

The present minireview provides a survey of literature reports on the application of 3,3,3-trifluoropropene derivatives bearing sulfur-containing functional groups at positions 1 or 2 in the synthesis of functionalized five-membered heterocycles – pyrroles, pyrrolines, pyrrolidines, tetrahydrothiophenes, pyrazoles, pyrazolines, pyrazolidines, isoxazoles, isoxazolines, isoxazolidines, and triazoles via [3+2] cycloaddition reactions with 1,3-dipolarophiles.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
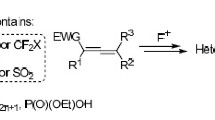
[3+2] Cycloaddition reactions are among the most versatile tools for the synthesis of five-membered heterocycles.1 Utilization of 3,3,3-trifluoropropene derivatives, bearing electron-withdrawing substituents – trifluoromethyl and sulfur-containing groups at the С=С bond, in combination with heteroatomic 1,3-dipoles in such cyclization reactions provides an efficient synthetic route to various types of five-membered heterocycles featuring both a CF3 group and a heteroatom moiety, as compounds with potential biological activity. The nature of substituents and their spatial arrangement in the alkene substrates of [3+2] cycloadditions directly affect the product structure, as well as the stereochemical outcome of the reactions. Furthermore, the ability of functionalities, such as a sulfonyl substituent, to act as a leaving group can produce a broader range of possible cycloaddition products, in contrast, for example, to the cyclizations of dipolarophiles containing an ester group. In such reactions, accompanied by the elimination of a sulfur-containing moiety, the starting substituted 3,3,3-trifluoropropene derivative can be considered as an equivalent of 3,3,3-trifluoropropyne.
In this review article, the syntheses of five-membered heterocycles via [3+2] cycloaddition reactions of 3,3,3-trifluoropropene derivatives bearing sulfur-containing substituents at positions 1 or 2 are classified according to the obtained types of heterocyclic products formed. In some cases, synthetic or biological applications of the obtained heterocycles are also discussed.
Cycloaddition to azomethine ylides. Synthesis of pyrrolidines
The cycloaddition reaction of N-benzyl azomethine ylide 4, generated in situ at room temperature from (methoxymethyl)(trimethylsilylmethyl)-N-benzylamine (3) in the presence of catalytic amounts of TFA in СН2Cl2, to 3,3,3-trifluoropropene derivatives bearing a sulfur-containing functional group at position 1 (compound 1)2 or 2 (compound 2)3 led to the formation of N-benzyl-4-(trifluoromethyl)- and N-benzyl-3-(trifluoromethyl)-pyrrolidines 5, 6 in 85–93 and 65–84% yields, respectively (Scheme 1). In the case of trans-alkenes 1, the cycloaddition leading to the formation of adducts 5 proceeded stereospecifically. Reductive debenzylation of compounds 5 using molecular hydrogen on palladium catalyst provided the respective NH-pyrrolidines.2

Scheme 1
When 1-phenylsulfonyl-3,3,3-trifluoropropene (1) was reacted with azomethine ylide – N-benzylideneglycinate 8, generated from imine 7 by the action of LDA in THF at –78°С, regioselective formation of 2,3,4,5-tetrasubstituted NH-pyrrolidine 9 was observed, giving rise to a mixture of stereoisomers in 58% yield (Scheme 2).4

Scheme 2
Chiral Cu(II) complexes and (S)-Tol-BINAP were used in asymmetric cycloaddition protocol involving reactions of various (β-polyfluoroalkyl/alkenyl)arylsulfones 1 in the presence of Et3N in THF with glycine and alanine iminoesters 10 as precursors of azomethine ylide 11. This proved to be a practical approach for the generation of a wide range of polysubstituted polyfluoroalkyl pyrrolidines 12 bearing four adjacent chiral centers in high yields (82–97%) and good enantio- and diastereoselectivity (Scheme 3).5

