
Abstract
Angiogenesis is a highly regulated physiological process that has been studied in considerable detail given its importance in several chronic pathologies. Many endogenous factors and hormones intervene in the regulation of angiogensis and classical as well as targeted drugs have been developed for its control. Angiogenesis inhibition has come off the bench and entered into clinical application for cancer therapy, particularly for metastatic disease. While the clinical benefit is currently in terms of months, preclinical data suggest that novel drugs and drug combinations could lead to substantial improvement. The many targets of endogenous angiogenesis inhibitors reflect the complexity of the process; in contrast, current clinical therapies mainly target the vascular endothelial growth factor system. Cancer chemopreventive compounds can retard tumor insurgence and delay or prevent metastasis and many of these molecules hinder angiogenesis, a mechanism that we termed angioprevention. Angiopreventive drugs appear to prevalently act through the inhibition of the pro-inflammatory and anti-apoptotic player NFκB, thus contrasting inflammation dependent angiogenesis. Relatively little is known concerning the effects of these angiogenesis inhibitors on gene expression of endothelial cells, the main target of many of these molecules. Here we provide an exhaustive list of anti-angiogenic molecules, and summarize their effects, where known, on the transcriptome and functional genomics of endothelial cells. The regulation of specific genes can be crucial to preventive or therapeutic intervention. Further, novel targets might help to circumvent resistance to anti-angiogenic therapy. The studies we review are relevant not only to cancer but also to other chronic degenerative diseases involving endothelial cells, such as cardiovascular disorders, diabetes, rheumatoid arthritis and retinopaties, as well as vessel aging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tumors are the second cause of death after cardiovascular/cerebrovascular diseases in industrialized nations, and cancer incidence is rapidly increasing among developing nations. Notwithstanding massive efforts and investments in improving cancer therapy, most of the progress made towards reducing overall mortality has been achieved through early diagnosis. For most cancer patients with metastatic disease, the improvements made in therapeutic efficacy in recent years can be measured on an average of months. Most current therapeutic strategies are based largely on directly killing tumor cells with cytotoxic agents, a traditional approach that inherently harbors extensive systemic side effects, now increasingly used in combination with more targeted therapies. In contrast to oncology, the mortality rates for cardiovascular disease have been declining over the last decades, a success attributable in large part to an active prevention approach. If we are to gain further improvement on treatment of cancer patients, we need to: (1) target the tumor microenvironment, by interrupting the tumor-host interactions that fuel tumor growth and metastatic spread, and (2) apply more effective prevention approaches. One approach that fits both of these categories is targeting tumor angiogenesis [1].
During development, vessels are formed by proliferation of endothelial progenitor cells (EPCs) that are derived from hematopoietic stem cells of the bone marrow (vasculogenesis). Tumor vascularization depends, however, mainly on sprouting from pre-existing vessels in the vicinity of the tumor through the release of the strongly angiogenic growth factors such as VEGF and bFGF (angiogenesis). Hence, tumor induced angiogenesis does not rely on EPC activation but rather on reactivation of quiescent endothelial cells. The incidence of tumors correlates with age. Endothelial cells of elder subjects may have accumulated many genotoxic insults since almost all carcinogens pass in the blood before they are destroyed in the liver or the kidney. Endothelial cells therefore need a particularly tight control of genome integrity since upon activation, the effects of mutations could lead to aberrant proliferation. In fact, malignant transformation of endothelial cells is extremely rare. A tight genome surveillance in endothelial cells most probably corresponds to a pre-commitment to apoptosis which in turn becomes an important target of anti-angiogenic therapy.
Anti-angiogenesis: clinical efficacy and resistance
Targeting angiogenesis clinically is currently largely limited to interruption of a single pathway, the VEGF pathway, yet has shown significant improvement on survival for several cancers, and provided novel therapeutic efficacy that was lacking for some difficult to manage cancers such as renal cancer. However, since to date the clinical benefit is largely in terms of months, there is vast room for improvement. Targeting pathways other than VEGF must be evaluated [2–5], as is also needed for the role of components of the tumor microenvironment, in particular inflammation [6–8]. Leukocyte recruitment and inflammatory cytokine induction have been suggested to precede even VEGF induced angiogenesis [9].
The successful prevention approaches in cardiovascular disease give further support to the concept of cancer chemoprevention, an active intervention to prevent cancer insurgence and progression [1, 10]. Cancer chemoprevention is defined as use of molecular approaches to inhibit or delay tumor onset and growth [10]. Many chemoprevention agents and substances (including dietary habits) associated with cancer prevention appear to target angiogenesis, a concept known as angioprevention [1, 11]. Angioprevention may represent a feasible and efficacious approach to preventing cancer, however, more effective drugs will be necessary for putting this into clinical practice. Angiopreventive drugs may have the potential to repress angiogenesis during early steps of carcinogenesis where they might retard the angiogenic switch, preventing unrestrained tumor growth. The identification of common molecular hubs targeted by the structurally diverse angioprevention compounds identified to date will provide insight into drug design and drug combinations to provide effective prevention with minimal or no deleterious side effects, as discussed below, leading to reduction of angiogenesis, thus delaying tumor growth, progression and metastasis.
The scope of this review is to summarize what we have learned on the response of the endothelial cell to anti-angiogenic and angiopreventive treatments with special regard to changes in the endothelial gene expression profile. The transcriptome of treated endothelial cells can help to (1) identify new molecular targets, (2) design combination therapies, (3) prevent or stimulate sencescence of vascular cells, (4) circumvent resistance to anti angiogenic therapy and (5) design array specific diagnostic tools.
Endogenous inhibitors of angiogenesis
Not surprisingly, a range of endogenous negative regulators of angiogenesis have been found, as expected from the angiogenesis “balance” concept in homeostasis where normally inhibitors of angiogenesis dominate over angiogenic stimuli. However, many of these are in surprising forms, in particular a number of proteolytic fragments have been found to harbor anti-angiogenic activity even though the parental intact protein is not involved in regulation of angiogenesis. These are often fragments of extracellular matrix proteins, such as collagen type IV chains, including Arresten (COL4A1) [12], Canstatin (COL4A2) [13, 14] and Tumstatin (COL4A3) [15]; the Collagen XVIII fragment Endostatin [16]; the perlecan fragment Endorepellin [17] and the fibronectin fragment Anastellin [18]. One of the most potent and physiologically relevant angiogenesis inhibitors is the intact extracellular matrix protein Thrombospondin [19, 20]. Long pentraxin 3, a molecule associated with inflammation and matrix assembly in the ovary [21], is a potent inhibitor of FGF2 induced angiogenesis that appears to be responsible for the paucity of angiogenesis in systemic sclerosis pateints [22, 23]. In contrast, the activity of the Tissue inhibitors of metalloproteinases [24] (TIMPs 1–4) also show repression of angiogenesis by inhibiting degradation and processing of extracellular matrices, in particular the basement membrane, necessary for endothelial cell migration and invasion during angiogenesis. Another class of parental molecules giving rise to angiogenesis inhibitory fragments are involved in regulation of thrombosis, including the first member identified, Angiostatin [25], a fragment of plasminogen, and a peptide derived from Antithrombin III [26], while Vasostatin [27] is instead derived from the endocrine modulator Chromogranin-A. The predominance of extracellular matrix and proteolysis products among known endogenous inhibitors of angiogenesis is striking, suggesting that endothelial cells may have inherent mechanisms for sensing areas of extensive tissue damage where revascularization may well be too premature. One particularly important defense mechanism whereby tissue degradation should inhibit angiogenesis would be in areas of infection with an intense immune reaction in progress; revascularization of the hypoxic infected tissue is blocked so as to prevent systemic dissemination of the infection. As we will see below, some of these inhibitors appear to target immune cells, suggesting this may be a key regulatory mechanism. The immune system is capable of sensing at least some forms of proteolytically generated peptides [28], a role for the immune system in the function of this class of angiogenesis inhibitors could be speculated, in keeping with the immunomodulatory properties of the calreticulin fragment vasostatin [29]. This also suggests that there may be a pattern recognition system involved in mediating the response to these proteolytic fragments.
