
Abstract
Background
H19 gene has been proved to be essential for human tumor growth which contains CpG rich regions. Imprinted gene expression in many cancers is usually associated with the function of methylation. We performed this study to better understand wether H19 DMR methylation correlates to the progression of esophageal squamous cell carcinoma through IGF2 imprinting pathway.
Methods
LOI of IGF2 was detected in 276 samples, which were determined as heterozygote with ApaI polymorphism in exon 9 of IGF2 by PCR–RFLP and RT-PCR–RFLP. Methylation status of H19 DMR in informative samples was analyzed by bisulfite sequencing PCR. IGF2 expression was examined by real-time PCR and IHC.
Results
208 ESCC patients were informative for ApaI polymorphism. 92 tumor and 30 normal tissues showed IGF2 LOI. Methylation status of H19 CBS6 was higher in patients with IGF2 LOI compared to patients with IGF2 MOI (p < 0.05). IGF2 expression in patients with IGF2 LOI was higher than patients with IGF2 MOI (p < 0.05) which was correlated with lymph node involvement, neoplastic grade and metastasis (p < 0.05).
Conclusions
Our results suggested that H19 CBS6 hypermethylation is related to the LOI of IGF2 which usually leads to an overexpression of IGF2, playing important roles in the occurrence, development as well as metastasis of ESCC. Therefore, H19 CBS6 methylation potentially represents a novel clinically relevant epigenetic marker to identify individuals at increased risk for the occurrence, progression and prognosis of ESCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the past decades, esophageal cancer (EC) is one of the most common cancers in the world with an extremely poor prognosis due to late tumor presentation and rapid progression, causing more than 40, 000 deaths each year [1]. The most prevalent type of EC is esophageal squamous cell carcinoma (ESCC), and China is among the highest risk areas [1, 2]. Epigenetic, genetic, or posttranscriptional factors may be important in ESCC development and the identification of such factors may improve the prevention of this malignancy and reduce mortality.
The H19 gene, located on the chromosome 11p15.5 and 135 kb toward the telomere, is oppositely imprinted and is expressed only from the maternal allele which has been proved to be essential for human tumor growth [3, 4]. Moreover, the H19 DMR located 4 kb upstream of the transcription start of H19, which contains six zinc finger CTCF binding sites (CBS), acts as methylation-sensitive insulator [5, 6]. These changes have been observed in growth disorders such as Beckwith–Wiedemann and Silver–Russell syndrome [7, 8].
Genomic imprinting is an epigenetic control in which one allele is expressed and other allele silenced based on the parental (maternal or paternal) origin of DNA. IGF2 displays genomic imprinting and is paternally imprinted in most tissues called maintenance of imprinting (MOI) [9]. Loss of genomic imprinting (LOI) is seen when there is relaxation of the silenced allele and monoallelic expression changes to biallelic expression. After being first reported in Wilms’ tumor [10], LOI of IGF2 has been reported in several human solid tumors, including gastrointestinal tract carcinomas [11–14]. There are two reports of IGF2 LOI in patients with ESCC, showing the prevalence of IGF2 LOI to range from 54 % (7 of 13) to 21 % (5 of 24) [11, 12]. Moreover, in esophageal adenocarcinoma, Zhao R and his colleagues have shown that LOI of IGF2 is present not only in cancer tissue, but also consistently in adjacent, histologically normal colorectal mucosa suggesting that LOI of IGF2 could be an early epigenetic event in carcinogenesis with potential clinical application for early diagnosis and screening [11]. To date, although IGF2 LOI in patients with ESCC has been reported in many studies, the mechanism of IGF2 LOI and IGF2 expression in ESCC is still unclear.
Some studies have suggested that imprinted genes are associated with CpG rich regions that have allele-specific DNA methylation known as differentially methylated regions (DMRs) [15, 16]. Human IGF2 located on the chromosome 11p15.5 was close to H19 [11], but the relationship between them had never been studied in ESCC. So we performed this study to better determine whether H19 DMR methylation correlates to the progression of esophageal squamous cell carcinoma through IGF2 imprinting pathway.
Materials and methods
Study population
Matched sets of paraffin-embedded adjacent histologically normal esophageal tissues and ESCC tumor samples were obtained from 276 patients at Nanjing First Hospital Affiliated to Nanjing Medical University. The patients’ age varied from 39 to 82 years old, with a mean age of 62. The study was conducted according to our institutional guidelines under the supervision of the Institutional Review Board of Nanjing First Hospital Affiliated to Nanjing Medical University, and written informed consent was obtained from all participants.
