
Abstract
Human activities have a major impact on habitat connectivity and landscape structure. In this context, it is critical to better understand animal movements and gene flow to develop appropriate conservation and land management measures. It is also important to better understand difference between sexes in space use and spatial scale of dispersal. We studied the common adder (Vipera berus), an elusive snake species with low mobility that is facing a substantial decline in Europe. A systematic sampling was carried out to clarify the dispersal pattern at a fine spatial scale (10 × 7 km2) in a rural landscape with both semi-natural (preserved heathlands, hedgerow networks) and degraded (crops, roads) habitats. Based on 280 captured adults and using 11 microsatellite markers, we detected no marked genetic differentiation, however, we detected relatively strong isolation-by-distance (IBD). Under IBD, we quantified a low neighborhood size (Ns ≈ 50) associated with limited natal dispersal (σ ≤ 1 km). We detected sex-biased dispersal in favor of males, but the pattern was dependent on the spatial scale considered. Our results also suggest that there is higher genetic diversity in the preserved habitat, notably among males. Overall, our study underlines the importance of sex variation in dispersal, and the spatial scale of landscape effects. This contrast between sexes should be considered to improve functional connectivity at fine spatial scales for reptile conservation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Anthropogenic pressures on the environment have grown exponentially over the last century and negatively impact biodiversity (Brook et al. 2008). Land-uses changes are responsible for major habitat loss and fragmentation (Newbold et al. 2015; Powers and Jetz 2019). Landscape degradation has multiple implications since it can reduce individual movement, reduce gene flow, and may ultimately lead to local extinctions (Fischer and Lindenmayer 2007). Species with low mobility are particularly vulnerable to anthropogenic disturbances because of their limited dispersal capacities (Stow et al. 2001; Sumner et al. 2004; Moore et al. 2008). However, important variations exist in activity capacities and mobility among terrestrial vertebrates (Hillman et al. 2014a). Low mobility is typically observed in amphibians and reptiles because thermal conditions can determine their activity levels and they have limited aerobic capacities (Hillman et al. 2014a, b). Not surprisingly, these two groups are facing substantial declines in relation to habitat degradation (Arntzen et al. 2017; Doherty et al. 2020).
Dispersal is defined as all individual movements with potential consequences for gene flow across space (Ronce 2007). The dispersal process can be influenced by multiple factors including individual traits (e.g. sex, behavior, condition-dependence) and environmental attributes (e.g. habitat quality, landscape structure; Matthysen 2012). Sex-biased dispersal appears to be ubiquitous in vertebrates (Trochet et al. 2016; Li and Kokko 2019) and related to the mating system (Greenwood 1980; Lawson Handley and Perrin 2007; Clutton-Brock and Lukas 2012). Several hypotheses have been proposed to explain sex-biased dispersal such as competition between individuals for local mates, competition for resources, and inbreeding avoidance (Motro 1991; Perrin and Mazalov 2000). All of them predict male-biased dispersal in polygynous mating systems, whereas only competition for resources induces female-biased dispersal in monogamous species (Greenwood 1980; Lawson Handley and Perrin 2007; Clutton-Brock and Lukas 2012). Because males and females may respond differently to habitat degradation (Stow et al. 2001; Li and Kokko 2019), sex biased dispersal should be considered when addressing genetic structuring and diversity in human-transformed habitat. Moreover, the selection of a relevant spatial scale is of primary importance to identify at what spatial scale the (sex-biased) dispersal occurs (Gauffre et al. 2009; Lechner et al. 2012). Such information is required to investigate patterns of genetic relationships (Jackson and Fahrig 2012, 2015) and improve conservation measures in fragmented landscapes (Pernetta et al. 2011; Keller et al. 2015).
Squamate reptiles (lizards and snakes) are often found in human-altered landscapes (Graitson et al. 2020), and habitat alteration and destruction have caused significant declines over previous decades (Doherty et al. 2020). This pattern is clear in Europe with a growing number of species considered as near-threatened (NT) in the IUCN Red List (Böhm et al. 2013) and a marked decline notably in snake populations (Reading et al. 2010). Snakes often have elusive behaviors and low detectability (Kéry 2002; Durso et al. 2011). Consequently, there is a general lack of data in this group, notably on dispersal (Weatherhead and Madsen 2009; Böhm et al. 2013). Genetic exchanges are influenced by different factors including mating systems and species spatial ecology. For instance snakes display a wide range of foraging strategies (Lourdais et al. 2014) and gene flow is often reduced in sit-and-wait foragers compared to active foragers (de Fraga et al. 2017). Breeding strategy often involve active mate searching during the breeding season and male-biased dispersal has been described in different snakes species (Hofmann et al. 2012; Folt et al. 2019). Yet, available data suggest that snakes have limited dispersal capacities both at juvenile and adult stages (e.g. Webb and Shine 1997), important philopatry (e.g. Sumner et al. 2001), and strong isolation-by-distance (IBD) patterns (e.g. Pernetta et al. 2011; Meister et al. 2012).
The common European adder (Vipera berus) is a cold-specialist viperid snake with a wide Eurosiberian range (Saint Girons 1980). This viviparous species is highly philopatric (Madsen and Shine 1992a; Luiselli 1993) and an ambush forager (Bea et al. 1992). In Western and Central Europe, this species often occurs in small populations isolated in remnant natural habitat patches. Previous studies have demonstrated that there is significant isolation and strong genetic structuring among populations (Ursenbacher et al. 2009; Ball et al. 2020), with deleterious consequences in fragmented/degraded habitats (e.g. inbreeding and inviable offspring; Madsen et al. 1996; Madsen et al. 1996). This species is threatened by increased land use and other anthropogenic disturbances (Gardner et al. 2019). However, there is a lack of data about gene flow and dispersal distances (Viitanen 1967; Prestt 1971), notably concerning sex differences. This is important because males and females strongly differ in their movement patterns during reproduction (higher mobility of males; Neumeyer 1987).
Our aim was to investigate the genetic structure, genetic diversity, and dispersal ability in V. berus at a fine spatial scale in a rural landscape in western France. The study area (70 km2) was composed of a preserved zone (natural heathland with rocky outcrops) surrounded by an agricultural landscape (fragmented habitat). Our main hypothesis was that dispersal would be limited and sex-dependent in the adder. We applied an individual-based sampling scheme to measure genetic structuring, diversity and also estimate effective dispersal (i.e. only leading to gene flow; see Broquet and Petit 2009). We tested the following predictions:
-
(1)
Because of limited dispersal, the genetic structure should be linked to main landscape boundaries and follow an isolation-by-distance pattern
-
(2)
Because of the polygynous mating system with males actively searching for females, we expected to find a male-biased gene flow pattern for all spatial scales
-
(3)
Due to reduced anthropogenic disturbances, we predicted that genetic diversity would be higher in the preserved zone than in the surrounding agricultural zone.
This approach should help conservation management decisions by providing valuable information on the dispersal ability of V. berus, and consequently help define the size of management units and the creation of corridors between populations.
