

(E)-3,3,3-Trichloro-1-nitroprop-1-ene undergoes [3+2] cycloaddition reaction with N-aryl(pyridin-3-yl) nitrones under mild conditions. The reaction is fully regio- and stereoselective and gives expected nitro-substituted nicotine analogs with 3,4-cis-4,5-trans-stereoconfiguration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Nicotine, a major alkaloid found in the tobacco plant Nicotiana tabacum,1 which is also present in such vegetables as potatoes, tomatoes, and eggplants,2 exhibits a wide pattern of biological and pharmacological activities that makes its application attractive in medicine.3 It is used as a neuronal nicotine acetylcholine receptor ligand in treating Alzheimer's and other central nervous system diseases.4,5 This compound is also used, usually in form of its salts, as a component of well-known insecticides.6 Nicotine is an easily biodegradable compound, and, in consequence, constitutes an eco-friendly component of plant protection products.7
Nowadays, simultaneously with the isolation of nicotine from natural matrices, large attention is devoted to obtaining of nicotine analogs in the synthetic way. In fact, synthesis of racemic nicotine had already been optimized on industrial scale,8,9,10 but industrial stereoselective synthesis still remains challenging since expensive chiral auxiliaries and sophisticated reaction conditions should be applied in order to achieve a reasonable level of selectivity.11,12,13 In addition, the separation of isomers by fractional crystallization does not seem to be especially practical.14,15
A few attempts to develop modified nicotine analogs have also been made.16,17 It should be noted that the presence of a nitro group in organic molecule generally stimulates additive functions of bioactivity.18,19 Moreover, nitro group forms an exceedingly attractive starting point for further functionalization into many interesting organic groups and moieties.20,21 Unfortunately, nitro-functionalized analogs of nicotine have not been prepared or characterized yet. Next, it is generally known that the presence of trihalomethyl group in organic molecules stimulates effectively their bioactivity.22
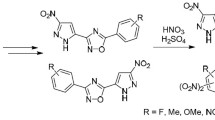
This work is a summary of our preliminary research on the fully selective synthesis of nitro-functionalized nicotine analogs (Fig. 1). Although the considered reactions may theoretically proceed giving four regio- and stereoisomeric cycloadducts, a single isomeric product has been obtained exclusively. For this purpose, [3+2] cycloaddition processes involving (E)-3,3,3-trichloro-1-nitroprop-1-ene (1) and N-aryl(pyridin-3-yl) nitrones 2a–e were applied. It should be underlined that the proposed strategy is very attractive due to: (a) full atomic economy toward [3+2] cycloaddition,23 (b) retention of the primary configuration of dipolarophile in the course of [3+2] cycloaddition,24 (c) high regio- and stereoselectivity in the cycloaddition involving conjugated nitroalkenes,25,26,27,28,29 (d) application of nitroprop-1-ene 1 gives the possibility to introduce two functional groups (NO2 and CCl3) into 1,2-oxazolidine cycle in one step.

Nicotine and its 1,2-oxazolidine analogs.
First we explored the reaction involving nitrone 2a (Scheme 1). It was found that this reaction proceeds easily in PhH solution under mild conditions. The HPLC analysis of the reaction mixture shows the presence of a single reaction product 3a which was isolated by column chromatography. Its constitution was elucidated by the means of spectral techniques. First, we analyzed HRMS data. It was found that the isolated compound gave a pseudo-molecular ion with m/z 388.0015 [M+H]+ which corresponds to the proposed molecular formula C15H13Cl3N3O3. Next, absorption bands typical to the nitro group,27 pyridine ring,30 1,4-disubstituted benzene ring,17 and 1,2-oxazolidine ring25 were identified in the IR spectra. Further information on the structure of the isolated compound 3a was obtained from the NMR spectra. In particular, in the 1H NMR spectrum three signals corresponding to the protons of the heterocyclic ring were identified along with those from the aromatic/ heteroaromatic ring protons. The signal from the 3-CH proton (a doublet) is in the highest field, while those corresponding to the 4-CH and 5-CH protons are in a relatively lower field. The values of the coupling constants prove, in turn, that protons 3-CH and 4-CH are located on the same side of the virtual plane of the heterocyclic ring (J3-CH,4-CH = 8.2 Hz) while protons 4-CH and 5-CH are located on the opposite sides (J4-CH,5-CH = 4.7 Hz). Therefore the configuration of 3-[(3RS,4SR,5SR)-4-nitro- 2-phenyl-5-(trichloromethyl)-1,2-oxazolidin-3-yl]pyridine (3a) may be assigned. For the full characterization of compound 3a, its 13C NMR spectrum was also recorded. Similarly, we examined analogous reactions involving nitrones 2b–e. In all cases, respective 3-[(3RS,4SR,5SR)- 2-(4-aryl)-4-nitro-5-(trichloromethyl)-1,2-oxazolidin-3-yl]- pyridines 3b–e were identified in the reaction mixtures as the only cycloaddition products.