Several molecules involved in cell signaling appear to interfere with angiogenic cell signaling, including chemokines (Platelet factor 4 [30–32]), Soluble Fms-like tyrosine kinase-1 (S-Flt-1) [33], Pigment epithelium derived factor (PEDF) [34] and Angiopoietin 2 [35]. Interestingly, an alternate splice variant of VEGF that is produced by normal tissues, designated VEGFxxxB [36], appears to inhibit VEGF signaling.
Angiostatin is a large peptide fragment of plasminogen endowed with anti-angiogenic properties originally isolated from the urine of tumor-bearing mice [37, 38]. Angiostatin and related forms consisting of the first 1–5 kingles in plasminogen are generated by the action of diverse proteases, including metalloproteases (MMP2, MMP12, MMP9) and serine proteases (PSA, neutrophil elastase) [39, 40].
Following identification with in vivo studies, numerous in vitro studies have sought to identify the effects of angiostatin on endothelial cells. Angiostatin has been demonstrated to produce an array of events ranging from apoptosis/activation of endothelium to inhibition of endothelial cell migration, [41–44] and tube formation [45]. Potential endothelial cell surface angiostatin receptors identified to date include cell surface ATP synthase, angiomotin and various integrins (see [40] for review).
Angiostatin inhibits migration of neutrophils and macrophages in vitro and neutrophil mediated angiogenesis in vivo, [44] and inhibits neutrophil and monomyeloid cell adhesion [46]. It also inhibits angiogenesis induced by HIV-tat, a molecule with chemokine-like and VEGF-like properties [47], reduces macrophage numbers in atherosclerotic plaques [48], and tumor-associated macrophage infiltration in vivo [49]. This activity was associated with repression of macrophage infiltration into matrigel sponges in vivo and inhibition of macrophage migration in vitro.
The effects of angiostatin on cellular infiltrates could dictate the alterations in the cytokine profile at the local microenvironment or systemic levels following angiostatin treatment. Using microarray analyses, we noted that exposure of endothelial cells to angiostatin in vitro strikingly limited effects on gene expression profiles as compared to that generated by cytokines such as interferons (unpublished observations and [50]). The range of modulation observed was modest (maximum 2.8-fold); only few genes found to be up-regulated over twofold being MMP14 (2.8-fold), IL-15 (2.6-fold), a lymphoid-specific helicase (HELLS) and its interacting protein protein MX2 (myxovirus resistance 2) and HSF2BP, a heat shock transcription factor 2 binding protein. PCDH7 (brain–heart-protocadherin, was the only gene substantially (0.5-fold) down-regulated. However, these data are based on two technical replicates and the lack of biological replicates does not permit to firmly conclude that these genes were indeed modulated by angiostatin. Further, modulation of IL-15 to a similar extent (3.4-fold) was also observed on endothelial cells treated with IL-12; however, analyses of numerous microarray analyses show that these human umbilical vein endothelial cells (HUVEC) express low or absent levels of the IL-12Rb1 and IL-12Rb2 subunits, consistent with previous observations [51] and the lack of biological effects of IL-12 on HUVE cells in vitro examining growth, migration or invasion [52, 53]. Altogether, the limited transcriptional effects observed suggested that non-endothelial cell types, such as immune cells, are primary targets in vivo.
Previous array studies found substantial modulation of the endothelial transcriptome when using diverse angiostatin forms (K1–3, the “canonical” K1–4, and k1–5) that were transduced into A549 lung carcinoma cells using and adenoviral vector. The effects of supernatants from the transfected cells, as compared to the null vector control, was the tested on HUVECs treated for 4 h containing 75% of the transduced A549 cell supernatants and final concentrations of 5% heat inactivated FBS, 5 ng/ml VEGF-A and 0.1% BSA [54]. Unfortunately, these authors did not investigate the effects of angiostatin transduction on the transcriptome of the A549 cells themselves, which may have undergone significant alterations as well, thus skewing the results. Further, the platform used, cDNA chips of 6,388 human unigenes, was limited in scope and quality control, although 3 biological replicates were used. Treatment of HUVE cells with these supernatants resulted in inhibition of proliferation, migration, tube formation and induction of apoptosis. Induction of apoptosis appeared to be related to FasL induction and was blocked by antibodies targeting fas, the α-ATP synthase and the αvβ3 integrin. The array analyses indicated 189 genes were differentially induced or repressed by at least twofold, most (161) of these were induced. These induced genes were dominated (70%) by functional groups related to growth/proliferation, inflammation, apoptosis/survival, extracellular matrix/adhesion and migration/cytoskeleton (in order of predominance). Of note, one gene up-regulated by angiostatin in this approach was E-selectin, both expression and function [54]. Expression of E-selectin, a downstream target of NF-κB, is generally associated with angiogenesis [55], and we have found that E-selectin is repressed by angioprevention agents [56]. These data could reflect the action of cytokines such as IL-1β that could have been induced by angiostatin in the A549 cells.
Endostatin was the next endogenous anti-angiogenic fragment to be discovered [16]. The effects of on HUVE cells of endostatin treatment for 30 min, 1, 2, 4, and 8 h was examined by microarray analyses [57] using a human cDNA 10 k array (Hs-UniGem2, produced by the NCI/NIH ATC) and compared to that of fumagillin, an anti-angiogenic fungal metabolite. The authors focused on a series of genes that showed rapid up-regulation (at 1–2 h of treatment) followed by down-regulation. These were transcription factors, in particular KLF4 and ID1, that have been shown to be involved in regulating angiogenesis, consistent with the evidence that endostatin influences signal transduction [58–60].
The group of O’Reilly and Folkman, following their discoveries of angiostatin and endostatin, found that tumor cells in culture generated a fragment of bovine antithrombin III that strongly inhibited angiogenesis [26]. Transcriptome analyses of the effects of the anti-thrombin fragment on endothelial cells based on Stanford 43 K human cDNA microarrays initially showed a dramatic down-regulation of perlecan [61], a heparan sulfate proteoglycan [62] involved in presentaion of angiogenic growth factors to their receptors [63] that also gives rise to an anti-angiogenic fragment [17]. More in-depth analyses showed modulation of 128 transcripts over twofold, in particular those of proteins associated with the extracellular matrix [64], including up-regulation of TIMPs 1, 2 and 3 and the proteoglycans agrin and vitronectin, and down-regulation of biglycan and syndecan 3. Syndecan 1, whose involvement in angiogenic signaling is complex [63] instead showed a unique rapid up-regulation followed by down-regulation. Interestingly, most of the transcripts were down-regulated.