Scheme 3
Cycloaddition to thiocarbonyl ylide. Synthesis of tetrahydrothiophenes
Cycloaddition of thiocarbonyl ylide 14 generated in situ from chloromethyl trimethylsilyl methyl sulfide (13) in the presence of CsF in MeCN at reflux temperature, to (Е)-3,3,3-trifluoropropene derivatives 1 containing a sulfonyl, sulfamide, or sulfoximine substituent at position 1, proceeded in a stereospecific manner, with the formation of 4-(trifluoromethyl)tetrahydrothiophenes 15 in 65–83% yields (Scheme 4).6 Subsequent oxidation and oxidative imination reactions of thiolanes 15 allowed to synthesize functionalized cyclic 4-(trifluoromethyl)tetrahydrothiophenyl S-oxides, S,S-dioxides, as well as S-imino-S-oxides.6

Scheme 4
Cycloaddition to isocyanomethylides and nitrile ylides. Synthesis of pyrrolines and pyrroles
The reaction of 1-sulfonyl- and 1-sulfamoyl-(Е)-3,3,3-trifluoropropenes 1 with isocyanoacetic ester 16 in MeCN, with a catalytic amount of AgOAc added, proceeded regioselectively at room temperature, to give 3-(trifluoromethyl)-2,3-dihydro-1H-pyrroles (Δ2-pyrrolines) 17 in 48–75% yields (Scheme 5).7 In the case of a reaction between alkene 1 bearing an iminosulfonyl substituent (R1 = S(O)(NCO2Et)Me), the intermediate sulfoximine-substituted pyrroline 17 underwent ring aromatization with elimination of methane(N-carbethoxy)imidosulfinic acid, resulting in the formation of ethyl 2-pyrrolecarboxylate 18 (Scheme 5). When the reaction of alkenes 1 and isocyanide 16 was performed in the presence of a base (t-BuOK) in THF, the in situ elimination of sulfur-containing fragment from cycloadducts 17 resulted in one-pot formation of pyrrole 18 (Scheme 5).7 Pyrazoline derivative 17 containing a methylsulfonyl group was used in the synthesis of 4-methanesulfonyl-3-(trifluoromethyl)pyrrolidine-2-carboxylic acid that can be considered as a fluorineand sulfur-containing analog of proline.7

Scheme 5
The reactions of alkenes 1 with isocyanomethylide anion obtained from tosylmethyl isocyanide 19 in the presence of a base (t-BuOK, 2 equiv) in THF proceeded with the formation of 4-(trifluoromethyl)-1H-pyrroles 21 in 57–88% yields (Scheme 6).7 α-Tosylpyrroline intermediates 20 formed in this reaction underwent in situ elimination of toluenesulfonate, yielding pyrroles 21 (the Van Leusen reaction) (Scheme 6).

Scheme 6
Cycloaddition of 1-phenylsulfonyl-3,3,3-trifluoropropene (1) with nitrile ylide 23 generated from methyl 4-chloro-N-[(trimethylsilyl)methyl]benzimidothioilate (22) upon heating in hexamethylphosphoramide (HMPA) in the presence of H2O gave 2-(4-chlorophenyl)-4-(trifluoromethyl) pyrrole (25) in 28% yield as a desulfonylation product derived from the sulfonylated pyrroline intermediate 24 (Scheme 7).8

Scheme 7
Cycloaddition to diazo compounds, nitrile imines, and azomethine imines. Synthesis of pyrazolines, pyrazoles, and pyrazolidines
Cycloadditions of diazomethane (26) with 3,3,3-trifluoropropene derivatives 2 bearing sulfanyl, sulfinyl, or sulfonyl group at position 2 proceeded regioselectively in Et2O at room temperature, affording isolable 3-substituted 3-(trifluoromethyl)-4,5-dihydro-3H-pyrazole derivatives (Δ1-pyrazolines) 27, which differ in thermal stability depending on the nature of sulfur-containing substituent (Scheme 8).3 Pyrazoline thioether 27 (R2 = SPh) is so stable that it can be distilled. Sulfinylated pyrazoline 27 (R2 = S(О)Ph) underwent elimination of phenylsulfenate on thermolysis at 80°С, affording 3-(trifluoromethyl)pyrazole (28). Sulfonylpyrazoline 27 (R2 = SО2Ph) can be isolated as individual compound, but the thermolysis led to a complex mixture of products.3

