetic and thermodynamic analysis of potential energy surface (PES) was presented for reaction between (Z)-1-(anthracen-9-yl)-N-methyl nitrone (1) with trans-β-nitrostyrene (2b) (Table 2). By analogy, the results for reaction of nitrone 1 with the rest of nitroalkenes 2a,c are given in Tables S1 and S2 (Supplementary information file). The computational study was performed using the MPWB95/6-311G(d,p) theory level. The solvent effect was included via polarizable continuum model (PCM) for PhMe and EtOH solutions.
The analysis of Eyring parameters shows that for the reaction between (Z)-1-(anthracen-9-yl)-N-methyl nitrone (1) and trans-β-nitrostyrene (2b) in PhMe (ε 2.4) the cycloaddition's enthalpies are negative: –2.76 (MC3b), –1.65 (MC4b), –4.84 (MC5b), –3.25 (MC6b). This is due to formation of the molecular complex (MC) at the first reaction stage (Table 2, Fig. 5). For all of the possible reaction paths MCs are created without the necessity of crossing an activation barrier. Therefore, MCs may not exist as stable complexes.

Enthalpy profile for 32CA reaction of (Z)-1-(anthracen-9-yl)-N-methyl nitrone (1) with trans-β-nitrostyrene (2b) in PhMe, according to MPWB95/6-311G(d,p) (PCM) calculations.
Further MC conversion along the reaction proceeds to the transition states (TSs), irrespective to the 32CA pathway. Results of intrinsic reaction coordinate (IRC) analysis indicate the appearance of TSs with the one imaginary eigenvalue in the Hessian. The IRC calculations connect transition state directly with the energy minimum of MC and product in any reaction pathways. It also confirms the absence of more energy minimum connected with the presence of other critical structures. Therefore, we conclude that the mechanism of reaction between (Z)-1-(anthracen-9-yl)-N-methyl nitrone (1) and trans-β-nitrostyrene (2b) should be considered as one-step. The TSs forming is associated with increase of the Gibbs free energy of the process to 14.35 kcal·mol–1 (Table 2, Fig. 5). Data in Table 2 suggests that the forming of (3RS,4RS,5SR)-3-(anthracen-9-yl)-2-methyl-4-nitro-5-phenylisoxazolidine (3b) is the most preferable product from the kinetic point of view. The activation barrier in this case is 32.46 kcal·mol–1 (Table 2). The obtained results correspond well with the the CDFT analysis results discussed above. In turn, the creation of (3RS,4SR,5RS)-3-(anthracen-9-yl)-2-methyl-4-nitro-5-phenylisoxazolidine (4b) as the second regioisomer should be considered as unfavorable from the kinetic point of view with the highest activation barrier 39.26 kcal·mol–1 (Table 2, Fig. 5). On the other hand, the activation barriers of both regioisomers 5b and 6b are comparable (34.92 and 33.57 kcal·mol–1, respectively) and the formation of these two products is of similar probability under the 32CA conditions in PhMe (Table 2).
In the case of more polar solvent EtOH (ε 24.8), solution is included as dielectric media to DFT calculations as the result the reaction profiles do not change qualitatively, but only quantitatively to a small extent comparing to the results calculated for PhMe solution. In particular, the increase of ΔH and ΔS values for all MCs are observed as well as activation barriers are significantly higher.34 Nevertheless, the paths preference of 1 + 2b reaction based on kinetic aspects for EtOH is identical as in a case of PhMe solution (Table 2).
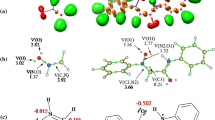
Finally, the last calculation part includes the diagnostic analysis for all critical structures of the reaction. Similarly to kinetic and thermodynamic studies, the MPWB95/6-311G(d,p) (PCM) theory level was applied for calculation. The results include key parameters35 such as interatomic distance between reaction centers for significant structures (r), development of new single bond (l), asymmetry index Δl and the global electron density transfer (GEDT) values and are given in average number of electrons in PhMe (Table 3) and in EtOH (Table 4). Due to similar results, the data for 32CA reactions of methyl nitrone 1 with nitroalkenes 2a,c are given in Tables S3 and S4 (Supplementary information file). The visualisation of key critical structures is shown only for the most favorable 1 + 2b reaction path leading to oxazolidine 3b in PhMe solution (Fig. 6).

Critical structures for 32CA reaction path of 1 + 2b → 3b in PhMe solution according to MPWB95/6-311G(d,p) (PCM) calculations.