The Tissue Inhibitors of MetalloProteinases, or TIMPs, have a complex relationship with angiogenesis, metastasis, and several other cell regulatory pathways. While TIMPs have anti-angiogenic activities, largely assigned to their capacity to block invasion, the effects of TIMPs have not been tested on endothelial cells. The effects of TIMP1 on differential gene expression in JD38 Burkitt lymphoma cells using NIH ATC 10 K microarrays [65] indicated most transcripts were repressed. The effects were centered on gene categories such as those regulating B-cell growth and differentiation, transcription and cell cycle regulators, again suggesting the complex signaling role of these molecules dominated. We note that CD44, a gene associated with matrix and metastasis, was among the most up-regulated, while the alpha 4 integrin subunit was strongly down-regulated. A quite different picture was obtained with TIMP3 transfected into Gli36 human glioma cells using Affymetrix GeneChip HG-U133A microarrays [66]. Here most genes were up-regulated, 3 major gene classes could be identified: (1) A series of disintegrin metalloprotease ADAM proteins (including ADAM-9, ADAM-10, ADAM-17, ADAM-19, ADAM-21, and ADAM-23), probably related to the regulatory role that TIMP3 is known to exert on membrane-anchored proteinases, (2) Numerous proteins associated with apoptosis, most likely related to the apoptotic activity associated to TIMP3, (3) Matrix/angiogenesis associated genes, including up-regulation of MMP2 and MMP9, chains of collagen IV, V and VI, laminins, PDGFs, FGF2&5 and FGFR1, several interleukins and other cytokines, TNF and its receptors, and finally VEGFA, B, C and neuropilin-1. This would imply induction of angiogenesis and a finite risk of enhancement of tumor growth, however, in vivo assays demonstrated that the TIMP3 transfected tumors grew slower [66], although histological analyses on tumor vascularization, apoptosis, and eventual microenvironment effects were not reported.
Hormones with anti-angiogenic activity
The endogenous metabolite of estrogen, 2-methoxyestradiol, has been described as a potent inhibitor of endothelial cell proliferation, migration and angiogenesis in vitro [67]. The metabolite is a strong inhibitor of superoxide dismutase [68] and this activity might be related to the anti-angiogenic effect inasmuch as superoxides stimulate inflammation as well as endothelial cell activation and proliferation.
Human beta-chorionic hormone (βHCG) has been studied following initial reports of its activity against the endothelial cell derived Kaposi’s sarcoma [69]. βHCG inhibits MMPs [70] but the precise mechanism has not been elucidated. Gene expression profiling of βHCG treated breast cancer cells using dedicated arrays with about thousand genes hints at cell proliferation, apoptosis, cell trafficking, and DNA repair without a clear relation to anti-angiogenic functions [71].
Somatostatin inhibits angiogenesis by binding to its receptors on the surface of endothelial cells that leads to the activation of the nitric oxide synthase and the MAP-kinase pathway [72]. Gene expression profiling of somatostatin treated pancreatic cancer cells led to the identification of several angiogenesis related genes including angiogenin, a potent pro-angiogenic RNase [73].
Prolactin, the lactation stimulating hormone that almost exclusively acts on the mammary gland, can give rise to a 16kD fragment with a strong anti-angiogenic activity that is mediated by the inhibition of MAP-kinases [74, 75]. Similar fragments are also released from the other members of the prolactin/human growth hormone family of proteins [75]. Interestingly, a study of the response of endothelial cells to the treatment with the fragment revealed induction of an NFκB dependent response including chemokines and the endothelial activation marker, E-selectin [76] which is one of the major down-regulated genes by the anti-angiogenic anti-oxidants N-acetyl cysteine and epigallocatechin gallate that repress NFκB signaling [77] (see also below). The authors suggest that enhanced leukocyte infiltration might be responsible for the anti-tumoral effects observed in the melanoma model [76] but it is not clear how enhanced NFκB activity may determine anti-angiogenic effects. Full length prolactin also induces NFκB leading to the induction of the anti-angiogenic cytokines CXCL-9, -10, and -11 [78]. The prolactin fragment might therefore share the anti-angiogenic mechanism with interferons (see below).
Anti-angiogenic cytokines
Despite the fact that several cytokines may interfere with angiogenesis in some experimental settings (see Table 1), only a few of them can be considered endogenous angiogenesis inhibitors. Interferon-α (IFN-α), a cytokine with marked therapeutic activity in transplantable tumor models, is possibly the prototype of anti-angiogenic cytokines. Initial evidence for anti-angiogenic effects of type I IFNs stems from observations in immuno-competent DBA/2 mice bearing IFN-resistant erythroleukemia, or ESb lymphoma cells; results indicated that IFN-α/β exerted an anti-tumor effect by damaging tumor blood vessels, thus causing disruption of tumor blood flow, which led to ischemic tumor necrosis [79]. Moreover, it was suggested that IFNs modulated the signal for angiogenesis produced by the tumor cells [80]. Later on, experiments of IFN-α gene transfer by various delivery systems confirmed and extended these findings (reviewed in [81]). In most of these studies, immuno-deficient mice lacking mature CD8+ lymphocytes and generally also NK cells were transplanted with IFN-α-resistant tumor cells, thus ruling out involvement of immune-based mechanisms or direct anti-proliferative effects in the anti-tumor effects observed.
The anti-angiogenic activity of class I IFN has classically been attributed to inhibition of basic fibroblast growth factor (bFGF) overproduction by tumor cells [82] or down-regulation of IL-8 and vascular endothelial growth factor (VEGF) gene expression [83, 84]. However, reduced production of pro-angiogenic factors is not likely to explain IFN-α-induced anti-vascular effects in experiments involving injection of murine IFN-α into mice bearing human tumor cells [85, 86], due to the relatively strict species-specificity of class I IFN. Moreover, even in the absence of these interspecies barriers, reduced production of VEGF or bFGF was barely observed in TRAMP mice—a transgenic model of prostatic cancer—treated with murine IFN-α, in spite of clear-cut anti-angiogenic effects in the tumors [87]. Thus, suppression of pro-angiogenic factors synthesis by IFNs is one plausible yet not exclusive explanation for the anti-angiogenic effects of this cytokine, and direct effects on endothelial cells (EC) can be envisioned. In this regard, microarray data have shown dramatic transcriptional effects of IFNs in EC, which are accompanied by modulation of some endothelial cell functions, such as in vitro proliferation and migration [88–91]. A key intracellular mediator of these effects could be guanylate binding protein 1 (GBP-1), whose expression in vivo has been almost exclusively associated with EC, where it may exert specific functions (including inhibition of endothelial cell proliferation and migration) in response to class I IFNs and other inflammatory cytokines through MMPs and other as yet unknown mechanisms [92, 93]. Moreover, anti-vascular effects of IFN-α may partially depend upon up-regulation of angiostatic chemokines [94, 95]. In vitro, treatment of human endothelial cells with IFN-α leads to marked transcriptional up-regulation of CXCL10–11, uncoupled from CXCL9 or IFN-γ [88]: these chemokines, which are released at low level by IFN-α-stimulated endothelial cells, could in theory act as biological amplifiers of the primary anti-angiogenic effects of IFN-α.