Scheme 8
The reactions of diazomethane (26) with sulfur-containing 1-substituted 3,3,3-trifluoropropene derivatives 1 in t-BuOMe at room temperature led to the formation of regioisomeric cycloadducts – 4-(trifluoromethyl)- and 3-(trifluoromethyl)-4,5-dihydro-1Н-pyrazoles (Δ2-pyrazolines) 29 and 30 (Scheme 8).9 The sulfur-containing substituent in regioisomers 30 was eliminated under the reaction conditions, giving pyrazole 28 (Scheme 8).9
Cycloaddition of diazoacetic ester 31 (R3 = CO2Et) with alkenes 2 in Et2O or PhH at room temperature proceeded regioselectively, with the formation of 5-(trifluoromethyl)-4,5-dihydro-1H-pyrazole derivatives (Δ2-pyrazolines) 32, which underwent ring aromatization upon heating, forming pyrazole 33 (Scheme 9).3

Scheme 9
The reactions of alkenes 1 with diazoacetic ester 31 (R3 = CO2Et) in Et2O and with 2,2,2-trifluorodiazoethane 31 (R3 = CF3) that was generated in situ from 2,2,2-trifluoroethylamine hydrochloride by the action of NaNO2 in biphasic CH2Cl2–H2O system proceeded with the formation of isomeric 5(3)-substituted 4-trifluoromethyl-3,4(4,5)-dihydro-2(1)Н-pyrazoles 34 and 4-substituted 5-(trifluoromethyl)-4,5-dihydro-1H-pyrazoles 35. Yet, depending on the nature of the sulfur-containing substituent, the initial cycloadducts could undergo further transformations. Pyrazolines 34 and 35 bearing a sulfonyl substituent were stable, while pyrazolines 34 and 35 having a sulfamide or sulfoximine substituent underwent spontaneous ring aromatization in the reaction mixture, leading to the formation of pyrazole derivatives 36 and 33, respectively (Scheme 9).9
Cycloaddition of 1-sulfonyl- and 1-sulfamoyl-(Е)-3,3,3-trifluoropropenes 1 with N-phenylnitrile imine 38 generated in situ from phenylhydrazinylidene chlorides 37 in the presence of an excess of Et3N, was performed by heating in PhMe and resulted in the formation of regioisomeric cyclodducts – 5-(trifluoromethyl)- and 4-(trifluoromethyl)-4,5-dihydro-1Н-pyrazole derivatives (Δ1-pyrazolines) 39 and 40. The latter, under the reaction conditions, were converted to 1-phenyl-4-(trifluoromethyl)-1Н-pyrazoles 41 via the loss of the sulfur-containing moiety (Scheme 10).9 When nitrile imine 38 was reacted with alkene 1 bearing an iminosulfonyl substituent (R1 = S(O)(NCO2Et)Me), both intermediate regioisomeric pyrazolines 39 and 40 underwent elimination of methane(N-carbethoxy)imidosulfinic acid to give pyrazoles 42 and 41, respectively (Scheme 10).9

Scheme 10
Cycloaddition reactions of (Е)-α,β-unsaturated β-fluoroalkyl-2-pyridyl- and phenylsulfones 1 with a series of aromatic, heteroaromatic, and aliphatic azomethine imines 43 were accomplished under mild conditions (heating in MeCN) in the absence of catalysts, resulting in regioselective formation of trifluoromethylated N,N'-bicyclic pyrazolidinones 44 that were isolated as racemic mixtures in high yields, with a good to high degree of exo selectivity (Scheme 11).10

Scheme 11
Cycloaddition to nitrones and nitrile oxides. Synthesis of isoxazolidines, isoxazolines, and isoxazoles
Cycloaddition of (E)-1,1,1-trifluoro-3-phenyl- and 2-pyridylsulfonylpropene 1 to nitrones 45 proceeded regioselectively upon refluxing in PhMe11 or heating in MeCN10 and led to highly stereoselective formation of substituted 4-sulfonyl-5-(trifluoromethyl)isoxazolidines 46 that were isolated in 39–99% yields (Scheme 12). Analogous reactions involving 1-(methanesulfonyl)-1-(trifluoromethyl)allenes 47 as active 1,3-dipolarophiles occurred readily without catalyst in PhH at room temperature providing high yields (86–94%) of polysubstituted isoxazolidine derivatives 48 containing an exocyclic С=С bond as single isomers (Scheme 12).12 Isoxazolidines 46 have been used as precursors of various acyclic compounds for the preparation of the respective trifluoromethylated syn-3-amino alcohols through a sequence of reductive desulfonylation and catalytic hydrogenation reactions.11