Materials and methods
Study site
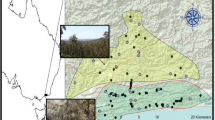
Our study site (70 km2) was located south-west of Rennes (France; Fig. 1A and B) within a rural landscape composed of agricultural fields (61.6% of crops and 1.2% of pastures) and a relatively dense network of semi-natural habitats (15.7% of forest, 8.9% of suitable habitats for V. berus and 1.5% of aquatic habitats). Habitats were considered as suitable when they included one of the two following components: (i) a dense hedgerow network, and (ii) remnants of heathlands or medio-European thickets. Urbanization (11.1%) mainly consisted of small villages with a road network (ca. 300 km including main roads and agricultural tracks; Fig. 1C). The study site also encompassed a preserved zone, the Canut valley, which is a 4.4 km2 protected conservation area (Fig. 1C; Natura 2000 site; see https://ec.europa.eu/environment/nature/natura2000/index_en.htm), mainly composed of forests (45.9%), suitable habitats (27.5% of heathland), and dry grasslands (3.5%; See Fig. S1D in the Supporting Information).

a Location of the study site in western France and b in the Ille-et-Vilaine department. c Study site and locations of DNA samples collected from 280 adders (Vipera berus). Number of individuals caught per sampling area is provided. Fourteen a priori groups were considered depending on the potential effects of large landscape boundaries
Sampling design and field work
A previously published study suggested that genetic differentiation is limited for V. berus populations below 3 km and strong beyond 6 km (Ursenbacher et al. 2009). Therefore, we selected a fine spatial extent (10 × 7 km2) to determine how and at which scale the dispersal process occurred, as recommended by Rousset (1997, 2000). The species seemed to be continuously distributed within the study area, allowing us to obtain a pairwise distance gradient from continuously sampled statistical units. As a consequence, we chose individuals as statistical units (Broquet and Petit 2009) and implemented an individual-based sampling scheme (ISS; Prunier et al. 2013). We designed our ISS on a one-square-kilometer-unit grid applied on-site. Within each square kilometer, we focused on suitable habitats to optimize sampling success, repeatedly if possible (Fig. 1C). Last, we also subdivided the study site into several units based on distinct main landscape boundaries (main roads, topological barriers; see Fig. 1.C) to carry out subsequent analyses on the genetic markers (see below).
Sampling took place in 2015 and 2016 between April and June when individuals are the easiest to detect during basking activities. A sampling area was defined as a location with a 200 m radius where individuals could be spatially close (local movements inferior to 200 m; Prestt 1971) and found within a continuous habitat. For each sampling area, the Visual Encounter Survey (VES) session was stopped as soon as four to five individuals were sampled, as recommended by Prunier et al. (2013). Otherwise, additional VES sessions were conducted until (i) the required number of samples was obtained or (ii) it was the end of field season. Additionally, all individuals that were easily detected and caught after the end of a VES were also sampled, resulting occasionally in more samples than strictly needed. Each captured individual was sexed visually and by manual eversion of the hemipenes. Snout-vent-length (SVL) was used to differentiate adults from subadults and yearlings (Guiller and Legentilhomme 2015) before collecting DNA samples from buccal and cloacal swabs (plus 2 skins). For each capture, we recorded GPS coordinates and took pictures of the head for identification (treated with Aphis software version 1.0.0) to avoid duplicate DNA samples (Sheldon and Bradley 1989; Moya et al. 2015).
Laboratory methods
Genomic DNA was extracted from buccal and cloacal swabs with a Qiagen DNeasy Blood & Tissue Kit (QIAGEN, Hombrechtikon, Switzerland) following the manufacturer’s protocol, except that the swab was removed before adding the washing buffer (before step 5). Fourteen microsatellite loci were amplified to analyze genetic diversity, eleven specifically developed for V. berus (Vb-64 and Vb-71 from Carlsson 2003; Vb-A8, Vb-A11, Vb-B’2, Vb-B’9, Vb-B10, Vb-B’10, Vb-B18, Vb-D’10 and Vb-D17 from Ursenbacher et al. 2009), two for Vipera ursini (Vu-29 and Vu-58; Metzger et al. 2011), and one for Vipera aspis (Va-35; Geser et al. 2013) following the PCR conditions described in the respective publications. Microsatellite PCR products were analyzed on an ABI 3130xl automated sequencer (Applied Biosystems, Waltham, USA), visualized with the dyed forward primers, and allele sizes were determined using Peakscanner 3.1 (Applied Biosystems).
Analysis of genetic markers and metrics
Observed and expected heterozygosities, and departures from Hardy–Weinberg equilibrium (HWE) were calculated with χ2 tests at each locus from (i) the entire dataset and (ii) groups of individuals based on main landscape boundaries (see above), to avoid detecting a deficit in heterozygosity linked to a Wahlund effect. These analyses were performed with the packages adegenet (Jombart 2008), pegas (Paradis 2010), and hierfstat (Goudet and Jombart 2015) implemented in R 4.0.0 (R Core Development Team 2020). The presence of null alleles and miss-scoring was checked using the Micro-checker 2.2.3 (Van Oosterhout et al. 2004), and Fstat 2.9.3.2 (Goudet 1995) was used to test for linkage disequilibrium among loci.
Genetic pairwise distances were calculated based on two genetic metrics: (i) âr metric from Spagedi 1.5a (Hardy and Vekemans 2002) and (ii) Bray–Curtis Dissimilarity (Bc) from the ecodist R package (Goslee and Urban 2007). Almost all analyses were performed with âr, a metric that is explicitly related to dispersal, where higher values indicate a greater dissimilarity between two genotypes (Rousset 1997, 2000). However, we had to use Bc in spatial autocorrelation analyses, but the two metrics were strongly correlated (Pearson correlation coefficients = 0.874). Euclidean distances between individuals were computed into matrices from Arcgis from geographical coordinates (10.1 ©Esri Inc.).
Genetic clustering analyses
Bayesian assignment tests were used to infer genetic delineations from individuals using Structure (Pritchard et al. 2000). We chose to make the genetic cluster (K) vary from one to 30, considering that a greater K would be unlikely at that spatial scale. A 200,000-step burn-in followed by 400,000 MCMC was parameterized for each run without available geographical information. Next, Structure harvester (Earl and vonHoldt 2012) was used to compute L(K) (also known as ln Pr(X|K)) and ΔK (Evanno et al. 2005) to determine the best K. Ten independent runs for each genetic cluster (K) were carried out using both no-admixture and admixture models.
Isolation-by-distance (IBD)
Distance matrices were log-transformed according to D = ln(d + 1) before using them in a Mantel test performed with restricted permutations per blocks (i.e. sampling area level) to preserve correlations among spatially close individuals and to avoid pseudo-replications (see Prunier et al. 2013 for more details). Ideally, the number of individuals in the blocks should be balanced, but a slightly unbalanced sampling is considered to have a limited impact on results (Prunier et al. 2013). Therefore, we implemented this methodology in the mantel function of the vegan R package (Oksanen et al. 2016) in order to apply 10,000 restricted permutations depending on our unbalanced blocks (i.e., sampling areas). See Fig. S2 in the Supporting Information for the plot depending on sex.
