
Abstract
Accumulating evidences indicate that long non-coding RNAs (lncRNAs) play important roles in several biological processes and dysregulated lncRNAs are involved in different kinds of cancer and are associated with carcinogenesis, metastasis, and prognosis of cancer. The role of a new lncRNA LOC100130476 in gastric cardia adenocarcinoma (GCA) has remained unknown. The present study investigated the role and methylation status of LOC100130476 in the pathogenesis of GCA, and further evaluated the potential prognostic role of LOC100130476 in GCA. Significant downregulation of LOC100130476 was detected in SGC-7901 and BGC-823 cell lines and primary GCA tissues. Methylation frequency of LOC100130476 was gradually increased from exon 1 to exon 2 both in tumor tissues and corresponding normal tissues; however, methylation status of region 1 closing to the transcription start site was more tumor-specific among the three regions examined. The findings of the association between LOC100130476 expression, methylation and TNM stage, pathological differentiation, and GCA patients’ survival further identified the role of LOC100130476 as a tumor suppressor gene. Furthermore, the hypermethylation of LOC100130476 was also detected in peripheral white blood cells of GCA cases. Thus, LOC100130476 may be act as a tumor suppressor gene in GCA carcinogenesis and aberrant methylation at the CpG sites near the transcription start site within exon 1 may be critical for gene silencing. In addition, aberrant methylation of LOC100130476 in peripheral white blood cells and GCA tissues may be used as a potential valuable biomarker in GCA diagnosis and prognosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Based on GLOBOCAN estimates, about 14.1 million new cancer cases and 8.2 million deaths occurred in 2012 worldwide. In less developed countries, liver and gastric cancers among males are leading causes of cancer death [1]. Arising in the transformation zone between the esophagus and the stomach, gastric cardia adenocarcinoma (GCA), formerly registered as gastric cancer or esophageal cancer, has been diagnosed independently in very recent years due to the distinct epidemiological and biological characteristics and improvement in classification. Although a steady decline in noncardia gastric cancer incidence and mortality rates has been observed in the majority of more developed countries in Northern America and Europe and also in areas with historically high rates, including several countries in Asia (Japan, China, and Korea), Latin America (Colombia and Ecuador), and Europe (Ukraine), the incidence rate for GCA has risen steeply in North America and Western European countries during the past few decades [1]. In China, GCA is one of the prevalent fatal malignancies and shares very similar geographic distribution with esophageal squamous cell carcinoma (ESCC), especially in the highest incidence areas of ESCC in the world, such as in some counties bordering Gansu, Henan, Hebei, and Shanxi Provinces [2, 3]. The incidence rate of GCA increased gradually over the past few years and a variable proportion of GCA cases strongly point to upper gastrointestinal cancers (UGIC) family history especially in the high incidence regions [4]. However, unlike chronic infection with H. pylori is the strongest identified risk factor for noncardia gastric cancer [5], the exact pathogenesis, mechanisms, and related risk factors for GCA tumorigenesis have not been well characterized.
Over the last few decades, intensive investigations have focused on the role of protein-coding genes in the pathogenesis of cancer. However, the ~20,000 protein-coding genes only represent <2 % of the total genome sequence. At least 90 % of the genome are actively transcribed into non-coding RNAs (ncRNAs), implicating the potential significant regulatory roles of ncRNAs in complex organisms [6]. NcRNAs are loosely grouped into two major classes based on transcript size; small ncRNAs and long non-coding RNAs (lncRNAs) [7]. As a new class of ncRNAs, with length ranged from 200 nt to 100 kb, lncRNA has recently caught increasing attention. Although they were initially thought to be spurious transcriptional noise resulting from low RNA polymerase fidelity, recent studies have demonstrated that lncRNAs may play important roles in several biological processes such as chromatin modification, cellular differentiation, regulation of gene expression, mRNA decay, cell cycle regulation, and nuclear-cytoplasmic trafficking [8, 9]. Function in the cytoplasm or nucleus, or in both, lncRNAs are highly conserved and may act as key regulators in gene regulation via various ways in the cytoplasm and the nucleus [10]. Increasing evidences have demonstrated that dysregulated lncRNAs are involved in many complicated human diseases including cancer [11]. LncRNAs have cell-type-specific expression patterns and dysregulated lncRNAs in various types of human cancer may be used as potential early tumor diagnostic, metastatic or prognostic markers and molecular-targeted therapy sites in the future [12]. Although the number of articles about lncRNAs increased greatly, only a handful of lncRNAs have been characterized. For example, dysregulated HOTAIR may affect epigenetic information and provide a cellular growth advantage and result in the progressive and uncontrolled growth of tumor [13]. Another lncRNA, MALAT1 regulates the invasive potential of metastatic tumor cells through interaction with and modulation of splicing factor proteins and act as an oncogenic lncRNA [14].