Theoretically possible and established routes of regio- and stereoselective [3+2] cycloaddition reaction between (E)-3,3,3-trichloro-1-nitroprop-1-ene (1) and N-aryl(pirydin-3-yl) nitrones 2a–e
It should be emphasized, that other disubstituted nitrones (such as C,N-diphenyl nitrone,31N-(4-methylphenyl)( 3,4,5-trimethoxyphenyl) nitrone,32 and N-methyl- (3,4,5-trimethoxyphenyl) nitrone33) react with (E)-3,3,3- trichloro-1-nitroprop-1-ene (1) under similar conditions. These reactions, however, are not stereoselective, and lead to the mixtures of respective 3,4-cis- and 3,4-trans-4-nitrosubstituted adducts.
Thus, [3+2] cycloaddition reactions between (E)-3,3,3- trichloro-1-nitroprop-1-ene and N-aryl(pyridin-3-yl) nitrones proceed with full regio- and stereoselectivity and give the expected nitro-substituted nicotine analogs with (3RS,4SR,5SR)-stereoconfiguration.
Experimental
IR spectra were registered on a FT-IR NICOLET 6700 apparatus in KBr pellets. 1H and 13C NMR spectra were recorded on a Bruker AMX-500 spectrometer (500 and 125 MHz, respectively) in CDCl3. TMS was used as internal standard. Mass spectra were obtained on a Sciex Q-TRAP 4000 series hybrid quadrupole mass spectrometer equipped with an electrospray ion source. The sprayed liquid was fed with a Harvard Apparatus syringe pump at 10 μl/min. Fragmentation of molecular ions was done at the 10 eV collision energy. Reaction completion and compound purity were monitored using a Knauer liquid chromatograph (UV detector, LiChrospher 100-10 RP18 4 × 240 mm column, eluent 70% aqueous MeOH, flow rate of the eluent 1.5 ml/min, detection at λ 254 nm).
(E)-3,3,3-Trichloro-1-nitroprop-1-ene (1) was prepared by deacylation of an acetic ester of the respective nitroalcohol according to the known procedure.34 Nitrones 2a–e were prepared by condensation reaction between pyridine-3-carbaldehyde and appropriate arylhydroxylamine according to the known procedure.35
Synthesis of 3-[(3RS,4SR,5SR)-2-aryl-4-nitro-5-(trichloromethyl)- 1,2-oxazolidin-3-yl]pyridines 3a–e (General method). A solution of (E)-3,3,3-trichloro-1-nitroprop- 1-ene (1) (0.02 mol) and appropriate nitrone 2a–e (0.01 mol) in dry PhH (25 ml) was mixed at room temperature for 24 h. The reaction mixture was filtered, and the solvent was evaporated under reduced pressure. Products were isolated at room temperature on the Knauer HPLC apparatus, using a semi-preparative column (LiChrospher 100-10 RP 18 mm, 16 × 240 mm) and 70% aqueous MeOH as the eluent, flow rate 15 ml/min, detection at λ 254 nm.