Dispersal parameters
As âr metrics have been explicitly connected to natal dispersal (Rousset 1997, 2000), a linear model was applied to semi-linearized distance matrices. No transformation was applied to genetic distances, âr being an analogue of FST / (1- FST), whereas a logarithmic transformation was applied to Euclidean distances (Rousset 1997, 2000). Neighborhood size (Ns) was computed from this linear model according to the inverse of the slope coefficient (b) that we used to quantify the variance of the axial dispersal distance (σ2) according to σ2 = 2 / (b × 4Dπ) (Rousset 1997, 2000). However, this calculation required prior information about population density (D), which is currently unknown at the study site. So, we chose to use a proxy of D, to which we applied several multiplier coefficients: D × 1, 2, 4, 10, 20, and 75 (pers. com. Robledo-Arnuncio; Dupont et al. 2015). These coefficients were chosen to range between (i) the minimum D value (i.e. the total number of individuals found during our field survey divided by the extent of the study site), assuming that we captured all individuals inhabiting on our study site, and (ii) the maximum value (75D) corresponding to three individuals per hectare (21,000/70 km2), which was observed in a more optimal subalpine environment (Neumeyer 1987). Lastly, we took the effect of short Euclidean distances into account (Rousset 2000): all pairs of individuals closer than the distance σ1 computed from the complete linear model were removed before extracting σ2 from the new model.
Genetic spatial autocorrelation
Spatial autocorrelation analyses (Oden and Sokal 1986; Sokal 1986) were carried out with Mantel correlograms from all individuals and separately for each sex. We used this approach because it can detect a pattern of spatial autocorrelation under IBD conditions (Borcard et al. 2011; Borcard and Legendre 2012; Legendre and Legendre 2012). Positive spatial autocorrelation is then expected for the first distance classes, but only considered significant if its previous values were also significant (Legendre and Legendre 2012). Correlograms were implemented with several distance steps (100 m, 200 m, 250 m, 300 m, 400 m and 500 m) chosen from field observations (distances between captures-recaptures of some individuals). However, we will only detail the correlogram providing the best trade-off between a minimum size of distance class and the number of pairwise values within each distance class in the Results section (Banks and Peakall 2012). Other distance steps are available in the Supporting Information (See Fig. S3 to Fig. S7 in the Supporting Information). Each correlogram was parameterized with a cut-off (i.e. limited to the first half of the existing distance classes) and 10,000 permutations with Holm’s correction to test the significance for each distance class using the vegan R package. The mantel.correlog function was implemented to carry out restricted permutations with unbalanced blocks. Moreover, 95% confidence intervals were implemented by bootstrapping with 1,000 replicates of the Mantel statistic for each distance class using the boot R package (Davison and Hinkley 1997; Canty and Ripley 2017).
Test of sex-biased dispersal
Inter-individual genetic distances measured with ar were used to determine the relative gene flow level within each sex. Comparisons were carried out at several spatial scales. For each of them, only the ar of individuals closest to a threshold distance were used. Several threshold distances were defined up to 3,000 m according to the distance step used for the best correlogram (see above and Results sections). In the first spatial scales, individuals of the farther dispersing sex (Ds) are less likely to remain in the vicinity of their relatives than individuals of the more philopatric sex (Ps). As a consequence, we expected a mean pairwise genetic differentiation to be higher between Ds (males V. berus) than between those of Ps (female V. berus). At broader spatial scales, we expected these differences to cancel out because a higher gene flow within the Ds should lead to a homogenization of genotypes at our study site.
Additionally, we applied the procedure of Coulon et al. (2006) by carrying out 10,000 “random resamplings without replacement of the relevant half-matrices such that each individual occurred only once in a given resampled set”. Mean values for each set were calculated depending on sex. The difference Δi between the means of the two sexes was then derived, where i is the number of the resampling set. We chose to apply a one-sided test by performing calculations in the direction following our predictions (Δi = ith resampling mean value of male–ith resampling mean value of female). Under the null hypothesis (i.e. no sex-dispersal), Δi should follow a normal distribution centered on 0; under the alternative hypothesis (i.e. male-biased dispersal), most of Δi values should be positive. Significance was considered when more than 95% of the Δi values were greater than 0. As a consequence, p-values were calculated as the number of negative Δi values divided by 10,000.
Genetic diversity
Differences in genetic diversity were tested at the individual level (i) with the entire dataset and (ii) according to the sex, depending on the location. For the latter aspect, we distinguished three landscape categories: (i) a preserved zone, (ii) a buffer zone (1,000 m around preserved) and (iii) an agricultural zone. Among the available genetic metrics, we chose to calculate and compare the homozygosity by locus (HL) from genhet (Coulon 2010) for the different landscape categories. This index was developed by Aparicio et al. (2006) to weight the contribution of loci depending on their allelic variability; it varies between 0 (all loci are heterozygous) to 1 (all loci are homozygous). As a consequence, genetic diversity is negatively correlated to HL.
Results
Sampling and genetic markers
We genotyped 280 adders (244 adults, 36 sub-adults, yearlings were excluded) corresponding to 148 females, 130 males, and two unsexed individuals sampled from shed skin distributed across the three landscape categories (Table 1). Sampling of individuals was also well-distributed across units based on main landscape boundaries (14 a priori groups; number of individuals per group: mean ± SE = 20.00 ± 1.77; min. = 10; max. = 31; Fig. 1; Table S1).
Among the 14 microsatellites, the Va-35 locus was monomorphic and the Vb-D’10 locus had around 60% of missing data (Fig. S8 in the Supporting Information), so we removed them from subsequent analysis. Out of the entire data set, HW disequilibrium was detected for 5 loci on the remaining 12 microsatellites (Table S2). However, only Vb-B18 showed a HW disequilibrium for more than one a priori group (Fig. S9 in the Supporting Information). Additionally, we also detected the presence of null alleles for Vb-B18, so we removed it from the analyses (Table S2). No linkage disequilibrium was discovered among the remaining 11 microsatellites.
Genetic differentiation: IBD & dispersal parameters
We detected only one genetic cluster (K = 1) from the clustering analysis implemented with Structure when using a non-admixture model. On the contrary, by using Evanno’s method, we detected that the best-supported values were K = 28 or 29 when using admixture models. However, we also noted the presence of one or more unstable runs for the high values of K and that the best supported genetic cluster was K = 1 from L(K) representation. Moreover, we observed an IBD pattern among individuals (Mantel test with restricted permutations, Pearson’s r = 0.0987, p = 0.0001). The pattern was slightly higher when spatially close individuals (i.e. distances < σ1, Tableau 14) were excluded (for D = 1 and with exclusion: slope = 0.0213, Mult. R2 = 0.0047, df = 34,258; without exclusion: slope = 0.0185, Mult. R2 = 0.0112, df = 39,058; Fig. 2). Depending on the estimate of D, dispersal parameters for models with exclusion of close individuals varied according to σ ranging from ca. 165 m to 1,365 m and Ns around 50 individuals (46.86 to 53.95; Table 2).