What are the consequences of these effects of IFN-α on tumor vessels? In vivo, anti-vascular activity in subcutaneous tumor xenografts has been associated to induction of areas of ischemic necrosis [85, 86, 96–99], confirming initial observations [79, 80, 100]. It appears that disruption of tumor microvessels by IFN-α leads to increased hypoxia in these models, a feature shared by other anti-angiogenic drugs [101]. It is important to note that hypoxia and necrosis were generally reported following implantation of tumor cells engineered with the IFN-α gene, a modality that likely favours anti-tumor effects because the cytokine is released early during tumor formation and in a sustained manner. In general, treatment of established tumors by gene therapy or high-dose cytokine administration led to less dramatic results, possibly due to resistance of established tumors to anti-angiogenic therapy or to the fact that certain biological activities of IFNs, such as anti-angiogenic effects, could follow a bell-shaped response and are thus not seen following conventional dosage [102, 103].
It is too early to understand whether these discoveries on the transcriptional signature of IFNs will have translational implications. In this regard, however, it is interesting to note that the INTERFEROME database of interferon regulated genes is now available to investigate, among others, the presence of the IFN signature in biological samples [104]. This could be very useful to unravel the endogenous IFN signature in tumor samples (and correlate it to vascularization) or monitor at the transcriptional level the outcome of IFN therapy.
A second cytokine whose effects are recurrently associated with angiogenesis inhibition is IL-12, which was initially reported to inhibit new vessel formation induced by the pro-angiogenic factor bFGF in a mouse corneal vascularization assay [105], and in Matrigel plug assays [106]. It has been shown that IFN-γ and, at least in part, CXCL10, appear to play a critical role as mediators of its anti-angiogenic effects [106, 107]. NK cells may contribute to inhibition of angiogenesis by IL-12 not only through the secretion of anti-angiogenic chemokines but apparently also through NK cell-mediated cytotoxicity against endothelial cells [108]. More recently, however, angiogenesis inhibition was observed in IFN-γ(−/−) and CXCR3(−/−) knockout mice treated with IL-12, indicating that NK− and/or T cell-initiated IFN-γ-chemokine cascades were not involved in angiogenesis inhibition in vivo [53, 109]. Whatever the underlying mechanism, results from all these studies agree on the identification of endothelial cells as a major downstream target of the biological effects of IL-12. However, it has been reported that HUVECs do not respond to IL-12 because they lack both subunits of the IL-12 receptor [51]. Microarray data on IL-12 effects on HUVE cells indicates essentially no effects (see above), in line with the lack of receptors on these cells. However, other endothelial cell types could behave differently would be extremely useful to investigate the issue as to whether microvascular endothelial cells express receptors for or respond to IL-12, unfortunately not yet available.
“Classic” anti-angiogenic drugs
The concept of anti-angiogenesis as an anti-cancer strategy has led to the development of many classic drugs such as Anecortave, an angiostatic steroid [110], the tubulin binding agent combretastatin A-4 phosphate [111] and analogues of fumagillin, a naturally secreted antibiotic of Aspergillus fumigatus fresenius [112]. No data exist, however, on the effect of these compounds on gene expression profiles. The anti-angiogenic effects of the sedative thalidomide are apparently linked to its teratogenic actions [113]. Several mechanisms of action involving VEGF, IGF1, bFGF [114] as well as NFκB [115] have been proposed. The effects of the drug on the transcription profile of endothelial cells have been analyzed using microarrays, however, the expression values have been reported for only six genes (VEGF, bFGF, HGF, IGF-1, IGFBP-3 and Ang1) [114]. These data indicate that thalidomide acts on activated endothelial cells such as those present in multiple myelomas. It would be of great interest to further exploit these data for the identification of other anti-angiogenesis related genes that respond to thalidomide.
Ferrari and colleagues reported anti-angiogenic effects of the synthetic retinoid N-(4-hydroxyphenyl)retinamide, 4-HPR. These effects were mediated by the member of the TGFβ-family members BMP1 and MIC as revealed by gene expression profiling of drug exposed human umbilical endothelial cells followed by functional assays [116]. This is particularly interesting in the light of cancer chemoprevention with 4-HPR [117].
The chemopreventive anti-estrogen, tamoxifen, also has been proposed to exert anti-angiogenic effects [118]. In addition to the abrogation of pro-angiogenic, estrogen receptor α mediated effects, tamoxifen apparently exerts anti-angiogenic effects that do not depend on the receptor [118]. Two studies have applied expression profiling to the analysis of tamoxifen effects, one analyzing breast cancer tissues of pre-surgically treated patients [119] and the other analyzing the estrogen receptor α positive breast cancer cell line MCF7 after over-expression of the aromatase transgene [120]. None of the two studies directly addressed anti-angiogenesis and the models are not suited to distinguish between ERα dependent and independent effects.
Targeted anti-angiogenic drugs
One of the key approaches in ant-angiogenesis has been development of targeted agents, which has been successful in the clinic. The classic examples are agents that directly target VEGF, with the most widely known and the first clinically successful anti-angiogenesis agent being Bevacizumab [121], a humanized antibody that recognizes most isoforms of VEGF, thus blocking its capacity to interact with cell surface receptors. Bevacizumab is clinically effective in enhancing disease-free survival in combination with chemotherapy (as monotherapy it is not effective) for a constantly expanding list of cancers, starting with colon. A variant of the same structural components is the use of an antibody fragment of Bevacizumab, known as Ranibizumab, specifically developed for ocular pathologies where it shows notable efficacy in treating wet age-related macular degeneration. The second key approach has been multi-tyrosine kinase inhibitors that block VEGFR1 and VEGFR2 as well as other tyrosine kinases, the classic examples of these are Sorafenib [122] and Sunitinib [123]. These two molecules have been particularly effective in treating renal cancer, where they are approved and have represented a major therapeutic advance. Recent studies suggest that targeting other components of the VEGF system may be even more effective with fewer side effects, in particular agents targeting PlGF [124] or specifically VEGFR1. Finally, targeting the key intracellular signaling hub, mTOR, with Rapamycin [125] or Rapamycin analogs is intensively under investigation and entering into the clinic.
Transcriptome analyses of the effects of bevacizumab, Sorafenib or Sunitinib on endothelial cells are not available, one would assume that this would be similar to that of VEGF deprivation. However, other effects of these molecules could also be encountered, for example transcriptome analyses of murine models found that bevacizumab treatment up-regulated the Sp1 transcription factor and associated genes [126]. These approaches could also shed light on the source of the hypertension response, which is a common and major collateral effect of this class of drugs. Expression analysis has been done on tumor tissues of bevacizumab treated breast cancer patients with a differential analysis of responders vs non-responders [127]. These data sets show a predominance of genes associated with the tumor microenvironment, including several gene ontology sets related to extracellular matrix and cellular mobility. We had previously noted that the tumor microenvironment signature could be predictive of metastasis [128]. Interestingly, VEGF responses seemed to represent a minor constituent of the response [127].
Transcriptome analysis of the effects of sorafenib in a pulmonary hypertension model indicated a much more complex response was induced by drug therapy [129]. Combinations of sorafenib and rapamycin have been suggested to be effective in preclinical models [130], however, microarray analyses of the effects of rapamycin also show a complex picture. A 12 h exposure of MCF-7 breast cancer cells to rapamycin alone produced relatively modest changes [131], these were enhanced by simultaneous treatment with a differentiating agent, cotylenin A. Interesting up-regulated genes included Transforming growth factor-β-induced gene (TGFBI) and Cyclin G2 and down-regulation of most other cyclins. A similar effect of rapamycin, with up-regulation of Cyclin G2 and down-regulation of cyclins D2 and F, was found in Jurkat T cells [132] and in developing myoblasts [133], although effects on differentiation state were also found in this latter set. The modulation of this group of genes targeted by mTOR inhibitors appears to act as a function of AKT activity [134]. Interestingly, down-regulation of a probable angiogenic factor, Heparin-binding EGF-like growth factor, was also observed [132].