As noted above, the first stage of 32CA reaction 1 + 2b in PhMe is the formation of a MC, independently from reaction path (Table 3, Fig. 6). The analysis of the structural aspects shows that within MC distances between reaction centres C3–C4 and C5–O1 remained outside the typical r range for bonds in transition state. It can be concluded that in all MCs no new single bonds start forming. GEDT parameters for each MC are equal to 0.0 e, which demonstrates that none of the MC forms electron density transfer complex (Table 3).36
The next reaction step is the formation of TS (Table 3, Fig.6). Distances C3–C4 and C5–O1 in the following TS are significantly shortened compared with the corresponding distances within the MC. The nature of TS depends on the relative orientations of complex subunits. In particular, for the TS3b the extent of C3–C4 and C5–O1 forming new bonds is identical with calculated distances equal to 0.79 and 0.72 Å, respectively (Table 3). In turn, for the same regioisomer but competitive stereoisomer 4b the predicted TS4b is more asynchronous. Thus, the distance C3–C4 is l 0.78 Å, while C5–O1 predicted bond length is 0.61 Å (Δl 0.17 Å). The two other possible TS5b and TS6b are also asynchronous with interatomic distance asymmetry index Δl 0.15 and 0.16 Å, respectively.
So, during the most kinetically favored reaction pathway, the formation of synchronic transition state is also more favorable. Noteworthy, that all TSs calculated for PhMe exhibit low polar nature in the range of GEDT 0.11–0.19 e (low polar GEDT parameter 0.05 < GEDT < 0.20, Table 3).30 However, the most preferred reaction pathway 1 + 2b → 3b in EtOH is characterized by GEDT 0.26 e and by the most synchronous TS3b (Table 4).37
The value changes of key parameters of the transformation in EtOH is only quantitative. Preferences regarding the length of interatomic distances, single bond development indices as well as synchronicity and polarity character of reactions are similar to those calculated for PhMe (Table 4). Still, in all of TSs in EtOH an increase of asynchronicity degree as well as the increase of the GEDT values can be observed, which is due to the more polar nature of the solvent.
To sum up, the presented quantum-chemical studies of [3+2] cycloaddition reactions between (Z)-1-(anthracen-9-yl)-N-methyl nitrone and trans-β-nitrostyrene analogs clearly indicate to a one-step asynchronous mechanism. The polar character of processes depends on the type of simulated solvent. The presented ELF and CDFT calculations show that the most favorable reaction path is determined by the nucleophilic attack of atom O of nitrone on the electrophilic Cα atom of nitroalkenes. Accordingly, the formation of regioisomer of 3-(anthracen-9-yl)-5-aryl-2-methyl-4-nitroisoxazolidine is the most probable. In turn, the analysis of kinetic and thermodynamic parameters as well as the diagnosis of critical structures for studied [3+2] cycloaddition reactions confirms the previous regioisomeric preference, and demonstrates the formation of (3RS,4RS,5SR)-3-(anthracen-9-yl)-5-aryl-2-methyl-4-nitroisoxazolidine as the most probable product among all possible.
Computational details
The calculations associated with the [3+2] cycloaddition reactions were performed using the Gaussian 09 package38 at the Prometheus computer cluster of the CYFRONET regional computational center in Cracow. DFT calculations were performed using the MPWB95/6-311g(d,p)39 theory level. The similar computational level has already been successfully used for the exploration of mechanistic aspects of different cycloaddition processes.40–43 Calculations of critical structures were performed at 298K temperature and 1 atm pressure. The localized stationary points were characterized using vibrational analysis. For optimized TS, intrinsic reaction coordinate (IRC)44 calculations have been performed to verify whether the located TSs are connected to the corresponding minimum stationary points associated with reactants and products.
The solvent effects were simulated using a standard selfconsistent reaction field (SCRF)45,46 based on the polarizable continuum model (PCM).47
The global electron density transfer (GEDT)37 values were estimated by the natural population analysis (NPA)24,25 using the equation GEDT(f) = charge qf, where q are the atoms of a framework (f) of the TSs.
Indices of single bond development (l) were calculated according to the formula:48
where rX–YTS is the distance between the reaction centers X and Y in the transition structure and rX–YP is the same distance in the corresponding product.