Genetic differentiation among 280 V. berus individuals according to the âr metric for genetic distances against log-transformed geographical distances. Two linear models were computed: one with all pairwise values (Eq. 1, dotted black line) and another with the exclusion of pairwise values corresponding to all pairs of individuals geographically closer than the distance σ1 (ln(σ1) = 6.94; Eq. 2, solid black line). Confidence intervals at a 95% level were not drawn for display reasons. These linear models showed an isolation-by-distance relationship according to the following equations: Eq. 1: \(\hat{a}_{r} \, = \,0.0185\, \times \,\ln \, \left( {geographical \, dis\tan ce} \right){-}0.0757\); Eq. 2: \(\hat{a}_{r} \, = \,0.0213\, \times \,\ln \;\left( {{\text{geographical}}\;{\text{distance}}} \right){-}0.0989\)
Genetic autocorrelation and sex-biased dispersal
A positive spatial autocorrelation up to ca. 2,225 m was detected (Fig. 3A), mainly due to females having a greater distance (up to 1,000–1,500 m; Fig. 3B) than males (not beyond 500-750 m; Fig. 3C). However, both sexes exhibited a similar pattern for the first distance class (250 m) without a difference in signal strength according to the bootstrap value with a 95% confidence interval (CI; Fig. 3A and B). At the scale of the study site (70 km2), the averages of the difference (Δi) between male and female genetic differentiation were not significantly greater than zero (p-value = 0.095; Fig. S10 in the Supporting Information). However, Δi was significantly greater than zero for spatial scales with threshold distances ranging from 1,000 m to 2,000 m (Fig. 4).

Mantel correlogram considering (a) all individuals (N = 280), b only females (N = 148) and c only males (N = 130), with distance classes following a 250-m step. Mantel tests were performed with 10,000 permutations and 95% confidence intervals from 1,000 bootstrap iterations. Holm’s correction was used for p-values, and significant values (i.e. spatial autocorrelation) were represented by full squares. The number of pairwise values between individuals per distance class was added above the x-axis

Average of the differences between males and females dispersal (Δi) obtained from each of the 10,000 random resampling dataset depending on the spatial scale tested. Spatial scales correspond with threshold distances for which only inter-individual genetic distance of individuals closer than the threshold were used. Averages of Δi and their 95% confidence interval were represented by full circles surrounded by an error bar in solid line. Mobile averages were represented according a dotted line. Significant Δi were highlighted with a grey box (p-value between 0.01 ‘*’ 0.05)
Influence of habitat type on genetic diversity
When the two sexes were pooled, no difference (Kruskal–Wallis chi-squared = 2.949, df = 2, p-value = 0.229) in the homozygosity per locus (HL) was found between the preserved zone (mean ± CI = 0.303 ± 0.029; N = 82), the buffer zone (mean ± CI = 0.341 ± 0.036; N = 66) and the agricultural zone (mean ± CI = 0.341 ± 0.029; N = 132). When sexes were analyzed separately (Fig. 5), homozygosity was significantly lower for males (Fig. 5) in the preserved zone (mean ± CI = 0.283 ± 0.0485; N = 42; Fig. 5 a) compared to the agricultural zone (mean ± CI = 0.373 ± 0.029; N = 57; Kruskal–Wallis chi-squared = 6.615, df = 2, p-value = 0.0366; Fig. 5b). Males sampled within the buffer zone did not differ in HL compared to those sampled within the other two zones (Fig. 5 a,b). In contrast, there were no differences in HL between any of the three zones for females.

Mean of genetic diversity at the individual level in the three landscape categories: (i) preserved (Natura 2000 sites dominated by forests and heathlands), (ii) buffer (1,000 m around preserved) and (iii) agricultural (dominated by crops and grasslands). Mean values and 95% confidence intervals were represented by points surrounded by solid error bars. Points were differently represented depending on sex: full circle for both females and males, full square for females and full triangle for males. Different letters indicate significant differences. Capital letters relate to both sex; lower letter relate to females (black background) and males (white background). The number of values used to calculate each mean was added above the x-axis
Discussion
Our results at a fine spatial scale clearly support that adder dispersal is limited and sex-dependent. The maximal natal dispersal distance (σmax) was estimated to 1,365 m and associated to a marked IBD pattern. Male-biased dispersal was validated at all spatial scales, even if it was only significant between 1,000 m and 2,000 m. The environmental quality impacted genetic diversity, with a higher genetic diversity of males in the preserved zone, whereas a lack of genetic structure within the whole studied area was detected. We discuss our results in detail below.
IBD and genetic differentiation
On the study site (70 km2), genetic differentiation seemed mainly driven by an IBD pattern in the adder. No a posteriori differentiated populations were found and the IBD level was marked. The IBD pattern was also supported by the spatial autocorrelation analysis, since neighboring individuals were genetically similar up to a distance of 2,225 m (Fig. 3A). The slope coefficients of the IBD models (Table 2) are similar to the highest values described in 11 studies also using an individual-based analytical approach and conducted on different vertebrate taxa (mean ± SE = 0.0111 ± 0.0020; see Table S3). The IBD models explained low variance (r2) in our study area, but this is expected under an individual-based model (Rousset 2000; Sumner et al. 2001). An IBD pattern has previously been reported in several snake species when the studies were conducted at a local or regional scale (< 20 km; Rivera et al. 2006; King 2009; Ursenbacher et al. 2009; Pernetta et al. 2011; Meister et al. 2012; DiLeo et al. 2013). However, IBD patterns can strongly differ among species coexisting on the same study site, as demonstrated for three Natricid species with different morphology and habitat usage (Thamnophis sirtalis, Storeria dekayi, and Nerodia sipedon; King and Lawson 2001). IBD patterns can also vary between sites and be detected at lower spatial scales (Manier and Arnold 2005). Therefore, the choice of the study area, the surface considered, and the sampling scheme must be carefully considered to fit the ecology of the target species. A recent study demonstrated that gene flow in snakes is lower in ambush foragers when compared to active foragers (de Fraga et al. 2017). Therefore, IBD patterns are more likely observed in sit-and-wait foragers, such as viperid snakes that are specialized for energy-saving (Lourdais et al. 2014).
From IBD models, the maximal natal dispersal distance of V. berus was evaluated to about 1,365 m (see σmax; Table 2). This indirect estimate should be considered with caution because local population size and the proportion of immigrants also contribute to genetic differentiation (Lowe and Allendorf 2010). These two parameters are complex, or even impossible, to precisely evaluate. For instance, population density is difficult to assess for snake because to their secretive behaviour and low detectability (Kéry 2002; Durso et al. 2011). Consequently, the real σmax is likely smaller (see Table 2) and field observations suggest a more realistic estimates around 500 m (field observations; pers. com. Guiller).
The dispersal is male-biased and scale-dependent
Multi scales approaches are particularly relevant to address the spatial dependence of landscape effects and genetic patterns (Jackson and Fahrig 2012, 2015) that may vary among sexes (Gauffre et al. 2009; Guerrini et al. 2014). We detected a male-biased dispersal at all spatial scales, but the difference was significant only for the spatial scale of 1,000–2,000 m (Fig. 4). Others studies that focus on snake species have detected male-biased dispersal regardless of the spatial scale (Rivera et al. 2006; Clark et al. 2008; Dubey et al. 2008; Pernetta et al. 2011; Hofmann et al. 2012). We found that females were more spatially autocorrelated, except in the first distance class, where males exhibited a similar partial autocorrelation. The discrepancy between sexes at a distance between 1000 and 2000 m could be explained by differences in space use during reproduction. During the breeding season, males intensively search for mates and can travel several hundred meters, or even more (Neumeyer 1987; Madsen and Shine 1993; Madsen et al. 1993). In contrast, females have substantial constraints during pregnancy, with limited displacements and increased thermoregulation (Madsen and Shine 1992b, 1993; Lorioux et al. 2016). Consequently, male-biased dispersal likely reflects sex differences in reproductive strategies (Bonte et al. 2012; Trochet et al. 2016). The absence of sex-biased dispersal at the very local scale could be explained by the strong philopatric behavior in this species outside of reproduction (Madsen and Shine 1992a; Luiselli 1993). The high relatedness pattern observed in mainland Britain confirms the philopatric behavior of the adder (Ball et al. 2020). The similar autocorrelation level between sexes at the local scale could also be related to variation among males in reproductive activities, with some individuals moving less for mating or individuals that are just coming back to their birthplace after mating. A similar case was observed in another Viperid snake (Crotalus horridus), which exhibited a strong level of philopatry and gene flow linked to seasonal male mating dispersal (Clark et al. 2008). Our findings in the adder provide additional support to the idea that male biased-dispersal in snakes reflects the active search of females for mating.