More recently, mounting evidences have shown that lncRNAs are involved in epigenetic processes [15]. DNA methylation was one of the first epigenetic alterations identified in cancer. The cancer epigenome is characterized by genome-wide hypomethylation and specific CpG islands hypermethylation. An lncRNA named AS1DHRS4 is transcribed from the locus of the DHRS4 gene and recruits DNA methyltransferases and other factors to the DHRS4 gene cluster, inducing DNA methylation at the DHRS4L2 promoter region [16]. Aberrant hypermethylation of specific CpG islands can lead to the silencing of tumor suppressor genes involved in key cellular pathways [17]. Promoter hypermethylation may be partly responsible for the downregulation of some lncRNAs including MEG3, H19, SRHC and CAHM in carcinoma [18–21]. To gain insight into the expression profile of lncRNA in GCA, we investigated the microarray expression profile of lncRNA transcriptome in 5 GCA tissues and 5 paired corresponding normal tissues. The GeneChip® Human Gene 2.1 ST Array (Affymetrix, CA, USA) was adopted to detect the expression profile of long intergenic non-coding RNA transcripts (lincRNA) and protein-coding mRNAs (including >11,000 long intergenic non-coding transcripts and >30,000 protein coding transcripts). Among these lincRNAs, 471 were consistently up-regulated and 879 were consistently downregulated (fold change ≥2.0, P < 0.05) (Supplementary Fig. 1). Among these differentially expressed lincRNAs, LOC100130476, which demonstrated 3.25 fold reduced expression in GCA tissues, is located on 6q23.3 (GRCh 38/hg38 database, from chr6: 137823670 to 137868233, NCBI: NR_049793.1) (Fig. 1a) and has three CpG islands spanning the regions from +208 to +1539 bp determined by MethPrimer program. We hypothesized that dysregulation of LOC100130476 may be associated with the occurrence and development of GCA and aberrant CpG island methylation may be partly responsible for the inactivation of LOC100130476 in GCA. In the present study, we investigated the function and methylation status of LOC100130476 in GCA, and further elucidate the role of LOC100130476 in the progression and prognosis of GCA.

The expression and effect of LOC100130476 on proliferation and invasion in human gastric cancer cell lines. a Genomic structure of LOC100130476. Its position on the chromosome is based on the GRCh38/hg38 database. b Relative expression of LOC100130476 in SGC-7901 and BGC-823 cell lines treated or untreated with 5-Aza-dC. * Compared with untreated cells, P < 0.05. c Overexpression of LOC100130476 inhibited SGC-7901 cell proliferation, as determined by CCK-8 assay. * Compared with empty vector, P < 0.05. d Overexpression of LOC100130476 inhibited SGC-7901 cell invasiveness, as determined by transwell invasion assay. * Compared with empty vector, P < 0.05
Materials and methods
Patients and specimens
Surgical primary GCA tissues and corresponding adjacent normal tissues were obtained from 121 GCA cases, which were all inpatients for surgical treatment in the Fourth Affiliated Hospital, Hebei Medical University between the years of 2007 and 2009. The GCA patients included 96 males and 25 females, mean age 57.8 years (ranged from 36 to 76 years). All gastric cardia carcinomas were adenocarcinomas with their epicenters at the gastroesophageal junction, i.e. from 1 cm above until 2 cm below the junction between the end of the tubular esophagus and the beginning of the saccular stomach [22]. All of the tissues were divided into two parallel parts, one part were frozen and stored at −80 °C until genomic DNA and RNA was extracted, the other part were formalin-fixed and paraffin-embedded. Fifty healthy controls were recruited from the cancer-screening program for early detection of upper gastrointestinal tumors in the same area and during the same period. All of the healthy controls were not diagnosed with cancer or precancerous lesions. Five ml of venous blood from GCA cases and healthy controls was drawn in Vacutainer tubes containing EDTA and stored at 4 °C. All study subjects were ethnically homogeneous Han nationality and residents of Hebei Province and its surrounding regions and informed consent was obtained from all recruited subjects. All subjects were interviewed by professional interviewers for their age, gender, histopathological diagnosis, and UGIC family history. Individuals with at least one first-degree relative or at least two second-degree relatives having esophageal/cardia/gastric cancer were defined as having family history of UGIC. Information on clinicopathologic characteristics of GCA cases was available from hospital recordings and pathological diagnosis. Recurrence and survival data were ascertained through the Tumor Registry and Hospital chart review (Supplementary Table 1). All patients were evaluated for recurrent disease by examinations of tumor markers or by diagnostic imaging, including computed tomography, ultrasonography, magnetic resonance imaging, and endoscopy, every 3–6 months. The patients were followed up for a minimum period of 1.5 years (range 1.5–6.5 years), with a median follow-up of 5.2 years for survivors. Fourteen patients were lost to follow up. The study was approved by the Ethics Committee of the Fourth Affiliated Hospital, Hebei Medical University.