Global electronic properties of the reactants were estimated according to the equations recommended in references.49 The CDFT indices were calculated at the B3LYP/6-31G(d) computational level in gas phase also used to define the electrophilicity and nucleophilicity scales.26,31,50 Electrophilic Pk+ and nucleophilic Pk– Parr functions were obtained from the changes of atomic spin density (ASD) of the reagents.33,50
Topological analysis of the electron localization function (ELF)23 was performed with the TopMod51 program, using monodeterminantal wave functions over a grid spacing of 0.1 au. Calculation was performed on B3LYP/6-31G(d) level theory in gas phase.
GaussView program52 was used to visualize molecular geometries and 3D representations of the radical anion and the radical cations and the position of the ELF basin attractors. The ELF localization domains at an isovalue of 0.75 au were obtained with the Paraview software.53,54
Supplementary information file, containing the kinetic and thermodynamic data as well as the key parameters of the critical structures for the 32CA reactions 1 + 2a and 1 + 2c, and cartesian coordinates of critical key structures is available at the journal website http://springerlink.bibliotecabuap.elogim.com/journal/10593.
References
Fryźlewicz, A.; Łapczuk-Krygier, A.; Kula, K.; Demchuk, O. M.; Dresler, E.; Jasiński, R. Chem. Heterocycl. Compd. 2020, 56, 120.
Lakhvich, F. A.; Koroleva, E. V.; Akhrem, A. A. Chem. Heterocycl. Compd. 1989, 25, 359.
Rescifina, A.; Chiacchio, M. A.; Corsaro, A.; De Clercq, E.; Ianuazzo, D.; Mastino, A.; Piperno, A.; Romeo, G.; Romeo, R.; Valveri, V. J. Med. Chem. 2006, 49, 709.
Mochulskaya, N. N.; Nosova, E. V.; Charushin, V. N. Chem. Heterocycl. Compd. 2021, 57, 374.
Mirzayev, F.; Viney, K.; Linh, N. N.; Gonzalez-Angulo, L.; Gegia, M.; Jaramillo, E.; Zignol, M.; Kasaeva, T. Eur. Respir. J. 2021, 57, 2003300.
Landeck, H.; Ranke, G. US Patent 5413627A.
Vretik, L.; Ritter, H. Macromolecules 2003, 36, 6340.
Sirotkina, E. V.; Efremova, M. M.; Starova, G. L.; Kuznetsov, M. A.; Molchanov, A. P. Chem. Heterocycl. Compd. 2020, 56, 1193.
Jasiński, R. Chem. Heterocycl. Compd. 2009, 45, 748.
Markitanov, Y. N.; Timoshenko, V. M. Chem. Heterocycl. Compd. 2021, 57, 1149.
Padwa, A.; Bur, S. Chem. Heterocycl. Compd. 2016, 52, 616.
Kula, K.; Dobosz, J.; Jasiński, R.; Kącka-Zych, A.; Łapczuk-Krygier, A.; Mirosław, B.; Demchuk, O. M. J. Mol. Struct. 2020, 1203, 127473.
Xiang, J.; Zhu, T.; Dang, Q.; Bai, X. Chem. Heterocycl. Compd. 2016, 52, 601.
Kula, K.; Kącka-Zych, A.; Łapczuk-Krygier, A.; Wzorek, Z.; Nowak, A.; Jasiński, R. Molecules 2021, 26, 1364.
Martina, K.; Tagliapietra, S.; Veselov, V. V.; Cravotto, G. Front. Chem. 2019, 7, 95.
Żmigrodzka, M.; Sadowski, M.; Kras, J.; Dresler, E.; Demchuk, O. M.; Kula, K. Sci. Rad. 2022, 1, 24.
Ríos-Gutiérrez, M.; Domingo, L. R. Eur. J. Org. Chem. 2019, 2, 267.
Kula, K.; Dresler, E.; Demchuk, O. M.; Jasiński, R. Przem. Chem. 2015, 94, 1385.
Łapczuk-Krygier, A.; Kącka-Zych, A.; Kula, K. Curr. Chem. Lett. 2019, 8, 13.
Shvekhgeimer, G. A.; Zvolinskii, V. I.; Kobrakov, K. I. Chem. Heterocycl. Compd. 1986, 22, 353.
Boguszewska-Czubara, A.; Kula, K.; Wnorowski, A.; Biernasiuk, A.; Popiołek, Ł.; Miodowski, D.; Demchuk, O. M.; Jasiński, R. Saudi Pharm. J. 2019, 27, 593.
Domingo, L. R. Molecules 2016, 21, 1319.