Effect of the environment on population structure and genetic diversity
Even if roads and topological barriers are present in the study area, we only detected one genetic cluster. This result suggests that the putative barriers do not strongly shape the genetic structuring, at least not yet or not in a sufficient way to be currently detectable. Past agricultural landscapes in western France were very suitable for V. berus until the removal of hedgerows in the late 1950s (Burel and Baudry 1990). Thus, it is very likely that the signal of genetic structuration is not yet able to be detected. Indeed, a lag time effect between landscape changes and the genetic response is frequently observed (Flavenot et al. 2015). Furthermore, detection of new genetic barriers takes longer for species with limited dispersal abilities (Landguth et al. 2010), especially in the presence of an IBD pattern (Blair et al. 2012). Consequently, using the analyses in this study, we cannot currently say whether or not the environmental elements in the study area are reducing the gene flow.
We found that genetic diversity was higher (lower level of homozygosity) in the preserved zone (and to a lesser extent in the buffer zone) than in the agricultural zone, but this pattern was significant only in males. The protected status of the nature preserve area is recent and the valley is composed of well-preserved heathland habitats with rocky outcrops. The topography and geology of the area has limited human disturbances over time. This contrasts with the nearby rural landscape which has various farming activities, including crop farming and cattle grazing. Therefore, the history of the site likely provides an explanation to the observed pattern, rather than the recent benefits of conservation status. High genetic diversity is important for the conservation of V. berus populations, as inbreeding within a small isolated population of adders can lead to a strong reduction in population size (Madsen et al. 1999, 2004). Very recently, Ball et al. (2020) observed a high relatedness of individuals in small isolated populations, but without loss of genetic diversity at the population level. The fact that higher genetic diversity was observed only in males is intriguing and deserves further work.
What matters for the conservation of V. berus?
Our results provided important insights on the dispersal and IBD pattern in the adder and are relevant for conservation management. First, the distance between individuals strongly affects the genetic differentiation within a population, even at a small spatial scale (a few kilometers) where there is limited dispersal. Second, we revealed that dispersal was sex-biased and scale-dependent: both sexes had a similar dispersal at a local scale, whereas males dispersed farther at a larger scale. These findings underline the importance of considering how genetic variation is affected depending on the sex (Guerrini et al. 2014) and the spatial scale of the effects (Gauffre et al. 2009; Keller et al. 2013). Moreover, genetic diversity of males varied with the quality of the environment, which was not detected in females. Habitat loss or alteration are direct threats for species with low mobility and our study provides additional support to the fact that sexes may differ in their responses to landscape changes (Stow et al. 2001; Amos et al. 2014; Niebuhr et al. 2015).
Based on these results, we recommend that conservation measures should be implemented within a buffer area of 500 m surrounding managed habitats, and that an optimal conservation management unit should be set in place, similar to those of the preserved area (4.4 km2). Keller et al. (2015) recommended to scale the surface of conservation units to several times that of the maximal dispersal distance. Among the actions to implement, we recommend maintaining or creating suitable habitats with diversified microhabitats (including shelters and basking site). This includes (i) legal protection for important habitats (e.g. linear edges, heathland patches, etc.), (ii) maintenance of open microhabitats for thermoregulatory activities (e.g. canopy opening) or connectivity between habitats (e.g. hedgerows, ditches or ditch banks) and (iii) to add suitable shelters to the habitat (e.g. pile of rocks, coarse woody debris; Mullin and Seigel 2009; Pike 2016).
Conclusion
Using muti-scale approaches is critically required to identify the spatial dependence of genetic and landscape effects (Angelone et al. 2011; Jackson and Fahrig 2012, 2015; Keller et al. 2013). Species with limited dispersal and high ecological specialization are more vulnerable to habitat loss and degradation (Öckinger et al. 2010). This is likely the case for Viperid snakes (e.g. Santos et al. 2006; Mullin and Seigel 2009) which should be better considered in conservation (genetic) studies, especially with regard to the dispersal process (Mullin and Seigel 2009).
Data availability
Data are stored in the file named “INPUT_R_Data” in.csv format with semicolon separator. These data were used to create the file named “INPUT_GENEALEX” in.xlsx format. Its “export data” function was used to generate the other software input files. Depending on the software, inputs files are stored according to “INPUT_ SOFTWARE” in.txt format with tabulation for Spagedi, in.txt format with space for Structure and in.dat or.lab format for Fstat. Euclidean distances between individuals are stored in the files named “OUTPUT_Arcgis” in.csv format with semicolon separator. The datasets generated during and/or analysed during the current study are not publicly available due to threats to the species (e.g. disturbance or destruction of habitats or individuals) and its protection status in Brittany, but are available from the corresponding author on reasonable request.
Code availability
The R code is stored in the “R_script_Francois_et_al” in.Rmd format but is only available from the corresponding author on reasonable request (see above).