Characteristics of patients and their pathology were recorded, including age, sex, body mass index (BMI), depth of invasion (T1: The cancer has grown through the muscularis mucosa and extends into the submucosa; T2: The cancer has grown through the submucosa and extends into the muscularis propria (thick outer muscle layer); T3: The cancer has grown through the muscularis propria and into the outermost layers of the colon or rectum but not through them. It has not reached any nearby organs or tissues; or T4: The cancer has grown through the serosa or through the wall of the colon or rectum and attached to or invades into nearby tissues or organs), lymph node involvement, neoplastic grading (Grade) of all the patients which were based on the guidelines of the American Joint Commission on Cancer, and the TNM Classification of Malignant Tumors (TNM) determined by the guidelines of the International Union Against Cancer (UICC).
Extraction of nucleic acids
Genomic DNA and total RNA were extracted simultaneously from paraffin-embedded tissues using E.Z.N.A.FFPE DNA Kit and E.Z.N.A.FFPE RNA Kit (omega, USA) following the manufacturers’ protocol. To avoid the possible DNA contamination, RNA was digested with RNase free DNaseI and cleaned with the RNA easy MinElute Cleanup Kit (Qiagen).
Identification of IGF2 polymorphism
Exon 9 of the human IGF2 gene is used to evaluate the IGF2 polymorphism by PCR which is known as an ApaI digestion single nucleotide polymorphism (rs680) [17]. The primer sequences were as follows: IGF2apFw: 5′-CCTTGGACTTTGAGTCAA ATT-3′ and IGF2apRev: 5′-GGTCGT GCCAATTACATTTCA-3′. The PCR conditions were initial denaturation for 5 min at 94 °C, followed by 35 PCR cycles of denaturing at 94 °C for 30 s, annealing at 55 °C for 30 s, and elongation at 72 °C for 30 s. A 2 % agarose gel electrophoresis was used to identify the 292 bp PCR product. The PCR products were then digested with ApaI at 30 °C overnight.
Analysis of IGF2 imprinting status
cDNA of the samples informative for ApaI polymorphism was synthesized by reverse transcription of 2 mg total RNA with random primers and Superscript Reverse Transcriptase III. Then, PCR was run on cDNA with the same primers used to determine informative status. The RT-PCR products were also digested by ApaI and electrophoresed on 2 % agarose gel. LOI of IGF2 was noted if there was biallelic transcription, which shows both 292 and 229 bp fragments. To assure no DNA contamination, a minus-RT tube, which contained all components except the reverse transcriptase, was set up for each sample.
Methylation analysis of H19 CBS6
Genomic DNA from informative samples was treated with bisulfite (EpiTect Plus DNA Bisulfite Kit 59124) to convert unmethylated cytosines to uracils, whereas methylated cytosines are unaffected. Bisulfite-treated DNA was subsequently amplified using the forward primer 5′-TGGGTATTTTTGGAGGTTTTTTT-3′ and reverse primer 5′-ATAAATATCCTATTCCCAAATAA-3′ [18]. These primers allowed the amplification of both the methylated and the unmethylated alleles by spanning a region with 18 differentially methylated CpGs. The region corresponded to GenBank nucleotides 7881–8100 (accession no. AF125183). The PCR condition was as follows: denatured at 95 °C for 15 min, followed by 95 °C for 30 s, 57 °C for 30 s and 72 °C for 20 s respectively for 50 cycles, and elongated at 72 °C for 5 min. Each PCR product (5 μl) was analyzed on 2 % agarose gel and the remaining 20 μl was purified using QIAquick purification kit (Qiagen) to be sequenced using the forward primer, and the sequencing results were analyzed using CpG viewer, a software tool for DNA methylation available by free download from http://dna.leeds.ac.uk/cpgviewer/download.php.