Cell culture, treatment, and transfection
Human gastric cancer cell lines SGC-7901 and BGC-823 were cultured in RPMI 1640 media (Invitrogen, Carlsbad, CA, USA) and supplemented with 10 % fetal bovine serum (Invitrogen, Carlsbad, CA, USA). Cells were seeded at a low density and incubated for 24 h prior to treatment with DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (5-Aza-dC). The gastric cancer Cells (2 × 105/mL) were treated with 5 μmol/L 5-Aza-dC (Sigma, St Louis, MO, USA) for 72 h and medium containing 5-Aza-dC was changed every 24 h. Control cells received no drug treatment. For overexpression of LOC100130476, the sequence of LOC100130476 was synthesized and subcloned into pcDNA3.1 (Invitrogen, Carlsbad, CA, USA). Exponentially grown SGC-7901 cells cultured in 24-well plates were transfected with LOC100130476 expression plasmid (pcDNA3.1-LOC100130476) or the empty vector (pcDNA3.1-EV) as control using FuGENE HD Transfection Reagent (Roche, Swiss) according to the manufacturer’s instructions. Transfection efficiency was tested by qRT-PCR.
Cell proliferation assay
The proliferation of transfected SGC-7901 Cells was measured by cell-counting kit-8 (CCK-8) assay. The cells were seeded into 96-well plates (5 × 103 cells per well) and 10 μl of CCK8 (Dojindo, Japan) was added to the 100 ul cul-tured cells for 2 h incubation. The absorbance of each well was detected at a wavelength of 450 nm (A450). The experiments were performed in triplicate.
Cell invasion assay
The invasiveness of transfected SGC-7901 Cells was evaluated in 24-well transwell chambers (Corning, Kennebunk, ME, USA). The transwell chambers were coated with 20 μL Matrigel and incubated at 37 °C for 1 h. The 5 × 104 cells/well at 48 h after transfected were seeded in the upper chambers, meanwhile the RPMI 1640 medium with 10 % FBS was injected into the lower chambers. After harvest, the bottom of the insert was stained with hematoxylin. The number of cells invaded through the membrane to the lower surface was counted in five microscopic fields (at × 100 magnification) per filter. The experiments were repeated in triplicate.
LOC100130476 expression via quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR) assay
Total RNA was extracted from gastric cancer cell lines, frozen GCA tumor, and corresponding normal tissues using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s recommended protocol and quantified by UV absorbance at 260–280 nm. Two μg RNA was used to synthesize single-stranded cDNA using the advantage RT-for-PCR kit (Clontech, Palo Alto, CA, USA) with random primer as primer which was recommended in the protocol. The cDNA from each sample was used as regular qRT-PCR template. The primers and reaction conditions for LOC100130476 were listed in Supplementary Table 2. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an internal control. Power SYBR Green PCR Master Mix (Life Technology, Foster City, CA, USA) was used as amplification reaction mixture according to the manufacturer’s instructions. The melting curve analysis was performed to confirm PCR product specificity and the expression levels of LOC100130476 were normalized with GAPDH using the 2−ΔΔCT method [23]. All the samples were run in triplicate.
DNA extraction and sodium bisulfite treatment
Genomic DNA was extracted from frozen GCA tumor and corresponding normal tissues using a simplified Proteinase K digestion method. Five ml of venous blood from 121 GCA cases (before surgery) and 50 healthy controls was drawn in Vacutainer tubes containing EDTA and stored at 4 °C. Genomic DNA was extracted within 1 week after blood collection by using proteinase K digestion method. To examine the DNA methylation patterns, 1 µg of genomic DNA was bisulfite modified using Epitect Fast Bisulfite Conversion Kits (Qiagen, Germany) according to the manufacturer’s instructions. After the standard sodium bisulfite DNA modification, unmethylated cytosine residues were converted to thymine, whereas methylated cytosine residues were retained as cytosine at CpG sites.
Methylation status of every CpG site of LOC100130476 via bisulfite genomic sequencing (BGS) method
The methylation status of every CpG site in the three CpG islands of LOC100130476 was verified by BGS assay in 4 matched tissue sets showing tumor-specific reduction of transcript levels. For BGS assay, primers were designed to recognize sodium bisulfite converted DNA and encompassing three CpG islands within the human LOC100130476 gene exon 1 to exon 2 regions (CpG island 1: from +193 to +467 bp, CpG island 2: from +550 to +1049 bp, CpG island 3: from +1315 to +1592 bp). Fifty ng of bisulfite-modified DNA was subjected to PCR amplification and the PCR products were cloned into pGEM-T vectors (Promega, Madison, WI, USA) and 8 to 10 clones of each specimen were sequenced by automated fluorescence-based DNA sequencing.