References
Amos JN, Harrisson KA, Radford JQ et al (2014) Species-and sex-specific connectivity effects of habitat fragmentation in a suite of woodland birds. Ecology 95:1556–1568. https://doi.org/10.1890/13-1328.1
Angelone S, Kienast F, Holderegger R (2011) Where movement happens: scale-dependent landscape effects on genetic differentiation in the European tree frog. Ecography 34:714–722. https://doi.org/10.1111/j.1600-0587.2010.06494.x
Aparicio JM, Ortego J, Cordero PJ (2006) What should we weigh to estimate heterozygosity, alleles or loci? Mol Ecol 15:4659–4665. https://doi.org/10.1111/j.1365-294X.2006.03111.x
Arntzen JW, Abrahams C, Meilink WRM et al (2017) Amphibian decline, pond loss and reduced population connectivity under agricultural intensification over a 38 year period. Biodivers Conserv 26:1411–1430. https://doi.org/10.1007/s10531-017-1307-y
Ball S, Hand N, Willman F et al (2020) Genetic and demographic vulnerability of adder populations: Results of a genetic study in mainland Britain. PLoS ONE 15:e0231809. https://doi.org/10.1371/journal.pone.0231809
Banks SC, Peakall R (2012) Genetic spatial autocorrelation can readily detect sex-biased dispersal. Mol Ecol 21:2092–2105. https://doi.org/10.1111/j.1365-294X.2012.05485.x
Bea A, Brana F, Baron J-P, Saint Girons H (1992) Régimes et cycles alimentaires des vipères européennes (Reptilia, Viperidae): étude comparée. Année Biol 31:25–44
Blair C, Weigel DE, Balazik M et al (2012) A simulation-based evaluation of methods for inferring linear barriers to gene flow. Mol Ecol Resour 12:822–833. https://doi.org/10.1111/j.1755-0998.2012.03151.x
Böhm M, Collen B, Baillie JEM et al (2013) The conservation status of the world’s reptiles. Biol Conserv 157:372–385. https://doi.org/10.1016/j.biocon.2012.07.015
Bonte D, Van Dyck H, Bullock JM et al (2012) Costs of dispersal. Biol Rev 87:290–312. https://doi.org/10.1111/j.1469-185X.2011.00201.x
Borcard D, Legendre P (2012) Is the Mantel correlogram powerful enough to be useful in ecological analysis? A simulation study. Ecology 93:1473–1481. https://doi.org/10.1890/11-1737.1
Borcard D, Gillet F, Legendre P (2011) Numerical ecology with R. Springer, New York
Brook BW, Sodhi NS, Bradshaw CJA (2008) Synergies among extinction drivers under global change. Trends Ecol Evol 23:453–460. https://doi.org/10.1016/j.tree.2008.03.011
Broquet PE (2009) Molecular estimation of dispersal for ecology and population genetics. Annu Rev Ecol Evol Syst 40:193–216. https://doi.org/10.1146/annurev.ecolsys.110308.120324
Burel F, Baudry J (1990) Structural dynamic of a hedgerow network landscape in Brittany France. Landsc Ecol 4:197–210. https://doi.org/10.1007/BF00129828
Canty A, Ripley BD (2017) boot: Bootstrap R (S-Plus) Functions
Carlsson M (2003) Phylogeography of the adder. University of Upssala, Vipera berus
Clark RW, Brown WS, Stechert R, Zamudio KR (2008) Integrating individual behaviour and landscape genetics: the population structure of timber rattlesnake hibernacula. Mol Ecol 17:719–730. https://doi.org/10.1111/j.1365-294X.2007.03594.x
Clutton-Brock TH, Lukas D (2012) The evolution of social philopatry and dispersal in female mammals. Mol Ecol 21:472–492. https://doi.org/10.1111/j.1365-294X.2011.05232.x
Coulon A (2010) genhet: an easy-to-use R function to estimate individual heterozygosity. Mol Ecol Resour 10:167–169. https://doi.org/10.1111/j.1755-0998.2009.02731.x
Coulon A, Cosson J-F, Morellet N et al (2006) Dispersal is not female biased in a resource-defence mating ungulate, the European roe deer. Proc R Soc B 273:341–348. https://doi.org/10.1098/rspb.2005.3329
Davison AC, Hinkley DV (1997) Bootstrap methods and their application. Cambridge University Press, Cambridge, NY
de Fraga R, Lima AP, Magnusson WE et al (2017) Contrasting patterns of gene flow for Amazonian snakes that actively forage and those that wait in ambush. J Hered 108:524–534. https://doi.org/10.1093/jhered/esx051
DiLeo MF, Rouse JD, Dávila JA, Lougheed SC (2013) The influence of landscape on gene flow in the eastern massasauga rattlesnake (Sistrurus c. catenatus): insight from computer simulations. Mol Ecol 22:4483–4498. https://doi.org/10.1111/mec.12411
Doherty TS, Balouch S, Bell K et al (2020) Reptile responses to anthropogenic habitat modification: a global meta-analysis. Glob Ecol Biogeogr 29:1265–1279. https://doi.org/10.1111/geb.13091
Dubey S, Brown GP, Madsen T, Shine R (2008) Male-biased dispersal in a tropical Australian snake (Stegonotus cucullatus, Colubridae). Mol Ecol 17:3506–3514. https://doi.org/10.1111/j.1365-294X.2008.03859.x
Dupont L, Grésille Y, Richard B et al (2015) Dispersal constraints and fine-scale spatial genetic structure in two earthworm species. Biol J Linn Soc 114:335–347
Durso AM, Willson JD, Winne CT (2011) Needles in haystacks: estimating detection probability and occupancy of rare and cryptic snakes. Biol Conserv 144:1508–1515. https://doi.org/10.1016/j.biocon.2011.01.020
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Fischer J, Lindenmayer DB (2007) Landscape modification and habitat fragmentation: a synthesis. Glob Ecol Biogeogr 16:265–280. https://doi.org/10.1111/j.1466-8238.2006.00287.x
Flavenot T, Fellous S, Abdelkrim J et al (2015) Impact of quarrying on genetic diversity: an approach across landscapes and over time. Conserv Genet 16:181–194. https://doi.org/10.1007/s10592-014-0650-8
Folt B, Bauder J, Spear S et al (2019) Taxonomic and conservation implications of population genetic admixture, mito-nuclear discordance, and male-biased dispersal of a large endangered snake, Drymarchon couperi. PLOS ONE 14:e0214439. https://doi.org/10.1371/journal.pone.0214439
Gardner E, Julian A, Monk C, Baker J (2019) Make the Adder Count: population trends from a citizen science survey of UK adders. Herpetol J 29:57–70. https://doi.org/10.33256/hj29.1.5770
Gauffre B, Petit E, Brodier S et al (2009) Sex-biased dispersal patterns depend on the spatial scale in a social rodent. Proc R Soc B 276:3487–3494. https://doi.org/10.1098/rspb.2009.0881
Geser S, Kaiser L, Zwahlen V, Ursenbacher S (2013) Development of polymorphic microsatellite loci markers for the Asp viper (Vipera aspis) using high-throughput sequencing and their use for other European vipers. Amphib-Reptil 34:109–113. https://doi.org/10.1163/15685381-00002861
Goslee SC, Urban DL (2007) The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw 22:1–19
Goudet J (1995) FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486. https://doi.org/10.1093/oxfordjournals.jhered.a111627
Goudet J, Jombart T (2015) hierfstat: estimation and tests of hierarchical F-statistics. R package version 0.04-22
Graitson E, Ursenbacher S, Lourdais O (2020) Snake conservation in anthropized landscapes: considering artificial habitats and questioning management of semi-natural habitats. Eur J Wildl Res. https://doi.org/10.1007/s10344-020-01373-2
Greenwood PJ (1980) Mating systems, philopatry and dispersal in birds and mammals. Anim Behav 28:1140–1162
Guerrini M, Gennai C, Panayides P et al (2014) Large-scale patterns of genetic variation in a female-biased dispersing Passerine: the importance of sex-based analyses. PLoS ONE 9:e98574. https://doi.org/10.1371/journal.pone.0098574