3-[(3RS,4SR,5SR)-4-Nitro-2-phenyl-5-(trichloromethyl)- 1,2-oxazolidin-3-yl]pyridine (3a). Yield 3.5 g (91%), lightyellow oil. IR spectrum, ν, cm–1: 3062, 2931, 1562, 1454, 1367, 1224, 935, 788, 761, 692. 1H NMR spectrum, δ, ppm (J, Hz): 8.76–8.74 (2H, m, H Ar); 8.56–8.55 (1H, m, H Ar); 7.95–7.93 (1H, m, H Ar); 7.38–7.32 (4H, m, H Ar); 7.19–7.18 (1H, m, H Ar); 5.87 (1H, d, J = 4.7, 5-CH); 5.77 (1H, dd, J = 8.2, 2J = 4.7, 4-CH); 5.61 (1H, d, J = 8.2, 3-CH). 13C NMR spectrum, δ, ppm: 151.0; 149.9; 146.0, 136.0; 129.0; 128.0; 126.5; 123.7; 121.1; 94.0; 88.3; 70.8. Found, m/z: 388.0015 [M+H]+. C15H13Cl3N3O3. Calculated, m/z: 388.0017.
3-[(3RS,4SR,5SR)-2-(4-Methylphenyl)-4-nitro-5-(trichloromethyl)- 1,2-oxazolidin-3-yl]pyridine (3b). Yield 3.7 g (92%), light-yellow oil. IR spectrum, ν, cm–1: 3016, 2933, 1559, 1428, 1370, 1233, 932, 792, 742, 667. 1H NMR spectrum, δ, ppm (J, Hz): 8.61–8.60 (2H, m, H Ar); 7.78–7.76 (1H, m, H Ar); 7.30 (1H, d, J = 4.9, H Ar); 7.14 (2H, d, J = 8.5, H Ar); 7.09 (2H, d, J = 8.5, H Ar); 5.75 (1H, d, J = 4.7, 5-CH); 5.70 (1H, dd, J = 8.2, 2J = 4.7, 4-CH); 4.98 (1H, d, J = 8.2, 3-CH); 2.28 (3H, s, CH3). 13C NMR spectrum, δ, ppm: 151.2; 150.0; 149.9; 135.5; 129.8; 128.5; 123.8; 121.9; 117.4; 94.3; 88.4; 72.2; 21.0. Found, m/z: 402.0174 [M+H]+. C16H15Cl3N3O3. Calculated, m/z: 402.0175.
3-[(3RS,4SR,5SR)-2-(4-Fluorphenyl)-4-nitro-5-(trichloromethyl)- 1,2-oxazolidin-3-yl]pyridine (3c). Yield 3.6 g (89%), light-yellow oil. IR spectrum, ν, cm–1: 2957, 2927, 1561, 1504, 1366, 1219, 964, 836, 797. 1H NMR spectrum, δ, ppm (J, Hz): 8.63–8.62 (2H, m, H Ar); 7.81 (1H, d, J = 8.0, H Ar); 7.39 (2H, d, J = 8.9, H Ar); 7.33– 7.31 (1H, m, H Ar); 7.04–7.01 (2H, m, H Ar); 5.76 (1H, d, J = 4.6, 5-CH); 5.71 (1H, dd, J = 8.1, 2J = 4.6, 4-CH); 4.96 (1H, d, J = 8.1, 3-CH). 13C NMR spectrum, δ, ppm (J, Hz): 157.1 (d, JCF = 164.2); 151.0; 149.4; 135.9; 130.9; 128.8; 124.0; 124.0; 94.1; 88.3; 72.5. Found, m/z: 405.9923 [M+H]+. C15H12Cl3FN3O3. Calculated, m/z: 405.9930.