Angiopreventive drugs
The publication of the first gene expression signatures [135, 136] stimulated a discussion on the validity of the multi-step carcinogenesis model [137, 138]. The possibility to predict the clinical outcome of a tumor through the analysis of the bulk of it has been extensively demonstrated for breast cancer. Yet this is in contrast with the multistep carcinogenesis model that predicts the metastatic subpopulation of the tumor to be very small, hidden in the bulk of the primary tumor. The apparent paradox can be resolved if the steps leading to a metastatic phenotype occur much earlier than so far assumed, long before the tumor becomes clinically overt [139]. Angiogenesis is of paramount importance for the acquisition of the metastatic phenotype inasmuch as it provides the route of dissemination of tumor cells to distant target tissues. Hence, metastasis could be blocked by blocking angiogenesis when the tumor is small, before the actual diagnosis of cancer.
Angiogenesis could therefore become a target of primary prevention in high risk subjects [11]. This means treating healthy people for a long time and compliance will depend on the absence of toxicity or side effects. The discovery of anti-angiogenic effects of the known anti-oxidant N-acetyl-cysteine [140] and the green tea gallate [141] opened a way to angioprevention [11]. The list of similar compounds with “angiopreventive” activity has considerably grown since (see Table 1). A common theme of their action is inhibition of the inflammation related transcription factor NFκB that also blocks apoptosis and is active in both, endothelial and tumor cells [77, 142–148] but other pathways play a role at least for triterpenoids [149–151] and artesunate [152] and perhaps for boswellic [153], gambogic [154] and α-lipoic acid [155].
Gene expression profiling has been performed for many of these compounds yet only very few studies have addressed effects on endothelial cells [77, 155, 156]. The effects of gallates on the transcriptome have been studied for rat liver with the aim to characterize its metabolism that appears to depend on the cytochrome p450 isoform 1A2 (CYP1A2) that also mediates caffeine, melatonin and theophyllin degradation [157]. Long term administration of gallate to rats is well tolerated even at high doses and the transcription of anti-oxidant enzymes, stress related genes as well as energy metabolism related genes is affected [158]. Breast cancer cells respond to the anti-oxidant by blocking ATK expression and phosphorylation and reduced MMP9 expression consistent with the observed inhibition of invasion [159]. Comparing wild-type and Nrf2 knockout mice, Shen and collaborators identified Nrf2-dependent and -independent effects of the gallate in mice liver and intestine after short term treatments and demonstrated a large number of responding genes [160]. Mammary cancers in carcinogen treated rats have an increased latency after treatment with EGCG, an effect that is apparently mediated by the regulation of the expression of genes involved in nuclear and cytoplasmic transport, transformation, redox signaling, response to hypoxia, and detoxification [161]. Interestingly, the growth inhibition exerted by ECGC on HER2/erbb2 positive mammary tumors is abolished through mutations in the receptor that lead to enhanced NFκB activity. EGCG resistant tumors show an activation of the MAP-kinase pathway as detected by microarray analyses [162]. Proliferation of normal human neonatal fibroblasts is reversibly blocked by EGCG in correspondence to the repression of transcription of several cell cycle related genes that also resumes after removal of the drug [163]. Endothelial cells, that in contrast to tumor cells do not undergo apoptotic effects at concentrations up to 50 μM EGCG, respond to the green tea component by the repression of endothelial activation through the reduction of NFκB translocation to the nucleus [77]. Regulation of NFκB and downstream genes such as MMPs thus appear to be at the core of the observed anti-angiogenic effects. ECGC transcriptomics has also been addressed in models of auto-immunity [164] and diabetes [165].
In a mouse model of inflammatory bowel disease Nones et al. showed that curcumin acts via an up-regulation of xenobiotic metabolism and a down-regulation of pro-inflammatory pathways, that the authors attribute to the activation of retinoid X receptor (RXR) by pregnane X receptor (Pxr) and peroxisome proliferator-activated receptor alpha (PPARα) [166]. Curcumin altered the expression of twelve genes in the livers of rats fed with a diet containing the poylphenol indicating a slight peroxisome proliferator acticvity [157]. In pancreatic tumor cells, curcumin induces miRNA22 leading to an up-regulation of the corresponding target mRNAs coding for the general transcription factor SP1 and the estrogen receptor α [167]. In human colon cancer cells, microarray analyses indicate an effect of curcumin on NFκB dependent expression of Cox2 and MMP2 in correlation to its effects on invasion [168]. Two studies have addressed the effects of Curcumin on breast cancer cells. Curcumin regulates several apoptosis related genes [169]. Complex microarrays reveal a series of NFκB downstream targets among the curcumin responsive genes including two pro-inflammatory cytokines, CXCL-1 and -2, that are downregulated by the polyphenol, but other pathways like ERG1 are also affected [170]. Taken together, these effects might well explain the considerable effect on breast cancer metastasis in the mouse model [171]. A single report addressed effects of the turmeric on endothelial cells analyzing the expression of cell cycle regulators and showing effects on several cell division cycle dependent kinases (CDKs) without an obvious link to the paramount curcumin target NFκB [156]. Reduced proliferation of endothelial cells could well contribute to the anti-angiogenic effects observed [172]. However, endothelial proliferation can be stimulated by inflammatory cytokines and reduced proliferation can therefore be secondary to NFκB inhibition [143].
α-Lipoic acid (α-LA) is an anti-oxidant in use for the treatment of peripheral neuropathies associated with non-insulin-dependent diabetes. It shows anti-angiogenic activities in vitro and in vivo that translate into a marked reduction of xenograft growth of vascularized tumors [155]. Gene expression studies of α-LA treated endothelial studies identified the pro-apoptotic death receptor ligand TRAIL and activin-A as major candidates for anti-angiogenic effects although a variety of metabolic processes are equally affected [155]. The anti-malaria drug, artesunate, also inhibits angiogenesis [173]. This drug has not been tested itself for effects on the transcriptome although it shows preferential effects on endothelial cells, yet the NCI panel of 60 cell lines has been analyzed with respect to angiogenesis related genes thirty of which significantly correlate with resistance to artesiminin [174].
Expression profiles of colon carcinoma cells that have been treated with ellagic acid, a dietary polyphenol present in berries, show differential expression of several genes of the MAP-kinase pathway including the tyrosine kinase receptors FGFR2 and EGFR [175]. It is not yet clear to which extent this polyphenol exerts effects similar to those of other compounds of the family.
The transcritpome analysis of H22 transplants in mice treated with gambogic acid has led to the hypothesis that the anti-tumor effect observed are indirect and mediated by the immune system since a large portion of the genes induced are classified as immunity related genes [176].
Hyperforin has been demonstrated to be the active compound in St. John’s wort inasmuch as the expression profiles elicited by the whole extract and the purified compound in human hepatoma cells are much alike [177]. The anti-angiogenic effects of hyperforin [144] appear to be mediated by the regulation of hypoxia associated genes in addition to mediators of proliferation and apoptosis as well as a series of drug metabolism genes [177].