Analysis of IGF2 mRNA expression by RT-PCR
Expression levels of IGF2 in both tumor and normal tissues were determined by real-time quantitative RT-PCR. The primer of IGF2 was the same as before: IGF2 Fw: 5′-CCTTGGACTTTGAGTCAAATT-3′ and IGF2 Rev: 5′-GGTCGTGCCAATTACATTTCA-3′. We selected the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene, a housekeeping gene as an internal control: GAPDH Fw: 5′-GTCAACGGATTTGGTCGTATT-3′ and GAPDH Rev: 5′-AGTCTTCTGGGTGGCAGTGAT-3′. Relative quantification of gene expression changes was recorded after normalizing for 18S expression, computed using the \( 2^{{ - \Updelta \Updelta C_{\text{T}} }} \) method (user manual 2, ABI Prism 7700 SDS). ∆∆C T was calculated by the following formula:
Analysis of IGF2 expression by immunohistochemistry
Three 4-mm thick unstained tissue sections were cut from each paraffin-embedded tissue block for immunohistochemical (IHC) analysis of IGF2. The sections were deparaffinized in xylene and subsequently rehydrated in sequential ethanols (100–70 %). After washing three times with 10 mM phosphate-buffered saline (PBS) (pH 7.4), antigen retrieval was performed by heating in a microwave at 95 °C for 15 min, and then washing twice in PBS for 5 min. The sections were treated with peroxidase blocking solution (Dako Japan, Kyoto, Japan) for 5 min, and incubated with a primary antibody for 60 min at room temperature. The primary antibodies used were rabbit anti-human IGF2 antibody. We used Image-Pro Plus6.0 to score the expression of IGF2 in each sections, a software from http://www.mediacy.com/index.aspx?page=IP_Premier&gclid=CIy68orZnbMCFchU4godYEEAEg.
Statistical analysis
Differences in IGF2 imprinting status, IGF2 expression and the methylation status of H19 DMR were compared with a Chi-square test, Student’s t test or Mann–Whitney test. All analyses were performed using SPSS 18.0 for Windows and p values of less than 0.05 were considered significant.
Result
IGF2 imprinting status
Genotype analysis of DNA samples from the entire series of 276 ESCC patients revealed 208 (75.4 %) to be informative (Fig. 1a). Of these, 92 (44.2 %) tumor and 78 (37.5 %) normal tissues showed IGF2 LOI (Fig. 1b). Significant difference was not found between LOI in tumor and normal tissues by Chi-square test (p = 0.163). Selected clinicopathological features of the 208 patients informative for IGF2 polymorphism are shown in Table 1. No significant differences were found between patient groups (n = 92) whose tumors demonstrated IGF2 LOI and those (n = 116) that had normal imprinting status with respect to gender, age, cigarette, BMI, family history of ESCC, depth of invasion, tumor differentiation or stage (p > 0.05). However, patients with IGF2 LOI had higher degree of lymph node involvement and appeared to metastasis much more than patients with IGF2 MOI (p < 0.05).

IGF2 imprinting status analysis. a PCR product digested by ApaI on 2 % agarose gel, homozygous samples were marked as A or B; heterozygous (informative) samples were marked as A/B. b Gel photo of ApaI digested RT-PCR production 2 % agarose gel, sample with biallelic expression (LOI) was labeled as LOI; samples with monoallelic expression (imprinted) were labeled as A/A or B/B
Association between H19 CBS6 methylation and IGF2 LOI
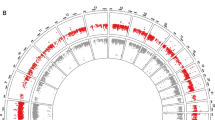
We developed bisulfite-PCR-pyrosequencing assay to measure DNA methylation at H19 CBS6 (the sixth CTCF binding site) (Fig. 2). All of the patients’ tumor tissues and normal tissues were analyzed. The comparison of H19 CBS6 methylation status between IGF2 LOI patients and IGF2 MOI patients is shown in Table 2, which demonstrated that the methylation status of H19 CBS6 were higher in patients with IGF2 LOI compared to patients with IGF2 MOI not only in tumor tissues but also in normal tissues (Fig. 3, p < 0.05). 72 tumor and 36 normal tissues of IGF2 LOI patients showed hypermethylation (>50 %) which was also higher than in IGF2 MOI patients (p < 0.05).