Methylation analysis of LOC100130476 via bisulfite conversion-specific and methylation-specific polymerase chain reaction (BS-MSP) method
The methylation status of LOC100130476 in GCA tissues and venous blood was then determined by BS-MSP method as previous description using bisulfite treated genomic DNA [24]. Three regions located in three CpG islands of LOC100130476 (region 1 located in exon 1: from +245 to +413 bp, region 2 located in intron 1: from +649 to +830 bp, region 3 located in exon 2: from +1418 to +1546 bp) were respectively analyzed. The primers and reaction conditions were listed in Supplementary Table 2. Genomic DNA, methylated in vitro by CpG methyltransferase (Sss I) following the manufacturer’s directions (New England BioLabs, Beverly, MA, USA), was used as a positive control and water blank was used as a negative control. BS-MSP products were analyzed on 2 % agarose gel with ethidium bromide staining, and were determined to have methylation if a visible band was observed in the methylation reaction. Tthe reactions were performed in duplicate with each of the samples.
Statistical analysis
Statistical analysis was performed with SPSS19.0 software package (SPSS Company, Chicago, Illinois, USA). The real-time RT-PCR results were expressed as the mean ± S.D. Student’s t test was used to compare the means between different groups. Chi square test was used to analyze the status of gene methylation between different groups. Relationships between variables were tested by Spearman correlation analysis. Survival curves were constructed by using the Kaplan–Meier method and the Log-rank or the Breslow tests were used as needed for the univariate comparison of LOC100130476 expression and methylation categories. Cox’s multivariate test applied in a stepwise forward method was used to adjust for potentially confounding variables (e.g., stage and UGIC family history) and to evaluate the role of LOC100130476 as independent predictors of patients’ prognosis. All statistical tests were two sided; and P < 0.05 was considered to be statistically significant for all tests.
Results
Downregulation of LOC100130476 in gastric cancer cell lines
We first analyzed the expression level of LOC100130476 in gastric cancer cell lines SGC-7901 and BGC-823 by qRT-PCR. As shown in Fig. 1b, the expression level of LOC100130476 was remarkably reduced in SGC-7901 and BGC-823 cells. After treatment with DNA methyltransferase inhibitor 5-Aza-dC, the expression of LOC100130476 was significantly upregulated, indicating the important role of aberrant methylation in the inactivation of LOC100130476 in gastric cancer cell lines.
Inhibition effect of LOC100130476 on SGC-7901 cell proliferation and invasiveness
As shown in Fig. 1c, cell proliferation assay showed that overexpression of LOC100130476 significantly inhibited the proliferation of SGC-7901 cells. Moreover, overexpression of LOC100130476 significantly suppressed the invasiveness of SGC-7901 (Fig. 1d).
Frequent inactivation of LOC100130476 in primary GCA tissues
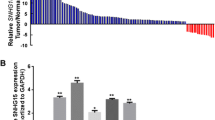
We further detected the expression of LOC100130476 in 121 pairs of GCA tissues and corresponding normal tissues by qRT-PCR method. As shown in Fig. 2a, the expression level of LOC100130476 in GCA tumor tissues was remarkably reduced compared to corresponding normal tissues (P < 0.01). There were 47 patients (38.8 %) whose LOC100130476 expression levels in their tumor tissues were less than 50 % of that in corresponding normal tissues.

The expression of LOC100130476 in GCA tissues. a Relative expression of LOC100130476 in normal tissues and corresponding GCA tumor tissues. * P < 0.05. b Relative expression of LOC100130476 in different subgroups. * P < 0.05
Association of LOC100130476 reduction with tumor progression
To explore the significance of LOC100130476 reduction in GCA tumor tissues, the LOC100130476 expression was further investigated in subgroups. As shown in Fig. 2b, when stratified for clinicopathologic characteristics, the expression level of LOC100130476 in GCA tumor tissues was not associated with age, gender, depth of invasion, and family history of UGIC. However, LOC100130476 expression was associated with TNM stage, pathological differentiation, LN metastasis, and distant metastasis or recurrence (P < 0.05).
Aberrant methylation of LOC100130476 in GCA tumor tissues
The MethPrimer program was used to determine the distribution of CpG islands of LOC100130476. As shown in Fig. 3a, three CpG islands spanning the regions from +208 to +1539 bp are found within exon 1 to exon 2. The methylation status of the CpG sites in the three CpG islands was verified by BGS assay (Fig. 3b). As shown in Fig. 3b, 4 tumor tissues with low LOC100130476 level showed frequent hypermethylation of the three CpG islands of the gene, whereas methylation was rare in the corresponding normal tissues.