Scheme 12
For the asymmetric variant of cycloaddition to nitrones 45, optically active α,β-unsaturated trifluoromethyl-arylsulfones 1, containing a chiral N,N-dialkylaminoethyl group at the ortho position, were used. In this case, the respective 5-(trifluoromethyl)isoxazolidines 49 were obtained regioselectively from nitrones 45 upon heating in PhMe, with 58–80% yields and 36–56% diastereoselectivity (Scheme 13).13

Scheme 13
The asymmetric version of cycloaddition has also been achieved by the use of chiral catalysts. Thus, a wide range of optically pure 5-(fluoroalkyl)isoxazolidines 50 were obtained in high yields (81–97%), as well as with high diastereo- and enantioselectivity (83–99% ее) via asymmetric cycloaddition reactions between a series of α,β-unsaturated fluoroalkyl-2-pyridylsulfones 1 and various aromatic and aliphatic nitrones 45 in the presence of Ni(II) bis(oxazoline) catalyst (S,S)-Ph-dbfox (Scheme 14).14 Products 50 have found applications in asymmetric synthesis of enantiomerically enriched α-trifluoromethylated γ-amino alcohols, as well as for the preparation of chiral trifluoromethyl-substituted 1,3-oxazinan-2-one derivatives as compounds with potential antiviral activity.14

Scheme 14
The cycloaddition reactions of β-fluoroalkylvinyl-pyridylsulfones 1 with various nitrile oxides 52 (R = Alk, Ar, Het) formed from the appropriate N-hydroxyimidoyl chlorides 51 by the action of a base (K2СО3) in Et2O at room temperature proceeded with the formation of 5-(trifluoromethyl)-4,5-dihydroisoxazole derivatives 53 in 48–85% yields with high regio- and diastereoselectivity (dr >99:1) (Scheme 15).15 Compounds 53 can be easily converted by treatment with DBU into the respective 5-fluoroalkyl-2-isoxazoles 55 via the cleavage of pyridylsulfonyl substituent (Scheme 15).15 In analogous reactions between (Е)-3,3,3-trifluoropropene derivatives 1 (RF = CF3) containing sulfonyl, sulfoximine, or sulfamide substituent at position 1 and ethyl cyanocarboxylate N-oxide 52 (R = CO2Et) generated in situ from ethyl oximinochloroacetate 51 upon refluxing in PhMe, a mixture of regioisomeric cycloadducts – 5-(trifluoromethyl)-and 4-(trifluoromethyl)-4,5-dihydroisoxazoles 53 and 54 was formed (Scheme 15).16 The latter were converted under the reaction conditions into isoxazole 56, which arose from spontaneous elimination of sulfur-containing fragment from isoxazolines 54 (Scheme 15).16

Scheme 15
Cycloaddition to azides. Synthesis of 1,2,3-triazoles
Cycloaddition of (E)-perfluoroalkyl-2-phenylsulfonylethenes 1 to 6-azido-α-D-galactose and -altrose 57 upon refluxing in PhMe was accompanied by desulfonylation of 1,2,3-triazoline intermediates 58 and formation of 1,2,3-triazole derivatives 59 as a single regioisomer, isolated in 40–75% yields (Scheme 16).17 These reactions were used for obtaining reverse nucleosides of fluoroalkyl-substituted 1,2,3-triazoles linked to the С-6 atom of sugars as potentially biologically active compounds.17