Methylation analysis of H19 CBS6. Variable methylation of H19 CBS6 in ESCC, Genomic DNA was treated with sodium bisulfite, and then was PCR-amplified and subcloned before sequencing. Ten clones were sequenced for each sample. Each line represents a separate clone. Filled circle methylated CpG sites; open circle unmethylated CpG sites. a Methylation analysis of H19 CBS6 in IGF2 LOI patients’ tumor tissues which showed hypermethylation; b Methylation analysis of H19 CBS6 in IGF2 MOI patients’ tumor tissues which showed normal methylation

Comparison of H19 DMR methylation status between IGF2 LOI patients and IGF2 MOI patients. The methylation status of H19 CBS6 was higher in patients with IGF2 LOI (56.57 ± 12.96 %) compared to patients with IGF2 MOI (46.62 ± 11.02 %, p = 0.004). The methylation status of H19 CBS6 was higher in IGF2 LOI normal tissues (49.09 ± 11.16 %) than normal tissues (41.03 ± 9.61 %, p = 0.007)
IGF2 expression in IGF2 LOI
IGF2 mRNA expression from adjacent histologically normal esophageal tissues and ESCC tumor samples was detected by real-time PCR, which demonstrated that IGF2 mRNA levels in tumor tissues from patients with IGF2 LOI (5.77 ± 5.01) were higher than patients with IGF2 MOI (2.37 ± 1.76, p = 0.017). IHC analysis was also used to detect IGF2 protein expression which confirmed that IGF2 protein levels in tumor tissues from patients with IGF2 LOI (0.064 ± 0.017) were higher than patients with IGF2 MOI (0.046 ± 0.010, p = 0.045, Fig. 4). Furthermore, the IGF2 protein expression in IGF2 LOI patients’ normal tissues was also detected by IHC (0.038 ± 0.018, Fig. 2) which showed that IGF2 levels in IGF2 LOI patients’ tumor tissues were higher than normal tissues (p = 0.02).

Analysis of IGF2 expression by immunohistochemistry. Immunohistochemical staining of IGF2 expression in ESCC tumor tissues and normal tissues from patients with IGF2 LOI and tumor tissues from patients with IGF2 MOI (×200). IGF2 protein levels in tumor tissues from patients with IGF2 LOI (0.064 ± 0.017) were higher than patients with IGF2 MOI (0.046 ± 0.010, p = 0.046) and also higher than normal tissues (0.038 ± 0.018, p = 0.02). IGF2 protein levels in normal tissues from IGF2 LOI patients with tumor metastasis (0.048 ± 0.016) were higher than those without metastasis (0.026 ± 0.011, p = 0.03). The yellow color shown by the marker was marked as IGF2 expression. a The IGF2 protein expression of tumor tissues from IGF2 LOI patients; b the IGF2 protein expression of tumor tissues from IGF2 MOI patients; c the IGF2 protein expression of normal tissues from IGF2 LOI patients without tumor metastasis: d the IGF2 protein expression of normal tissues from IGF2 LOI patients with tumor metastasis
IGF2 mRNA expression and clinicopathological features
Selected clinicopathological features of the 92 IGF2 LOI patients were used to compare with IGF2 mRNA expression (Table 3). Though differences were not found in comparison with depth of invasion and TNM, IGF2 mRNA expression were significantly related to lymph node involvement, neoplastic grade and metastasis (p < 0.05). Patients with higher lymph node involvement or neoplastic grade usually showed higher IGF2 mRNA levels (p < 0.05). Patients with tumor metastasis usually demonstrated higher IGF2 mRNA levels than those without tumor metastasis (p < 0.05).
Discussion
In the present study, we demonstrated that LOI of IGF2 was presented in 44.2 % ESCC patients’ tumor tissues and 37.5 % normal tissues. Patients with LOI of IGF2 in their tumor had deeper degree of lymph node involvement and were more likely to occur metastasis than those with MOI of IGF2. Then, we found that H19 CBS6 hypermethylation is related to LOI of IGF2 in human ESCC and IGF2 LOI usually leads to an overexpression of IGF2 which was correlated with lymph node involvement, neoplastic grade and tumor metastasis. To the best of our knowledge, the association between H19 CBS6methylation and IGF2 LOI in ESCC has never been reported in other studies.
After being first reported in Wilms’ tumor, LOI of IGF2 has been reported in several human solid tumors and suggested to be a reliable risk factor [19]. ESCC as the most common type of EC which is the 8th most common cancer and the 6th most frequent cause of cancer worldwide has also been proved to be associated with IGF2 LOI [11]. In this study, the frequency of IGF2 LOI in ESCC patients was 44.2 % which was consistent with earlier studies [11, 12]. But whether LOI of IGF2 was a risk factor for esophageal neoplasma, its presence should be proven to precede the development of disease and remain constant. Therefore, the correlation between IGF2 LOI and tumor differentiation or stage was studied which showed that IGF2 LOI in ESCC was not associated with either tumor differentiation or stage (p > 0.05). However, we found that ESCC patients with IGF2 LOI usually showed higher IGF2 expression than patients with IGF2 MOI which was correlated with lymph node involvement, neoplastic grade and tumor metastasis. We also found that IGF2 LOI was correlated with lymph node involvement and tumor metastasis. Both indicated that IGF2 LOI usually leads to the IGF2 overexpression which played an important role in ESCC tumor progression.