Methylation status of LOC100130476 in GCA tissues. a Schematic structure and sequence of LOC100130476 CpG islands. Three CpG islands of LOC100130476 are shown and the BS-MSP regions analyzed is indicated. Exons in upper case and introns in lower case. b High-resolution mapping of the methylation status of every CpG site in the LOC100130476 CpG islands by BGS assay in four GCA tissues and corresponding normal tissues. Each CpG site is shown at the top row as an individual number. Percentage methylation was determined as percentage of methylated cytosines from 8 to 10 sequenced colonies. The color of circles for each CpG site represents the percentage of methylation. c The methylation status of three regions of LOC100130476 determined by BS-MSP analysis in GCA tumor tissues. m methylated; u unmethylated. d Relative expression of LOC100130476 in the tumor tissues with and without methylation of the three regions. * P < 0.05
The primers of BS-MSP method were then designed according to the distribution of methylated CpG sites determined by BGS assay and the methylation status of LOC100130476 was successfully performed in all tissue specimens (Fig. 3c). As shown in Table 1, of primary tumor tissues and corresponding normal tissues, hypermethylation was observed in 62.0 % (75/121) and 4.9 % (6/121) at region 1, 68.6 % (83/121) and 17.4 % (21/121) at region 2, 79.3 % (96/121) and 42.1 % (51/121) at region 3, respectively. Among the three regions, methylation status of region 1 was more tumor-specific. When stratified for clinicopathologic characteristics, the methylation status of region 1 was associated with TNM stage, pathological differentiation, LN metastasis, and distant metastasis or recurrence; the methylation status of region 2 was only associated with TNM stage; while the methylation status of region 3 was not associated with any clinicopathologic features (Table 2).
Aberrant methylation of LOC100130476 in peripheral white blood cells
The methylation status of three regions of LOC100130476 was further detected in the peripheral white blood cell DNA of 121 GCA cases and 50 healthy controls. The methylation frequency of 3 regions of LOC100130476 in GCA WBC was 42.1 % (51/121), 45.5 % (55/121), and 61.2 % (74/121), respectively; while aberrant methylation of 3 regions of LOC100130476 was not detected in the WBC of healthy controls. Interestingly, we found that hypermethylation of 3 regions of LOC100130476 which occurred in GCA WBC also occurred in the corresponding tumor tissues. When stratified for clinicopathologic characteristics, the methylation status of region 1 of LOC100130476 in GCA WBC was associated with TNM stage, pathological differentiation, and distant metastasis or recurrence; while the methylation status of region 2 and region 3 of LOC100130476 in GCA WBC was not associated with any clinicopathologic features (Table 2).
Association between LOC100130476 expression and methylation status
The association between LOC100130476 expression and methylation was further detected in GCA tissues. As shown in Fig. 3d, the expression level of LOC100130476 in GCA tissues with hypermethylation of region 1 and region 2 was significantly lower than that with unmethylation of both regions, and the effect was more significant at region 1 (P < 0.05).
Survival analysis of LOC100130476 in GCA
As shown in Fig. 4a, b LOC100130476 expression was positively correlated with GCA patients’ survival, while LOC100130476 region 1 methylation was inversely correlated with GCA patients’ survival. GCA patients with relative low LOC100130476 expression (the expression level of LOC100130476 in the tumor tissues was less than 50 % of that in matched corresponding normal tissues) or region 1 hypermethylation of LOC100130476 demonstrated significant poorer 5-year survival rates as opposed to the GCA patients with relative high LOC100130476 expression or region 1 unmethylation of the gene (P < 0.05; Log-rank test). GCA cases with both low expression and region 1 hypermethylation of LOC100130476 showed worse survival rates compared to the GCA cases with both relative high expression and region 1 unmethylation of LOC100130476 (Fig. 4c). When TNM stage and LOC100130476 expression or region 1 methylation status were combined to analysis, GCA patients in stage III and IV, with low expression or region 1 hypermethylation of LOC100130476 were most likely to develop metastatic disease and also demonstrated the worse survival (Fig. 4d, e).

Kaplan–Meier univariate survival analysis of LOC100130476 expression and methylation in GCA cases. a Showing a direct correlation between low LOC100130476 expression and poor patient survival. b Showing consistently a direct correlation between LOC100130476 region 1 methylation and poor patient survival. c GCA cases with simultaneous low expression and region 1 methylation of LOC100130476 showing poor patient survival. d GCA cases in stage III and IV and with low expression of LOC100130476 showing poor patient survival. e GCA cases in stage III and IV and with region 1 methylation of LOC100130476 showing poor patient survival
Cox multivariate analysis was done using LOC100130476 expression, methylation, tumor stage, as well as other confounding variables such as patient age, gender and UGIC family history. As shown in Table 3, TNM stages, LOC100130476 expression, and region 1 methylation were independently associated with GCA patients’ survival.