3-[(3RS,4SR,5SR)-2-(4-Chlorophenyl)-4-nitro-5-(trichloromethyl)- 1,2-oxazolidin-3-yl]pyridine (3d). Yield 3.8 g (90%), light-yellow oil. IR spectrum, ν, cm–1: 2928, 2857, 1560, 1488, 1365, 1263, 933, 796, 764. 1H NMR spectrum, δ, ppm (J, Hz): 8.64–8.62 (2H, m, H Ar); 7.78 (1H, dt, J = 8.0, 2J = 1.9, H Ar); 7.33 (1H, ddd, J = 8.0, 2J = 4.8, 3J = 0.5, H Ar); 7.25–7.22 (2H, m, H Ar); 7.11– 7.08 (2H, m, H Ar); 5.76 (1H, d, J = 4.7, 5-CH); 5.73 (1H, dd, J = 8.0, 2J = 4.7, 4-CH); 5.0 (1H, d, J = 8.0, 3-CH). 13C NMR spectrum, δ, ppm: 151.3; 149.6; 144.2; 135.7; 129.3; 128.6 127.1; 124.0; 122.3; 93.9; 88.3; 71.8. Found, m/z: 421.9627 [M+H]+. C15H12Cl4N3O3. Calculated, m/z: 421.9639.
3-[(3RS,4SR,5SR)-2-(4-Bromophenyl)-4-nitro-5-(trichloromethyl)- 1,2-oxazolidin-3-yl]pyridine (3e). Yield 4.1 g (88%), light-yellow oil. IR spectrum, ν, cm–1: 2924, 2849, 1560, 1448, 1365, 1224, 933, 796, 761, 648. 1H NMR spectrum, δ, ppm (J, Hz): 8.65–8.63 (2H, m, H Ar); 7.78–7.75 (1H, m, H Ar); 7.39 (2H, d, J = 8.9, H Ar); 7.33–7.30 (1H, m, H Ar); 7.04 (2H, d, J = 8.9, H Ar); 5.76 (1H, d, J = 4.7, 5-CH); 5.74 (1H, dd, J = 7.9, J = 4.7, 4-CH); 5.01 (1H, d, J = 7.9, 3-CH). 13C NMR spectrum, δ, ppm: 151.3; 150.4; 149.6; 135.7; 133.9; 132.2; 124.1; 124.0; 122.4; 93.9; 88.3; 71.6. Found, m/z: 465.9122 [M+H]+. C15H12BrCl3N3O3. Calculated, m/z: 465.9133.
References
Sun, B.; Tian, Y.-X.; Zhang, F.; Chen, Q.; Zhang, Y.; Luo, Y.; Wang, X.-R.; Lin, F.-C.; Yang, J.; Tang, H.-R. Biomolecules2018, 8, 114.
Siegmund, B.; Leitner, E.; Pfannhauser, W. J. Agric. Food Chem.1999, 47, 3113.
Powledge, T. M. PLoS Biol.2004, 2, e404.
Bontempi, B.; Whelan, K. T.; Risbrough, V. B.; Lloyd, G. K.; Menzaghi, F. Neuropsychopharmacology2003, 28, 1235.
Khurana, N.; Ishar, M. P. S.; Gajbhiye, A.; Goel, R. K. Eur. J. Pharmacol.2011, 662, 22.
Nicotinoid Insecticides and the Nicotinic Acetylcholine Receptor; Yamamoto, I.; Casida, J. E., Eds.; Springer: Tokyo, 1999.
Sarker, S.; Lim, U. T. PLoS One2018, 13, e0198302.
Divi, M. K. P.; Padakandla, G. R.; Rao, M. A. N.; Katta, H. B. US Patent US20120209006A1, 2011.
Mainkar, P. S.; Sunder, K. S.; Kumar, T. P.; Chandrasekhar, S. US Patent US20190016699A1, 2016.
Ericsson, T.; Bultropp, L. L. E.; Govindji, V. M. EP Patent EP1951074A4, 2006.
Welter, C.; Moreno, R. M.; Streiff, S.; Helmchen, G. Org. Biomol. Chem.2005, 3, 3266.
Seeman, J. I.; Chavdarian, C. G.; Secor, H. V. J. Org. Chem.1985, 50, 5419.
Brunner, H.; Kürzinger, A.; Mahboobi, S.; Wiegrebe, W. Arch. Pharm.1988, 321, 73.