Guiller G, Legentilhomme J (2015) Classification de classes d’âges (nouveau-né, immature et mature) en fonction de la taille chez six espèces d’ophidiens du département de la Loire-Atlantique. Bull Soc Sci Nat Ouest Fr 37:135–142
Hardy OJ, Vekemans X (2002) spagedi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620. https://doi.org/10.1046/j.1471-8278.2002.00305.x
Hillman SS, Drewes RC, Hedrick MS, Hancock TV (2014a) Physiological vagility: correlations with dispersal and population genetic structure of amphibians. Physiol Biochem Zool 87:105–112. https://doi.org/10.1086/671109
Hillman SS, Drewes RC, Hedrick MS, Hancock TV (2014b) Physiological vagility and its relationship to dispersal and neutral genetic heterogeneity in vertebrates. J Exp Biol 217:3356–3364. https://doi.org/10.1242/jeb.105908
Hofmann S, Fritzsche P, Solhøy T et al (2012) Evidence of sex-biased dispersal in Thermophis baileyi inferred from microsatellite markers. Herpetologica 68:514–522. https://doi.org/10.1655/HERPETOLOGICA-D-12-00017
Jackson HB, Fahrig L (2012) What size is a biologically relevant landscape? Landsc Ecol 27:929–941. https://doi.org/10.1007/s10980-012-9757-9
Jackson HB, Fahrig L (2015) Are ecologists conducting research at the optimal scale? Glob Ecol Biogeogr 24:52–63. https://doi.org/10.1111/geb.12233
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Keller D, Holderegger R, van Strien MJ (2013) Spatial scale affects landscape genetic analysis of a wetland grasshopper. Mol Ecol 22:2467–2482. https://doi.org/10.1111/mec.12265
Keller D, Holderegger R, van Strien MJ, Bolliger J (2015) How to make landscape genetics beneficial for conservation management? Conserv Genet 16:503–512. https://doi.org/10.1007/s10592-014-0684-y
Kéry M (2002) Inferring the absence of a species: a case study of snakes. J Wildl Manag 66:330–338. https://doi.org/10.2307/3803165
King RB (2009) Population and conservation genetics. In: Mullin SJ, Seigel RA (eds) Snakes: ecology and conservation. Comstock Pub. Associates/Cornell University Press, Ithaca, pp 78–122
King RB, Lawson R (2001) Patterns of population subdivision and gene flow in three sympatric natricine snakes. Copeia 2001:602–614
Landguth EL, Cushman SA, Schwartz MK et al (2010) Quantifying the lag time to detect barriers in landscape genetics. Mol Ecol 19:4179–4191. https://doi.org/10.1111/j.1365-294X.2010.04808.x
Lawson Handley LJ, Perrin N (2007) Advances in our understanding of mammalian sex-biased dispersal. Mol Ecol 16:1559–1578. https://doi.org/10.1111/j.1365-294X.2006.03152.x
Lechner AM, Langford WT, Jones SD et al (2012) Investigating species–environment relationships at multiple scales: Differentiating between intrinsic scale and the modifiable areal unit problem. Ecol Complex 11:91–102. https://doi.org/10.1016/j.ecocom.2012.04.002
Legendre P, Legendre L (2012) Numerical ecology, 3rd English edn. Elsevier, Amsterdam
Li X-Y, Kokko H (2019) Sex-biased dispersal: a review of the theory. Biol Rev 94:721–736. https://doi.org/10.1111/brv.12475
Lorioux S, Angelier F, Lourdais O (2016) Are glucocorticoids good indicators of pregnancy constraints in a capital breeder? Gen Comp Endocrinol 232:125–133. https://doi.org/10.1016/j.ygcen.2016.04.007
Lourdais O, Gartner GE, Brischoux F (2014) Ambush or active life: foraging mode influences haematocrit levels in snakes. Biol J Linn Soc 111:636–645
Lowe WH, Allendorf FW (2010) What can genetics tell us about population connectivity? Mol Ecol 19:3038–3051. https://doi.org/10.1111/j.1365-294X.2010.04688.x
Luiselli L (1993) High philopatry can produce strong sexual competition in male adders, Vipera berus. Amphib-Reptil 14:310–311. https://doi.org/10.1163/156853893X00516
Madsen T, Shine R (1992a) Sexual competition among brothers may influence offspring sex ratio in snakes. Evolution 46:1549–1552. https://doi.org/10.2307/2409957
Madsen T, Shine R (1992b) Determinants of reproductive success in female adders, Vipera berus. Oecologia 92:40–47
Madsen T, Shine R (1993) Costs of reproduction in a population of European adders. Oecologia 94:488–495
Madsen T, Shine R, Loman J, Håkansson T (1993) Determinants of mating success in male adders, Vipera berus. Anim Behav 45:491–499. https://doi.org/10.1006/anbe.1993.1060
Madsen T, Shine R, Olsson M, Wittzell H (1999) Restoration of an inbred adder population. Nature 402:35–35. https://doi.org/10.1038/46944
Madsen T, Stille B, Shine R (1996) Inbreeding depression in an isolated population of adders Vipera berus. Biol Conserv 75:113–118. https://doi.org/10.1016/0006-3207(95)00067-4
Madsen T, Ujvari B, Olsson M (2004) Novel genes continue to enhance population growth in adders (Vipera berus). Biol Conserv 120:145–147. https://doi.org/10.1016/j.biocon.2004.01.022
Manier MK, Arnold SJ (2005) Population genetic analysis identifies source-sink dynamics for two sympatric garter snake species (Thamnophis elegans and Thamnophis sirtalis). Mol Ecol 14:3965–3976. https://doi.org/10.1111/j.1365-294X.2005.02734.x
Matthysen E (2012) Multicausality of dispersal: a review. In: Clobert J, Baguette M, Benton TG, Bullock JM (eds) Dispersal ecology and evolution, 1st edn. Oxford University Press, Oxford, pp 3–18
Meister B, Ursenbacher S, Baur B (2012) Grass snake population differentiation over different geographic scales. Herpetologica 68:134–145. https://doi.org/10.1655/HERPETOLOGICA-D-11-00036.1
Metzger C, Ferchaud A-L, Geiser C, Ursenbacher S (2011) New polymorphic microsatellite markers of the endangered meadow viper (Vipera ursinii) identified by 454 high-throughput sequencing: when innovation meets conservation. Conserv Genet Resour 3:589–592. https://doi.org/10.1007/s12686-011-9411-x
Moore JA, Miller HC, Daugherty CH, Nelson NJ (2008) Fine-scale genetic structure of a long-lived reptile reflects recent habitat modification. Mol Ecol 17:4630–4641. https://doi.org/10.1111/j.1365-294X.2008.03951.x
Motro U (1991) Avoiding inbreeding and sibling competition: the evolution of sexual dimorphism for dispersal. Am Nat 137:108–115
Moya Ó, Mansilla P-L, Madrazo S et al (2015) APHIS: A new software for photo-matching in ecological studies. Ecol Inform 27:64–70. https://doi.org/10.1016/j.ecoinf.2015.03.003
Mullin SJ, Seigel RA (eds) (2009) Snakes: ecology and conservation. Comstock Pub. Associates/Cornell University Press, Ithaca
Neumeyer R (1987) Density and seasonal movements of the adder (Vipera berus L. 1758) in a subalpine environment. Amphib-Reptil 8:259–276. https://doi.org/10.1163/156853887X00306
Newbold T, Hudson LN, Hill SLL et al (2015) Global effects of land use on local terrestrial biodiversity. Nature 520:45–50. https://doi.org/10.1038/nature14324
Niebuhr BBS, Wosniack ME, Santos MC et al (2015) Survival in patchy landscapes: the interplay between dispersal, habitat loss and fragmentation. Sci Rep. https://doi.org/10.1038/srep11898
Öckinger E, Schweiger O, Crist TO et al (2010) Life-history traits predict species responses to habitat area and isolation: a cross-continental synthesis. Ecol Lett 13:969–979. https://doi.org/10.1111/j.1461-0248.2010.01487.x
Oden NL, Sokal RR (1986) Directional autocorrelation: an extension of spatial correlograms to two dimensions. Syst Zool 35:608–617
Oksanen J, Blanchet FG, Kindt R, et al (2016) vegan: Community Ecology Package. R package version 2.3–4