Discussion
As a new class of the non-coding RNAs, although tens of thousands of lncRNAs are encoded in the human genome, only a few of them have been characterized in detail. Dysregulation of lncRNAs have been found in a number of carcinomas, acting as tumor suppressors or oncogenes, thus suggesting the potential critical roles of lncRNAs as prospective novel diagnostic or therapeutic targets during tumor occurrence and development. Here, we found a new lncRNA LOC100130476 that may play tumor suppressor gene role in GCA progression. To our best knowledge, the roles and epigenetic inactivation mechanism of LOC100130476 on GCA tumor progression and prognosis have not been investigated and clarified so far. In the present study, we demonstrated that abnormal reduction of LOC100130476 was a tumor-specific phenomenon. Methylation frequency of LOC100130476 was gradually increased from exon 1 to exon 2 both in tumor tissues and corresponding normal tissues; however, Region 1 methylation status was more tumor-specific among the three regions examined, suggesting that the critical region for LOC100130476 silencing may lies in region 1 which located in exon 1 closing to the transcription start site. The most obvious correlation between LOC100130476 expression and region 1 methylation was further verified that aberrant methylation of the CpG sites closing to the transcription start site was more prone to influence its transcription. The findings of the association between LOC100130476 expression, methylation and TNM stage, pathological differentiation, and GCA patients’ survival further identified the role of LOC100130476 as a tumor suppressor gene and may be used as a useful marker of GCA tumor progression and prognosis. Furthermore, the hypermethylation of LOC100130476 in peripheral white blood cells of GCA cases further supported the potential for gene-specific methylation measured in peripheral WBC as a biomarker of cancer risk.
The downregulation of LOC100130476 in gastric cancer cell lines and GCA tumor tissues suggested its tumor suppressive role in GCA. As one of the epigenetic mechanisms, DNA methylation may play important roles in gene expression and regulation. However, with the steady progress of such research, the methylation frequency of the same gene in carcinomas was found to be different in different laboratories. DNA methylation may have tumor type specificity may be one of the reasons; the different CpG sites which they focused on may be another reason. In the present study, we found that methylation frequency of LOC100130476 was gradually increased from exon 1 to exon 2 both in tumor tissues and corresponding normal tissues, however, methylation near the transcription start sites (region 1, located in exon 1) appeared to be mostly cancer-specific. These findings are essentially the same as those for RUNX3 and RASSF2 in gastric cancer and DAPK in colorectal and gastric cancer [25–27]. They found that the 5′ and 3′ outskirt regions of the CpG island was more susceptible to methylation in both the primary tumor tissues and corresponding normal tissues than regions near or spanning the transcription start site which was more critical for gene inactivation. One attractive hypothesis is that methylation of the edge—for example, at simple repetitive sequences or at transposons such as Alu and B1 may serves as a trigger for spreading methylation into the centre of the region. Satoh et al. found that Alu sequences situated about 500 bp upstream of the DAPK 5′ CpG island are densely methylated in most of samples, indicating the role for repetitive elements as a trigger for 5′ CpG island methylation [27]. Furthermore, the lower frequency of methylation near the transcription start site suggests that physiological mechanisms may exist to protect the regions from undergoing methylation. For example, RUNX3 has two promoters and the distal promoter does not contain a CpG island, whereas the proximal promoter has a CpG island that includes binding sites for Sp1. Sp1 elements locate upstream and downstream of the transcription start site and have been shown to protect CpG islands from methylation [25]. Thus the pattern of spread of methylation, initially at the outskirts of CpG islands and then progressing to regions critical for gene silencing, might be common to various types of methylation-related gene silencing. Since methylation frequencies are influenced by the location of MSP primers, it is important for the determination of the critical CpG sites in a given tumor suppressor or tumor-related gene.
Circulating RNA in plasma or serum has become an emerging field for non-invasive diagnostic applications. At present, several up-regulated lncRNAs in carcinomas have been characterized as potential tumor biomarkers in plasma. For example, plasma level of H19 was significantly higher in gastric cancer patients compared with normal controls [28], upregulation of lncRNA HULC was detected in the plasma of hepatocellular carcinoma [29], plasma level of lncRNA PCAT18 was increased incrementally from healthy individuals to those with localized and metastatic prostate cancer [30], plasma levels of POU3F3 was significantly higher in ESCC patients [31]. Recent years, mounting evidences have demonstrated that aberrant DNA hypermethylation of tumor suppressor genes may be observed not only at tumor tissue level but also in plasma and white blood cells of tumor patients. Peripheral blood cell DNA may contain epigenetic information, which is a valuable predictive marker of an individual’s risk of developing cancer [32]. Aberrant hypermethylation of BRCA1, IGFII, SFRP1 in WBC has been detected in breast cancer and colon cancer patients [33–36]. However, few study investigated DNA methylation status of circulating lncRNA for early detection of tumor patients. Methylated CAHM sequences were detected in the plasma DNA of 40/73 (55 %) of colorectal cancer patients [21]. In the present study, hypermethylation of CpG sites in three regions of LOC100130476 was detected in peripheral WBC of GCA patients, the methylation status of region 1 of LOC100130476 in GCA WBC was associated with TNM stage, pathological differentiation, and distant metastasis or recurrence, while aberrant methylation of three regions of LOC100130476 was not detected in the peripheral WBC of healthy controls. These results support the potential for cancer-specific hypermethylation of certain lncRNA measured in peripheral WBC as a diagnostic biomarker for cancer.