N-acetyl-cysteine (NAC) is used as a mucolytic drug for many years and its anti-angiogenic effects have been described several years ago [140]. A single study has addressed the effect of the anti-oxidant on global gene expression showing that downstream targets of NFκB constitute the majority of NAC responsive genes in endothelial cells [77].
The triterpenoids or synthetic oleananes are a novel class of anti-angiogenic chemopreventive drugs [178] acting via PPARγ [149], JAK/STAT [150], NFκB [147] and NRF2 [151, 179]. Microarray analyses in wild type and Nrf2 knockout mice confirm the latter pathway to be responsible for the chemopreventive effect against aflatoxin induced DNA-adducts [179]. However, anti-angiogenic effects are most likely mediated by other pathways with NFκB being an obvious candidate.
For boswellic acid, pinitol, thymoquinone and xanthohumol no gene expression data are available and the data on artesunate are not derived from direct treatments with the drug. Xanthohumol [148] and pinitol [146] act via NFκB, and the same might hold true for thymoquinone, while similar data are lacking for boswellic acid.
It turns out that most of the angiopreventive drugs act through the inhibition of NFκB with IκK being a preferential target, although the mechanism has not always been described in detail. Gene expression studies reflect this mechanistic analogy to a certain extent. A major problem with comparing such data derives from the many different cellular systems analyzed as well as from the fact, that there is no certainty on downstream targets of NFκB which most likely differ from cell type to cell type. If, for example, gene expression analysis reveals hypoxia as a central process that is targeted by anti-angiogenic drugs, this most probably corresponds to NFκB mediated effects since most hypoxia related genes are also controlled by NFκB. A single study analyzed different anti-oxidants in parallel on endothelial cells showing the high similarity of the NFκB dependent effects elicited by the drugs in endothelial cells [77].
More comparative studies are needed to identify drugs that obtain the desired anti-angiogenic effect at the lowest dosage possible in a specific cellular system. This would also help to identify the drugs best suited for clinical (angio-)prevention trials.
Anti-angiogenic phytoestrogens
There is compelling evidence that endogenous estrogens have pro-angiogenic actions [180–182], which probably participate in the beneficial actions of hormone replacement therapy on the myocardium [183]. Anti-angiogenic activities of estrogens are generally attributed to the estrogen receptor β [184] and the partial antagonist, tamoxifen, shows anti-angiogenic activity in ERα knock-out mice [118]. Phytoestrogens are plant derived drugs with a certain affinity for ERα that compete with the endogenous hormone for binding to the receptor and therefore exert a partial antagonistic effect. The effect of the phytoestrogens therefore strongly differs between pre- and post-menopause. Given the wide use of hormone replacement therapies and the identification of its contribution to the breast cancer risk in postmenopausal women [185] many groups seek safe substitutes for it. This has determined much consideration of phytoestrogens in the hope they might maintain the beneficial effects of estrogens on vascular health without the detrimental pro-carcinogenic effect.
Genistein, however, turned out to be anti-angiogenic [186]. A study on gene expression in endothelial cells revealed that genistein regulates several angiogenesis related genes such as endothelin-converting enzyme-1, endothelin-2, estrogen related receptor alpha and atrial natriuretic peptide receptor A precursor. Genistein also countered the effect of oxidized LDL on VEGFR [187]. Piao and colleagues identified cellular adhesion molecules as a major target for genistein in endothelial cells at concentrations that are, however, likely to induce apoptosis [188]. In PC3 cells that do not express estrogen receptors, Genistein regulates many genes involved in the processes of cell growth, cell cycle, apoptosis, cell signaling transduction, angiogenesis, tumor cell invasion and metastasis [189, 190], most probably consistent with its anti-NFκB activity [191, 192]. Pancreatic cells showed the involvement of EGFR and the putative NFκB target EGR1 in the response to the phytoestrogen [193]. Similar effects have been shown in several others cellular systems [194–196] and in mice [197–199]. Genistein effects clearly depend on the dosage with pro-estrogenic activities being dominant at lower and anti-apoptotic activities at higher concentrations [200], similar observations have been reported by Konstantakopoulos et al. [201]. The phytoestrogens resveratrol and quercetin present in the Mediterranean diet have also been reported to exert anti-angiogenic activities through the inhibition of endothelial cell proliferation yet only at high concentrations [202]. Both compounds are believed to act via the ERα [203–205] a fact that sheds some doubt on how much their anti-angiogenic activity can be generalized. Several microarray based studies have been carried out (see Table 1) yet there are no clues to the putative anti-angiogenic activities nor have endothelial cells been considered.
Conclusions
Gene expression profiling has provided important information on the identification of the pathways involved in cellular processes or in the response to external stimuli has not matched reality. Many microarray studies, however, fall short of rigorous experimental and statistical methods, often the simple “fold change” value is used without any statistics or simple t-tests are performed without consideration of the heavily multiparametric nature of global expression analyses. Only recently has array quality dramatically improved with widespread use of high quality commercial microarrays produced in a highly controlled manner. Yet there is a biological issue in addition to the quality aspect that makes the use of microarrays much less straightforward than expected. Biological systems are complex and variable. The actual state of transcription of a given depends on the number, the type, the position (distance to the transcription start or position relative to other regulatory elements) and the composition of transcription factor binding sites, the long and short range chromatin structure including post-translational histone modifications, the actual sequence, possible cytosine methylation and the availability of transcriptional co-activators and -repressors as well as the presence of specific non coding RNAs that influence mRNA stability. The same drug may therefore elicit very different effects on gene transcription in different cell lines or tissues and the activation of a specific signaling pathway or of a specific transcription factor may result in very different expression profiles depending on the cellular context as well as experimental, physiological or pharmacological concentrations.
In cancer research it is of paramount importance that, in addition to microarray studies on tumor cells and on transcriptional modulation in tumor cells, the influence of drugs on the microenvironment is studied with functional genomics. We have previously shown how breast cancer cells can be classified for their metastatic potential based on expression of genes related to matrix and stroma [128]. In this review we broadly analyze how gene expression of endothelial cells, based on microarray studies, can identify new pathways and explain response behaviour.
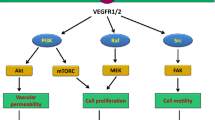
In particular, the NFκB, TNF-α, TGF-β, Akt and MAPK pathways emerge as central targets for anti-angiogenic effects that are shared by many of the drugs tested. Future gene expression studies should therefore study these pathways more systematically eventually choosing very few representative compounds to be studied at a wide range of concentrations and time intervals. Angiogenesis assays in vitro and in vivo may help to select the “best” angiogenesis inhibitor among a list of drugs with similar target specificities and eventually, the knowledge of the precise molecular mechanism may help to identify potentially synergistic drug combinations if, for example, different kinase inhibitory activities can be identified that inhibit NFκB activation at different steps.