Earlier studies had suggested that the IGF axis comprised two growth factors (IGF1 and IGF2), several high affinity binding proteins (IGFBPs1-6), IGFBP-related proteins, and two principal cell membrane receptors (IGF1R and IGF2R) homologous to the insulin receptor (IR) [20, 21]. In many tumor systems, IGFs had been found to stimulate cell proliferation, protect cells from apoptosis, and be associated with tumor metastasis mediated via paracrine or autocrine mechanisms [22]. Increased IGF2 expression resulting from LOI of IGF2 had been reported in murine models [23], and in patients with Wilms’ tumor [24] and colorectal cancer [25], likely a consequence of abnormal activation of the normally silenced maternal allele. Therefore, it was possible that a similar mechanism as described underlies ESCC metastasis, whereby LOI of IGF2 results in increased IGF2 expression, with potential modulation of IGF1R and other signaling pathways.
Epigenetic abnormalities identified subsequently include global genomic hypomethylation [26], promoter hypermethylation of CpG islands [27], and LOI [28], or loss of the normal parent of origin-dependent gene silencing. IGF2 LOI was an epigenetic alteration in human cancer that had been proved to be related to the aberrant methylation of H19 DMR. The function of H19 DMR methylation on IGF2 LOI had been observed in many studies which suggested the enhancer competition model that IGF2 and H19 promoters compete on the same chromosome for a shared enhancer, and access of the maternal IGF2 allele to this enhancer is blocked by the H19 DMR when unmethylated, likely because of the insulator activity of CTCF binding to the unmethylated H19 DMR. However, H19 DMR methylation blocks CTCF binding and permits the enhancers to access the IGF2 promoters resulting in IGF2 expression [13, 14].. Hyang-Min Byun’s study in bladder cancer and Sachin Bhusari’s study in prostate also approve the function of aberrant methylation of H19 DMR on IGF2 LOI [29, 30]. But Cui’s study on CRC with LOI demonstrated that the H19 DMR was hypomethylated on both alleles [19]. In this study, ESCC patients with IGF2 LOI usually showed higher methylation status of H19 CBS6, one of the six zinc finger CTCF binding sites in H19 DMR, and more IGF2 expression than patients with IGF2 MOI both in tumor and normal tissues which was consistent with the enhancer competition model.
In summary, our results suggested that H19 CBS6 hypermethylation is related to the LOI of IGF2 which usually leads to an overexpression of IGF2, playing important roles in the occurrence, development as well as metastasis of ESCC. Therefore, H19 CBS6 methylation potentially represents a novel clinically relevant epigenetic marker to identify individuals at increased risk for the occurrence, progression and prognosis of ESCC.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90.
Parkin DM, Bray FI, Devesa SS. Cancer burden in the year 2000. The global picture. Eur J Cancer. 2011;37(Suppl 8):S4–66.
Luo M, Li Z, Wang W, Zeng Y, Liu Z, Qiu J. Upregulated H19 contributes to bladder cancer cell proliferation by regulating ID2 expression. FEBS J. 2013;280(7):1709–16.
Sorin V, Ohana P, Gallula J, Birman T, Matouk I, Hubert A, et al. H19-promoter-targeted therapy combined with gemcitabine in the treatment of pancreatic cancer. ISRN Oncol. 2012;2012:351750.
Cui H, Onyango P, Brandenburg S, Wu Y, Hsieh CL, Feinberg AP. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002;62:6442–6.
Murphy SK, Huang Z, Wen Y, Spillman MA, Whitaker RS, Simel LR, et al. Frequent IGF2/H19 domain epigenetic alterations and elevated IGF2 expression in epithelial ovarian cancer. Mol Cancer Res. 2006;4:283–92.
Gicquel C, Rossignol S, Cabrol S, Houang M, Steunou V, Barbu V, et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver–Russell syndrome. Nat Genet. 2005;37:1003–7.
Eggermann T, Eggermann K, Schonherr N. Growth retardation versus overgrowth: Silver–Russell syndrome is genetically opposite to Beckwith–Wiedemann syndrome. Trends Genet. 2008;24:195–204.