Paine, J. B., III J. Org. Chem.2008, 73, 4939.
Willis, B.; Ahmed, M. M.; Freund, W.; Sawyer, D. US Patent US20160326134A1, 2015.
Andrade, M. M.; Barros, M. T.; Pinto, R. C. Tetrahedron2008, 64, 10521.
Singh, G.; Ishar, M. P. S.; Girdhar, N. K.; Singh, L. J. Heterocycl. Chem.2005, 42, 1047.
Kim, P.; Zhang, L.; Manjunatha, U. H.; Singh, R.; Patel, S.; Jiricek, J.; Keller, T. H.; Boshoff, H. I.; Barry, C. E., III; Dowd, C. S. J. Med. Chem.2009, 52, 1317.
Boguszewska-Czubara, A.; Lapczuk-Krygier, A.; Rykala, K.; Biernasiuk, A.; Wnorowski, A.; Popiolek, L.; Maziarka, A.; Hordyjewska, A.; Jasiński, R. J. Enzyme Inhib. Med. Chem.2016, 31, 900.
Perekalin, V. V.; Lipina, E. S.; Berestovitskaya, V. M.; Efremov, D. A. Nitroalkenes: Conjugated Nitro Compounds; Wiley: New York, 1994.
Barrett, A. G. M. Chem. Soc. Rev.1991, 20, 95.
Anisimova, N. A.; Slobodchikova, E. K.; Kuzhaeva, A. A.; Rybalova, T. V.; Stukan’, E. V.; Berestovitskaya, V. M. Russ. J. Gen. Chem.2014, 84, 834. [Zh. Obshch. Khim.2014, 84, 741.]
Methods and Applications of Cycloaddition Reactions in Organic Syntheses; Nishiwaki, N., Ed.; Wiley & Sons, Inc.: Hoboken, 2014.
Carruthers, W. Cycloaddition Reactions in Organic Synthesis; Pergamon Press, 2013.
Jasiński, R.; Mróz, K.; Kącka, A. J. Heterocycl. Chem.2016, 53, 1424.
Jasiński, R.; Jasińska, E.; Dresler, E. J. Mol. Model.2017, 23, article 13.
Łapczuk-Krygier, A.; Ponikiewski, Ł.; Jasiński, R. Crystallogr. Rep.2014, 59, 961.
Łapczuk-Krygier, A.; Jaśkowska, J.; Jasiński, R. Chem. Heterocycl. Compd.2018, 54, 1172. [Khim. Geterotsikl. Soedin.2018, 54, 1172.]
Kutyashev, I. B.; Barkov, A. Yu; Zimnitskiy, N. S.; Korotaev, V. Yu.; Sosnovskikh, V. Ya. Chem. Heterocycl. Compd.2019, 55, 861. [Khim. Geterotsikl. Soedin.2019, 55, 861.]
Silverstein, R. M.; Webster, F. X.; Kiemle, D. Spectrometric Identification of Organic Compounds; John Wiley & Sons, Inc., 2005.
Jasiński, R.; Barański, A. Pol. J. Chem.2006, 80, 1493.
Szczepanek, A.; Jasińska, E.; Kącka, A.; Jasiński, R. Curr. Chem. Lett.2015, 4, 33.
Jasiński, R.; Ziółkowska, M.; Demchuk, O. M.; Maziarka A. Cent. Eur. J. Chem.2014, 12, 586.
Brower, F.; Burkett, H. J. Am. Chem. Soc.1953, 75, 1082.
Kuzenkov, A. V.; Zakharychev, V. V.; Volkova, A. N. Russ. J. Org. Chem.2018, 54, 763. [Zh. Org. Khim. 2018, 54, 766.]
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fryźlewicz, A., Łapczuk-Krygier, A., Kula, K. et al. Regio- and stereoselective synthesis of nitrofunctionalized 1,2-oxazolidine analogs of nicotine. Chem Heterocycl Comp 56, 120–122 (2020). https://doi.org/10.1007/s10593-020-02631-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-020-02631-6