References
Albini A, Sporn MB (2007) The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer 7(2):139–147
Casanovas O, Hicklin DJ, Bergers G et al (2005) Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8(4):299–309
Mizukami Y, Jo WS, Duerr EM et al (2005) Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat Med 11(9):992–997
Rusnati M, Presta M (2007) Fibroblast growth factors/fibroblast growth factor receptors as targets for the development of anti-angiogenesis strategies. Curr Pharm Des 13(20):2025–2044
Viloria-Petit A, Crombet T, Jothy S et al (2001) Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res 61(13):5090–5101
Albini A, Tosetti F, Benelli R et al (2005) Tumor inflammatory angiogenesis and its chemoprevention. Cancer Res 65(23):10637–10641
Shojaei F, Wu X, Malik AK et al (2007) Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+ Gr1 + myeloid cells. Nat Biotechnol 25(8):911–920
Shojaei F, Wu X, Zhong C et al (2007) Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 450(7171):825–831
Aplin AC, Gelati M, Fogel E et al (2006) Angiopoietin-1 and vascular endothelial growth factor induce expression of inflammatory cytokines before angiogenesis. Physiol Genomics 27(1):20–28
Sporn MB, Suh N (2002) Chemoprevention: an essential approach to controlling cancer. Nat Rev Cancer 2(7):537–543
Tosetti F, Ferrari N, De Flora S et al (2002) Angioprevention’: angiogenesis is a common and key target for cancer chemopreventive agents. FASEB J 16(1):2–14
Sudhakar A, Nyberg P, Keshamouni VG et al (2005) Human alpha1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by alpha1beta1 integrin. J Clin Invest 115(10):2801–2810
Kamphaus GD, Colorado PC, Panka DJ et al (2000) Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J Biol Chem 275(2):1209–1215
Petitclerc E, Boutaud A, Prestayko A et al (2000) New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J Biol Chem 275(11):8051–8061
Maeshima Y, Yerramalla UL, Dhanabal M et al (2001) Extracellular matrix-derived peptide binds to alpha(v)beta(3) integrin and inhibits angiogenesis. J Biol Chem 276(34):31959–31968
O’Reilly MS, Boehm T, Shing Y et al (1997) Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88(2):277–285
Mongiat M, Sweeney SM, San Antonio JD et al (2003) Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J Biol Chem 278(6):4238–4249
Yi M, Ruoslahti E (2001) A fibronectin fragment inhibits tumor growth, angiogenesis, and metastasis. Proc Natl Acad Sci USA 98(2):620–624
Good DJ, Polverini PJ, Rastinejad F et al (1990) A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci USA 87(17):6624–6628
Taraboletti G, Roberts D, Liotta LA et al (1990) Platelet thrombospondin modulates endothelial cell adhesion, motility, and growth: a potential angiogenesis regulatory factor. J Cell Biol 111(2):765–772
Garlanda C, Bottazzi B, Bastone A et al (2005) Pentraxins at the crossroads between innate immunity, inflammation, matrix deposition, and female fertility. Annu Rev Immunol 23:337–366
Margheri F, Serrati S, Lapucci A et al (2009) Systemic sclerosis-endothelial cell antiangiogenic pentraxin 3 and matrix metalloprotease 12 control human breast cancer tumor vascularization and development in mice. Neoplasia 11(10):1106–1115
Alessi P, Leali D, Camozzi M et al (2009) Anti-FGF2 approaches as a strategy to compensate resistance to anti-VEGF therapy: long-pentraxin 3 as a novel antiangiogenic FGF2-antagonist. Eur Cytokine Netw 20(4):225–234
Moses MA, Sudhalter J, Langer R (1990) Identification of an inhibitor of neovascularization from cartilage. Science 248(4961):1408–1410
Rastinejad F, Polverini PJ, Bouck NP (1989) Regulation of the activity of a new inhibitor of angiogenesis by a cancer suppressor gene. Cell 56(3):345–355
O’Reilly MS, Pirie-Shepherd S, Lane WS et al (1999) Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science 285(5435):1926–1928
Pike SE, Yao L, Jones KD et al (1998) Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J Exp Med 188(12):2349–2356
Okamura Y, Watari M, Jerud ES et al (2001) The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem 276(13):10229–10233
Huegel R, Velasco P, De La Luz Sierra M et al (2007) Novel anti-inflammatory properties of the angiogenesis inhibitor vasostatin. J Invest Dermatol 127(1):65–74
Taylor S, Folkman J (1982) Protamine is an inhibitor of angiogenesis. Nature 297(5864):307–312
Maione TE, Gray GS, Petro J et al (1990) Inhibition of angiogenesis by recombinant human platelet factor-4 and related peptides. Science 247(4938):77–79
Sharpe RJ, Byers HR, Scott CF et al (1990) Growth inhibition of murine melanoma and human colon carcinoma by recombinant human platelet factor 4. J Natl Cancer Inst 82(10):848–853
Kendall RL, Thomas KA (1993) Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci USA 90(22):10705–10709
Dawson DW, Volpert OV, Gillis P et al (1999) Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science 285(5425):245–248
Maisonpierre PC, Suri C, Jones PF et al (1997) Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277(5322):55–60
Bates DO, Cui TG, Doughty JM et al (2002) VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res 62(14):4123–4131
O’Reilly MS, Holmgren L, Shing Y et al (1994) Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 79(2):315–328
Abad M, Arni R, Grella D et al (2002) The X-ray crystallographic structure of the angiogenesis inhibitor angiostatin. J Mol Biol 318:1009–1017
O’Reilly MS, Wiederschain D, Stetler SW et al (1999) Regulation of angiostatin production by matrix metalloproteinase-2 in a model of concomitant resistance. J Biol Chem 274(41):29568–29571
Paleari L, Brigati C, Anfosso L et al (2005) Anti-angiogenesis in search of mechanisms: angiostatin as a prototype. In: Weber GF (ed) Cancer therapy: molecular targets in tumor-host interactions. Horizon Scientific Press, Norfolk, pp 143–168
Ito H, Rovira II, Bloom ML et al (1999) Endothelial progenitor cells as putative targets for angiostatin. Cancer Res 59(23):5875–5877
Walter JJ, Sane DC (1999) Angiostatin binds to smooth muscle cells in the coronary artery and inhibits smooth muscle cell proliferation and migration In vitro. Arterioscler Thromb Vasc Biol 19(9):2041–2048
Moser T, Kenan D, Ashley T et al (2001) Endothelial cell surface F1–F0 ATP synthase is active in ATP synthesis and is inhibited by angiostatin. Proc Natl Acad Sci USA 98:6656–6661
Benelli R, Morini M, Carrozzino F et al (2002) Neutrophils as a key cellular target for angiostatin: implications for regulation of angiogenesis and inflammation. FASEB J 16:267–269
Wahl ML, Kenan DJ, Gonzalez-Gronow M et al (2005) Angiostatin’s molecular mechanism: aspects of specificity and regulation elucidated. J Cell Biochem 96(2):242–261
Chavakis T, Athanasopoulos A, Rhee JS et al (2005) Angiostatin is a novel anti-inflammatory factor by inhibiting leukocyte recruitment. Blood 105(3):1036–1043
Benelli R, Morini M, Brigati C et al (2003) Angiostatin inhibits extracellular HIV-Tat-induced inflammatory angiogenesis. Int J Oncol 22(1):87–91
Moulton KS, Vakili K, Zurakowski D et al (2003) Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci USA 100(8):4736–4741