Becke, A. D.; Edgecombe, K. E. J. Chem. Phys. 1990, 92, 5397.
Reed, A. E.; Weinstock, R. B.; Weinhold, F. J. Chem. Phys. 1985, 83, 735.
Reed, A. E.; Curtiss, L. A.; Weinhold, F. Chem. Rev. 1988, 88, 899.
Domingo, L. R.; Ríos-Gutiérrez, M.; Pérez, P. Molecules 2016, 21, 748.
Parr, R. G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989.
Domingo, L. R.; Kula, K.; Ríos-Gutiérrez, M.; Jasiński, R. J. Org. Chem. 2021, 86, 12644.
Parr, R. G.; Szentpaly, L. V.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922.
Domingo, L. R.; Chamorro, E.; Pérez, P. J. Org. Chem. 2008, 73, 4615.
Domingo, L. R.; Ríos-Gutiérrez, M. In Conceptual Density Functional Theory: Towards a New Chemical Reactivity Theory; Liu, S., Ed.; WILEY-VCH GmbH: Weinheim, 2022, vol. 2, p. 481.
Aurell, M. J.; Domingo, L. R.; Pérez, P.; Contreras, R. Tetrahedron 2004, 60, 11503.
Domingo, L. R.; Pérez, P.; Sáez, J. A. RSC Adv. 2013, 3, 1486.
Benchouk, W.; Mekelleche, S. M.; Silvi, B.; Aurell, M. J.; Domingo, L. R. J. Phys. Org. Chem. 2011, 24, 611.
Jasiński, R. Chem. Heterocycl. Compd. 2022, 58, 260.
Domingo, L. R.; Ríos-Gutiérrez, M. Org. Biomol. Chem. 2019, 17, 6478.
Domingo, L. R. RSC Adv. 2014, 4, 32415.
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, 2013.
Zhao, Y.; Truhlar, G. D. J. Phys. Chem. A 2004, 108, 6908.
Zawadzińska, K.; Ríos-Gutiérrez, M.; Kula, K.; Woliński, P.; Mirosław, B.; Krawczyk, T.; Jasiński, R. Molecules 2021, 26, 6774.
Kula, K.; Kącka-Zych, A.; Łapczuk-Krygier, A.; Jasiński, R. Pure Appl. Chem. 2021, 93, 427.
Kula, K.; Łapczuk-Krygier, A. Curr. Chem. Lett. 2018, 7, 27.
Demchuk, O. M.; Jasinski, R.; Strzelecka, D.; Dziuba, K.; Kula, K.; Chrzanowski, J.; Krasowska, D. Pure Appl. Chem. 2018, 90, 49.
Fukui, K. J. Phys. Chem. 1970, 74, 4161.
Tapia, O. J. Math. Chem. 1992, 10, 131.
Tomasi, J.; Perisco, M. Chem. Rev. 1994, 94, 2027.
Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Chem. Phys. Lett. 1996, 225, 327.
Mlostoń, G.; Jasiński, R.; Kula, K.; Heimgartner, H. Eur. J. Org. Chem. 2020, 2, 176.
Kula, K.; Zawadzińska, K. Curr. Chem. Lett. 2021, 10, 9.
Mlostoń, G.; Kula, K.; Jasiński, R. Molecules 2021, 26, 5562.
Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Comput. Chem. 1999, 23, 597.
Dennington, R.; Keith, T. A.; Millam, J. M. GaussView, Version 6.; Semichem, Inc.: Shawnee Mission, 2016.
Ahrens, J.; Geveci, B.; Law, C. In ParaView: An End-User Tool for Larga Data Visualization. The Visualization Handbook; Elsevier: Amsterdam, 2005.
Ayachit, U. The ParaView Guide: A Parallel Visualization Application; Kitware: New York, 2015.
This work was partially supported by PLGrid Infrastructure.
All calculations reported in this paper were performed on Prometheus supercomputer cluster in the CYFRONET computational center in Cracow.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Geterotsiklicheskikh Soedinenii, 2023, 59(3), 138–144
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kula, K., Sadowski, M. Regio- and stereoselectivity of [3+2] cycloaddition reactions between (Z)-1-(anthracen-9-yl)-N-methyl nitrone and analogs of trans-β-nitrostyrene on the basis of MEDT computational study. Chem Heterocycl Comp 59, 138–144 (2023). https://doi.org/10.1007/s10593-023-03175-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-023-03175-1