The expression and methylation status in region 1 of LOC100130476 were found to be closely related to TNM stage, pathological differentiation, LN metastasis, and distant metastasis or recurrence in the present study, indicating the potential role of LOC100130476 in GCA progression. In further survival analysis, LOC100130476 expression and region 1 methylation was found to be correlated with GCA patients’ survival. GCA patients with simultaneous low expression and hypermethylation of LOC100130476 demonstrated poor patient survival. Thus, LOC100130476 expression and hypermethylation may be used as valuable candidate prognostic markers for GCA.
In conclusion, the downregulation of LOC100130476 may be involved in the progression of GCA and aberrant methylation at the CpG sites near the transcription start site within exon 1 may be critical for gene silencing. In addition, aberrant methylation of LOC100130476 in peripheral WBC and GCA tissues may be used as a potential biomarker in GCA diagnosis and prognosis.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A (2015) Global cancer statistics, 2012. CA Cancer J Clin 65:87–108
Yang L (2006) Incidence and mortality of gastric cancer in China. World J Gastroenterol 12:17–20
Ma JL, Zhang L, Brown LM, Li JY, Shen L, Pan KF, Liu WD, Hu Y, Han ZX, Crystal-Mansour S, Pee D, Blot WJ, Fraumeni JF Jr, You WC, Gail MH (2012) Fifteen-year effects of Helicobacter pylori, garlic, and vitamin treatments on gastric cancer incidence and mortality. J Natl Cancer Inst 104:488–492
Gao Y, Hu N, Han X, Giffen C, Ding T, Goldstein A, Taylor P (2009) Family history of cancer and risk for esophageal and gastric cancer in Shanxi, China. BMC Cancer 9:269
Plummer M, Franceschi S, Vignat J, Forman D, de Martel C (2015) Global burden of gastric cancer attributable to pylori. Int J Cancer 136:487–490
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Röder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See LH, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guigó R, Gingeras TR (2012) Landscape of transcription in human cells. Nature 489:101–108
Sun M, Kraus WL (2015) From discovery to function: the expanding roles of long noncoding RNAs in physiology and disease. Endocr Rev 36:25–64
Mercer TR, Dinger ME, Mattick JS (2009) Long non-coding RNAs: insights into functions. Nat Rev Genet 10:155–159
Ponting CP, Oliver PL, Reik W (2009) Evolution and functions of long noncoding RNAs. Cell 136:629–641
Zhang K, Shi ZM, Chang YN, Hu ZM, Qi HX, Hong W (2014) The ways of action of long non-coding RNAs in cytoplasm and nucleus. Gene 547:1–9
Qiu MT, Hu JW, Yin R, Xu L (2013) Long noncoding RNA: an emerging paradigm of cancer research. Tumour Biol 34:613–620
Qi P, Du X (2013) The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod Pathol 26:155–165
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464:1071–1076
Tano K, Mizuno R, Okada T, Rakwal R, Shibato J, Masuo Y, Ijiri K, Akimitsu N (2010) MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS Lett 584:4575–4580
Beckedorff FC, Amaral MS, Deocesano-Pereira C, Verjovski-Almeida S (2013) Long non-coding RNAs and their implications in cancer epigenetics. Biosci Rep 33(4):e00061
Li Q, Su Z, Xu X, Liu G, Song X, Wang R, Sui X, Liu T, Chang X, Huang D (2012) AS1DHRS4, a head-to-head natural antisense transcript, silences the DHRS4 gene cluster in cis and trans. Proc Natl Acad Sci USA 109:14110–14115
Esteller M (2007) Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet 16:R50–R59
Zhou Y, Zhang X, Klibanski A (2012) MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol 48:R45–R53
Gao T, He B, Pan Y, Gu L, Chen L, Nie Z, Xu Y, Li R, Wang S (2014) H19 DMR methylation correlates to the progression of esophageal squamous cell carcinoma through IGF2 imprinting pathway. Clin Transl Oncol 16:410–417
Zheng H, Yang S, Yang Y, Yuan SX, Wu FQ, Wang LL, Yan HL, Sun SH, Zhou WP (2015) Epigenetically silenced long noncoding-SRHC promotes proliferation of hepatocellular carcinoma. J Cancer Res Clin Oncol 141:1195–1203