Paradis E (2010) pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26:419–420. https://doi.org/10.1093/bioinformatics/btp696
Pernetta AP, Allen JA, Beebee TJC, Reading CJ (2011) Fine-scale population genetic structure and sex-biased dispersal in the smooth snake (Coronella austriaca) in southern England. Heredity 107:231–238. https://doi.org/10.1038/hdy.2011.7
Perrin N, Mazalov V (2000) Local competition, inbreeding, and the evolution of sex-biased dispersal. Am Nat 155:116–127
Pike DA (2016) Conservation management. In: Dodd CKJ (ed) Reptile ecology and conservation: a handbook of techniques. Oxford University Press, Oxford, pp 419–435
Powers RP, Jetz W (2019) Global habitat loss and extinction risk of terrestrial vertebrates under future land-use-change scenarios. Nat Clim Chang 9:323–329. https://doi.org/10.1038/s41558-019-0406-z
Prestt I (1971) An ecological study of the viper Vipera berus in southern Britain. J Zool 164:373–418. https://doi.org/10.1111/j.1469-7998.1971.tb01324.x
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Prunier JG, Kaufmann B, Fenet S et al (2013) Optimizing the trade-off between spatial and genetic sampling efforts in patchy populations: towards a better assessment of functional connectivity using an individual-based sampling scheme. Mol Ecol 22:5516–5530. https://doi.org/10.1111/mec.12499
R Core Development Team (2020) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna
Reading CJ, Luiselli LM, Akani GC et al (2010) Are snake populations in widespread decline? Biol Lett 6:777–780. https://doi.org/10.1098/rsbl.2010.0373
Rivera PC, Gardenal CN, Chiaraviglio M (2006) Sex-biased dispersal and high levels of gene flow among local populations in the argentine boa constrictor, Boa constrictor occidentalis. Austral Ecol 31:948–955. https://doi.org/10.1111/j.1442-9993.2006.01661.x
Ronce O (2007) How does it feel to be like a rolling stone? Ten questions about dispersal evolution. Annu Rev Ecol Evol Syst 38:231–253. https://doi.org/10.1146/annurev.ecolsys.38.091206.095611
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Rousset F (2000) Genetic differentiation between individuals. J Evol Biol 13:58–62. https://doi.org/10.1046/j.1420-9101.2000.00137.x
Saint Girons H (1980) Biogéographie et évolution des vipères européennes. C R Soc Biogéogr 496:146–172
Santos X, Brito JC, Sillero N et al (2006) Inferring habitat-suitability areas with ecological modelling techniques and GIS: a contribution to assess the conservation status of Vipera latastei. Biol Conserv 130:416–425. https://doi.org/10.1016/j.biocon.2006.01.003
Sheldon S, Bradley C (1989) Identification of individual adders (Vipera berus) by their head markings. Herpetol J 1:392–395
Sokal (1986) Spatial data analysis and historical processes. In: Diday E, International Symposium on Data Analysis and Informatics, Institut National de Recherche en Informatique et en Automatique (eds) Data analysis and informatics, IV. North-Holland, Amsterdam, pp 29–43
Stow AJ, Sunnucks P, Briscoe DA, Gardner MG (2001) The impact of habitat fragmentation on dispersal of Cunningham’s skink (Egernia cunninghami): evidence from allelic and genotypic analyses of microsatellites. Mol Ecol 10:867–878. https://doi.org/10.1046/j.1365-294X.2001.01253.x
Sumner J, Rousset F, Estoup A, Moritz C (2001) ‘Neighbourhood’size, dispersal and density estimates in the prickly forest skink (Gnypetoscincus queenslandiae) using individual genetic and demographic methods. Mol Ecol 10:1917–1927. https://doi.org/10.1046/j.0962-1083.2001.01337.x
Sumner J, Jessop T, Paetkau D, Moritz C (2004) Limited effect of anthropogenic habitat fragmentation on molecular diversity in a rain forest skink, Gnypetoscincus queenslandiae. Mol Ecol 13:259–269. https://doi.org/10.1046/j.1365-294X.2003.02056.x
Trochet A, Courtois EA, Stevens VM et al (2016) Evolution of sex-biased dispersal. Q Rev Biol 91:297–320. https://doi.org/10.1086/688097
Ursenbacher S, Monney J-C, Fumagalli L (2009) Limited genetic diversity and high differentiation among the remnant adder (Vipera berus) populations in the Swiss and French Jura Mountains. Conserv Genet 10:303–315. https://doi.org/10.1007/s10592-008-9580-7
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Viitanen P (1967) Hibernation and seasonal movements of the viper, Vipera berus berus (L.), in southern Finland. Ann Zool Fenn 4:472–546
Weatherhead PJ, Madsen T (2009) Linking behavioral ecology to conservation objectives. In: Mullin SJ, Seigel RA (eds) Snakes: ecology and conservation. Comstock Pub. Associates/Cornell University Press, Ithaca, pp 149–171
Webb JK, Shine R (1997) A field study of spatial ecology and movements of a threatened snake species, Hoplocephalus bungaroides. Biol Conserv 82:203–217
Acknowledgements
This work was funded by the Ministère de l’Enseignement Supérieur et de la Recherche (Ph.D grant to DF) and the Service du Patrimoine Naturel du Département d’Ille-et-Vilaine which manages the Natura 2000 site of “Vallée du Canut” as an “Espace Naturel Sensible” (ENS). We address special thanks to all the field workers: Régis Morel (Chargé de mission Bretagne-Vivante), Manon Vasseur, Mélaine Rouault, Morgan Le Vot and Maxime Spagnol. We thank Valerie Zwahlen (laboratory technician) for carrying out the genetic analyses at the University of Basel. We also thank Juan José Robledo-Arnuncio and Jérôme Prunier for advice about statistics. Finally, we thank Jean-Pierre Vacher, Sydney Hope and the two anonymous reviewers for their helpful comments on the manuscript.
Funding
This work was financially supported by the Ministère de l’Enseignement supérieur et de la recherche (Ph.D grant to DF) and the Service du patrimoine naturel du Département d’Ille-et-Vilaine which manages the Natura 2000 site of “Vallée du Canut” as an “Espace naturel sensible” (ENS).
Author information
Authors and Affiliations
Contributions
DF and FY applied for grants for the project. DF designed the experiments with the help of SU and OL. DF and OL participated to field work and enhanced its implementation. SU supervised the laboratory work. DF and SU analysed the data. DF wrote most of the manuscript. AB, DF, FY, OL and SU discussed the results, the analyses, and commented or modified the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Ethical approval
This study was performed in accordance with laws relative to capture, transport and animal welfare on V berus (DREAL permit # N°2016-07).
Consent for publication
We confirm that neither the article nor portions of it have been previously published elsewhere. The manuscript is not under consideration for publication in another journal. All authors consent to the publication of the manuscript in ELA, should the article be accepted by the Editor-in-chief upon completion of the refereeing process.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
François, D., Ursenbacher, S., Boissinot, A. et al. Isolation-by-distance and male-biased dispersal at a fine spatial scale: a study of the common European adder (Vipera berus) in a rural landscape. Conserv Genet 22, 823–837 (2021). https://doi.org/10.1007/s10592-021-01365-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-021-01365-y