Ohlsson R, Hedborg F, Holmgren L, Walsh C, Ekstrom TJ. Overlapping patterns of IGF2 and H19 expression during human development: biallelic IGF2 expression correlates with a lack of H19 expression. Development. 1994;120(2):361–8.
Ogawa O, Eccles MR, Szeto J, McNoe LA, Yun K, Maw MA, et al. Relaxation of insulin-like growth factor II gene imprinting implicated in Wilms’ tumour. Nature (Lond.). 1993;362:749–51.
Mori M, Inoue H, Shiraishi T. Relaxation of insulin-like growth factor 2 gene imprinting in esophageal cancer. Int J Cancer. 1996;68:441–6.
Xu W, Fan H, He X, Zhang J, Xie W. LOI of IGF2 is associated with esophageal cancer and linked to methylation status of IGF2 DMR. J Exp Clin Cancer Res. 2006;25:543–7.
Cruz-Correa M, Cui H, Giardiello FM. Loss of imprinting of insulin growth factor II gene: a potential heritable bio-marker for colon neoplasia predisposition. Gastroenterology. 2004;126:964–70.
Liou JM, Wu MS, Lin JT. Loss of imprinting of insulin-like growth factor II is associated with increased risk of proximal colon cancer. Eur J Cancer. 2007;43:1276–82.
Leighton PA, Ingram RS, Eggenschwiler J, Efstratiadis A, Tilghman SM. Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature. 1995;375(6526):34–9.
Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405(6785):486–89.
Cui H, Horon IL, Ohlsson R, Hamilton SR, Feinberg AP. Loss of imprinting in normal tissue of colorectal cancer patients with microsatellite instability. Nat Med. 1998;4:1276–80.
Boissonnas CC, Abdalaoui HE, Haelewyn V, Fauque P, Dupont JM, Gut I, et al. Specific epigenetic alterations of IGF2-H19 locus in spermatozoa from infertile men. Eur J Hum Genet. 2010;18(1):73–80.
Cui H. Loss of imprinting of IGF2 as an epigenetic marker for the risk of human cancer. Dis Markers. 2007;23:105–12.
Yu H, Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst. 2000;92:1472–89.
Furstenberger G, Senn HJ. Insulin-like growth factors and cancer. Lancet Oncol. 2002;2:298–302.
Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20–47.
Sakatani T, Kaneda A, Iacobuzio-Donahue CA. Loss of imprinting of Igf2 alters intestinal maturation and tumori-genesis in mice. Science. 2005;307:1976–8.
Ravenel JD, Broman KW, Perlman EJ. Loss of imprinting of insulin-like growth factor-II (IGF2) gene in distinguishing specific biologic subtypes of Wilms tumor. J Natl Cancer Inst. 2001;93:1698–703.
Woodson K, Flood A, Green L. Loss of insulin-like growth factor-II imprinting and the presence of screen-detected colorectal adenomas in women. J Natl Cancer Inst. 2004;96:407–10.
Feinberg AP, Gehrke CW, Kuo KC, Ehrlich M. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res. 1988;48:1159–61.
Baylin SB, Hoppener JW, de Bustros A, Steenbergh PH, Lips CJ, Nelkin BD. DNA methylation patterns of the calcitonin gene in human lung cancers and lymphomas. Cancer Res. 1986;46:2917–22.
Rainier S, Johnson LA, Dobry CJ, Ping AJ, Grundy PE, Feinberg AP. Relaxation of imprinted genes in human cancer. Nature (Lond.). 1993;362:747–9.
Byun HM, Wong HL, Birnstein EA, Wolff EM, Liang G, Yang AS. Examination ofIGF2 and H19 loss of imprinting in bladder cancer. Cancer Res. 2007;67(22):10753–8.
Bhusari S, Yang B, Kueck J, Huang W, Jarrard DF. Insulin-like growth factor-2 (IGF2) loss of imprinting marks a field defect within human prostates containing cancer. Prostate. 2011;71(15):1621–30.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
T. Gao and B. He contributed equally to this work.
Rights and permissions
About this article
Cite this article
Gao, T., He, B., Pan, Y. et al. H19 DMR methylation correlates to the progression of esophageal squamous cell carcinoma through IGF2 imprinting pathway. Clin Transl Oncol 16, 410–417 (2014). https://doi.org/10.1007/s12094-013-1098-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-013-1098-x