Pedersen SK, Mitchell SM, Graham LD, McEvoy A, Thomas ML, Baker RT, Ross JP, Xu ZZ, Ho T, LaPointe LC, Young GP, Molloy PL (2014) CAHM, a long non-coding RNA gene hypermethylated in colorectal neoplasia. Epigenetics 9:1071–1082
Siewert JR, Stein HJ (1998) Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 85:1457–1459
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408
Sasaki M, Anast J, Bassett W, Kawakami T, Sakuragi N, Dahiya R (2003) Bisulfite conversion-specific and methylation-specific PCR: a sensitive technique for accurate evaluation of CpG methylation. Biochem Biophys Res Commun 309:305–309
Homma N, Tamura G, Honda T, Matsumoto Y, Nishizuka S, Kawata S, Motoyama T (2006) Spreading of methylation within RUNX3 CpG island in gastric cancer. Cancer Sci 97:51–56
Endoh M, Tamura G, Honda T, Homma N, Terashima M, Nishizuka S, Motoyama T (2005) RASSF2, a potential tumour suppressor, is silenced by CpG island hypermethylation in gastric cancer. Br J Cancer 93:1395–1399
Satoh A, Toyota M, Itoh F, Kikuchi T, Obata T, Sasaki Y, Suzuki H, Yawata A, Kusano M, Fujita M, Hosokawa M, Yanagihara K, Tokino T, Imai K (2002) DNA methylation and histone deacetylation associated with silencing DAP kinase gene expression in colorectal and gastric cancers. Br J Cancer 86:1817–1823
Zhou X, Yin C, Dang Y, Ye F, Zhang G (2015) Identification of the long non-coding RNA H19 in plasma as a novel biomarker for diagnosis of gastric cancer. Sci Rep 5:11516
Xie H, Ma H, Zhou D (2013) Plasma HULC as a promising novel biomarker for the detection of hepatocellular carcinoma. Biomed Res Int 2013:136106
Crea F, Watahiki A, Quagliata L, Xue H, Pikor L, Parolia A, Wang Y, Lin D, Lam WL, Farrar WL, Isogai T, Morant R, Castori-Eppenberger S, Chi KN, Wang Y, Helgason CD (2014) Identification of a long non-coding RNA as a novel biomarker and potential therapeutic target for metastatic prostate cancer. Oncotarget 5:764–774
Tong YS, Wang XW, Zhou XL, Liu ZH, Yang TX, Shi WH, Xie HW, Lv J, Wu QQ, Cao XF (2015) Identification of the long non-coding RNA POU3F3 in plasma as a novel biomarker for diagnosis of esophageal squamous cell carcinoma. Mol Cancer 14:3
Terry MB, Delgado-Cruzata L, Vin-Raviv N, Wu HC, Santella RM (2011) DNA methylation in white blood cells: association with risk factors in epidemiologic studies. Epigenetics 6:828–837
Widschwendter M, Apostolidou S, Raum E, Rothenbacher D, Fiegl H, Menon U, Stegmaier C, Jacobs IJ, Brenner H (2008) Epigenotyping in peripheral blood cell DNA and breast cancer risk: a proof of principle study. PLoS ONE 3:e2656
Kaaks R, Stattin P, Villar S, Poetsch AR, Dossus L, Nieters A, Riboli E, Palmqvist R, Hallmans G, Plass C, Friesen MD (2009) Insulin-like growth factor-II methylation status in lymphocyte DNA and colon cancer risk in the Northern Sweden Health and Disease cohort. Cancer Res 69:5400–5405
Iwamoto T, Yamamoto N, Taguchi T, Tamaki Y, Noguchi S (2011) BRCA1 promoter methylation in peripheral blood cells is associated with increased risk of breast can-cer with BRCA1 promoter methylation. Breast Cancer Res Treat 129:69–77
Wong EM, Southey MC, Fox SB, Brown MA, Dowty JG, Jenkins MA, Giles GG, Hopper JL, Dobrovic A (2011) Constitutional methylation of the BRCA1 promoter is specifically associated with BRCA1 mutation-associated pathology in early-onset breast cancer. Cancer Prev Res (Phila) 4:23–33
Acknowledgments
We thank the patients for taking part in this study. Supported by Grants from the National Natural Science Foundation (No. 81472335), Natural Science Foundation of Hebei Province (No. H2015206196), and Grants from the Ordinary University Considerable Distinctive Subjects Foundation of Hebei Province (No. 200552).
Author information
Authors and Affiliations
Corresponding author
Additional information
Wei Guo and Zhiming Dong contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Guo, W., Dong, Z., Shi, Y. et al. Methylation-mediated downregulation of long noncoding RNA LOC100130476 in gastric cardia adenocarcinoma. Clin Exp Metastasis 33, 497–508 (2016). https://doi.org/10.1007/s10585-016-9794-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-016-9794-x