Scheme 16
In the present review, we have summarized for the first time the literature data on the synthesis of trifluoromethyl-substituted sulfur-containing five-membered heterocycles using the methodology of 1,3-dipolar [3+2] cycloaddition of functionalized 3,3,3-trifluoropropene derivatives, as well as highlighted the effect of the nature of substituents in the initial substrates on the regioselectivity of reactions and the structure of cycloadducts. The practical value of this synthetic approach, by employing fluorine- and sulfur-containing alkene substrates as dipolarophiles, lies in the possibility of the simultaneous introduction of both a fluoroalkyl group and an exocyclic sulfur-containing fragment into the heterocycle molecule. The heterocycles obtained in this manner are promising targets for biological investigations, and some of them, due to the revealed biological activity, have already found practical application. To conclude, the wide range of available 1,3-dipoles suggests an undoubted development of the synthetic strategy illustrated in this review for the preparation of novel barely accessible heterocyclic compounds.
References
(a) Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A.; Pearson, W. H., Eds.; Wiley: New York, 2002. (b) Gothelf, K. V.; Jørgensen, K. A. Chem. Rev. 1998, 98, 863. (c) Stanley, L. M.; Sibi, M. P. Chem. Rev. 2008, 108, 2887. (d) Hashimoto, T.; Maruoka, K. Chem. Rev. 2015, 115, 5366.
Markitanov, Yu. M.; Timoshenko, V. M.; Shermolovich, Yu. G.; Mykhalchuk, V. L.; Grafova, I. A.; Grafov, A. V. Chem. Heterocycl. Compd. 2016, 52, 503. [Khim. Geterotsikl. Soedin. 2016, 52, 503.]
Plancquaert, M.-A.; Redone, M.; Janousek, Z.; Viehe, H. G. Tetrahedron 1996, 52, 4383.
Taguchi, T.; Tomizawa, G.; Kawara, A.; Nakajima, M.; Kobayashi, Y. J. Fluorine Chem. 1988, 40, 171.
Cheng, F.; Kalita, S. J.; Zhao, Z.-N.; Yang, X.; Zhao, Y.; Schneider, U.; Shibata, N.; Huang, Y.-Y. Angew. Chem., Int. Ed. 2019, 58, 16637.
Markitanov, Yu. M.; Timoshenko, V. M.; Rudenko, T. V.; Rusanov, E. B.; Shermolovich, Yu. G. J. Sulfur Chem. 2019, 40, 629.
Markitanov, Yu. N.; Timoshenko, V. M.; Rusanov, E. B.; Shermolovich, Yu. G. Chem. Heterocycl. Compd. 2021, 57, 253. [Khim. Geterotsikl. Soedin. 2021, 57, 253.]
Kuhn, D. G.; Kamhi, V. M.; Furch, J. A.; Diehl, R. E.; Lowen, G. T.; Kameswaran, V. Pestic. Sci. 1994, 41, 279.
Markitanov, Yu. N.; Timoshenko, V. M.; Mykhaylychenko, S. S.; Rusanov, E. B.; Khyzhan A. I.; Shermolovich, Yu. G. Chem. Heterocycl. Compd. 2021, 57, 1107. [Khim. Geterotsikl. Soedin. 2021, 57, 1107.]
Kou, Y.-D.; Zhao, Zh.-N.; Yang, X.; Kalita, S. J.; Chen, X.-J.; Xie, Zh.-Zh.; Zhao, Y.; Huang, Y.-Y. Asian J. Org. Chem. 2018, 7, 1830.
Tsuge, H.; Okano, T.; Eguchi, S. J. Chem. Soc., Perkin Trans. 1 1995, 2761.
Li, J.-L.; Yang, X.-J.; Jiang, M.; Liu, J.-T. Tetrahedron Lett. 2017, 58, 3377.
Tsuge, H.; Okano, T.; Eguchi, S.; Kimoto, H. J. Chem. Soc., Perkin Trans. 1 1997, 1581.
Yang, X.; Cheng, F.; Kou, Y.-D.; Pang, S.; Shen, Y.-C.; Huang, Y.-Y.; Shibata, N. Angew. Chem., Int. Ed. 2017, 56, 1510.
Ou, Zh.; Huang, Q.; Kou, Y.-D.; Cheng, F.; Kalita, S. J.; Zhao, Zh.-N.; Huang, Y.-Y. Asian J. Org. Chem. 2019, 8, 2184.
Markitanov, Yu. N.; Timoshenko, V. M.; Shermolovich, Yu. G. Chem Heterocycl Compd. 2018, 54, 89. [Khim. Geterotsikl. Soedin. 2018, 54, 89.]
Hager, C.; Miethchen, R.; Reinke, H. J. Fluorine Chem. 2000, 104, 135.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2021, 57(12), 1149–1154
Rights and permissions
About this article

Cite this article
Markitanov, Y.N., Timoshenko, V.M. Synthesis of five-membered heterocycles by [3+2] cycloaddition reactions of sulfur-containing 3,3,3-trifluoropropene derivatives. Chem Heterocycl Comp 57, 1149–1154 (2021). https://doi.org/10.1007/s10593-021-03035-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-021-03035-w