Perri SR, Nalbantoglu J, Annabi B et al (2005) Plasminogen kringle 5-engineered glioma cells block migration of tumor-associated macrophages and suppress tumor vascularization and progression. Cancer Res 65(18):8359–8365
Indraccolo S, Pfeffer U, Minuzzo S et al (2007) Identification of genes selectively regulated by interferons in endothelial cells. J Immunol 178(2):1122–1135
Torpey N, Maher SE, Bothwell AL et al (2004) Interferon alpha but not interleukin 12 activates STAT4 signaling in human vascular endothelial cells. J Biol Chem 279(25):26789–26796
Albini A, Brigati C, Ventura A et al (2009) Angiostatin anti-angiogenesis requires IL-12: the innate immune system as a key target. J Transl Med 7:5
Morini M, Albini A, Lorusso G et al (2004) Prevention of angiogenesis by naked DNA IL-12 gene transfer: angioprevention by immunogene therapy. Gene Ther 11(3):284–291
Chen YH, Wu HL, Li C et al. (2006) Anti-angiogenesis mediated by angiostatin K1-3, K1-4 and K1-4.5. Involvement of p53, FasL, AKT and mRNA deregulation. Thromb Haemost 95(4):668–677
Yu Y, Moulton KS, Khan MK et al (2004) E-selectin is required for the antiangiogenic activity of endostatin. Proc Natl Acad Sci USA 101(21):8005–8010
Vannini N, Pfeffer U, Lorusso G et al (2008) Endothelial cell aging and apoptosis in prevention and disease: E-selectin expression and modulation as a model. Curr Pharm Des 14(3):221–225
Mazzanti CM, Tandle A, Lorang D et al (2004) Early genetic mechanisms underlying the inhibitory effects of endostatin and fumagillin on human endothelial cells. Genome Res 14(8):1585–1593
Hanai J, Gloy J, Karumanchi SA et al (2002) Endostatin is a potential inhibitor of Wnt signaling. J Cell Biol 158(3):529–539
Schmidt A, Wenzel D, Thorey I et al (2006) Endostatin influences endothelial morphology via the activated ERK1/2-kinase endothelial morphology and signal transduction. Microvasc Res 71(3):152–162
Wickstrom SA, Alitalo K, Keski-Oja J (2002) Endostatin associates with integrin alpha5beta1 and caveolin-1, and activates Src via a tyrosyl phosphatase-dependent pathway in human endothelial cells. Cancer Res 62(19):5580–5589
Zhang W, Chuang YJ, Swanson R et al (2004) Antiangiogenic antithrombin down-regulates the expression of the proangiogenic heparan sulfate proteoglycan, perlecan, in endothelial cells. Blood 103(4):1185–1191
Noonan DM, Fulle A, Valente P et al (1991) The complete sequence of perlecan, a basement membrane heparan sulfate proteoglycan, reveals extensive similarity with laminin A chain, low density lipoprotein-receptor, and the neural cell adhesion molecule. J Biol Chem 266(34):22939–22947
Aviezer D, Iozzo RV, Noonan DM et al (1997) Suppression of autocrine and paracrine functions of basic fibroblast growth factor by stable expression of perlecan antisense cDNA. Mol Cell Biol 17(4):1938–1946
Zhang W, Chuang YJ, Jin T et al (2006) Antiangiogenic antithrombin induces global changes in the gene expression profile of endothelial cells. Cancer Res 66(10):5047–5055
Guedez L, Martinez A, Zhao S et al (2005) Tissue inhibitor of metalloproteinase 1 (TIMP-1) promotes plasmablastic differentiation of a Burkitt lymphoma cell line: implications in the pathogenesis of plasmacytic/plasmablastic tumors. Blood 105(4):1660–1668
Lam P, Sian Lim K, Mei Wang S et al (2005) A microarray study to characterize the molecular mechanism of TIMP-3-mediated tumor rejection. Mol Ther 12(1):144–152
Fotsis T, Zhang Y, Pepper MS et al (1994) The endogenous oestrogen metabolite 2-methoxyoestradiol inhibits angiogenesis and suppresses tumour growth. Nature 368(6468):237–239
Wood L, Leese MR, Leblond B et al (2001) Inhibition of superoxide dismutase by 2-methoxyoestradiol analogues and oestrogen derivatives: structure-activity relationships. Anticancer Drug Des 16(4–5):209–215
Albini A, Paglieri I, Orengo G et al (1997) The beta-core fragment of human chorionic gonadotrophin inhibits growth of Kaposi’s sarcoma-derived cells and a new immortalized Kaposi’s sarcoma cell line. AIDS 11(6):713–721
Pfeffer U, Bisacchi D, Morini M et al (2002) Human chorionic gonadotropin inhibits Kaposi’s sarcoma associated angiogenesis, matrix metalloprotease activity, and tumor growth. Endocrinology 143(8):3114–3121
Guo S, Russo IH, Lareef MH et al (2004) Effect of human chorionic gonadotropin in the gene expression profile of MCF-7 cells. Int J Oncol 24(2):399–407
Florio T, Morini M, Villa V et al (2003) Somatostatin inhibits tumor angiogenesis and growth via somatostatin receptor-3-mediated regulation of endothelial nitric oxide synthase and mitogen-activated protein kinase activities. Endocrinology 144(4):1574–1584
Patel SG, Zhou G, Liu SH et al (2009) Microarray analysis of somatostatin receptor 5-regulated gene expression profiles in murine pancreas. World J Surg 33(4):630–637
D’Angelo G, Struman I, Martial J et al (1995) Activation of mitogen-activated protein kinases by vascular endothelial growth factor and basic fibroblast growth factor in capillary endothelial cells is inhibited by the antiangiogenic factor 16-kDa N-terminal fragment of prolactin. Proc Natl Acad Sci USA 92(14):6374–6378
Struman I, Bentzien F, Lee H et al (1999) Opposing actions of intact and N-terminal fragments of the human prolactin/growth hormone family members on angiogenesis: an efficient mechanism for the regulation of angiogenesis. Proc Natl Acad Sci USA 96(4):1246–1251
Tabruyn SP, Sabatel C, Nguyen NQ et al (2007) The angiostatic 16 K human prolactin overcomes endothelial cell anergy and promotes leukocyte infiltration via nuclear factor-kappaB activation. Mol Endocrinol 21(6):1422–1429
Pfeffer U, Ferrari N, Dell’Eva R et al (2005) Molecular mechanisms of action of angiopreventive anti-oxidants on endothelial cells: microarray gene expression analyses. Mutat Res 591(1–2):198–211
Kanda N, Watanabe S (2007) Prolactin enhances interferon-gamma-induced production of CXC ligand 9 (CXCL9), CXCL10, and CXCL11 in human keratinocytes. Endocrinology 148(5):2317–2325
Dvorak HF, Gresser I (1989) Microvascular injury in pathogenesis of interferon-induced necrosis of subcutaneous tumors in mice. J Natl Cancer Inst 81(7):497–502
Sidky YA, Borden EC (1987) Inhibition of angiogenesis by interferons: effects on tumor- and lymphocyte-induced vascular responses. Cancer Res 47(19):5155–5161
Minuzzo S, Moserle L, Indraccolo S et al (2007) Angiogenesis meets immunology: cytokine gene therapy of cancer. Mol Aspects Med 28(1):59–86
Singh RP, Dhanalakshmi S, Agarwal C et al (2005) Silibinin strongly inhibits growth and survival of human endothelial cells via cell cycle arrest and downregulation of survivin, Akt and NF-kappaB: implications for angioprevention and antiangiogenic therapy. Oncogene 24(7):1188–1202
Oliveira IC, Sciavolino PJ, Lee TH et al (1992) Downregulation of interleukin 8 gene expression in human fibroblasts: unique mechanism of transcriptional inhibition by interferon. Proc Natl Acad Sci USA 89(19):9049–9053
von Marschall Z, Scholz A, Cramer T et al (2003) Effects of interferon alpha on vascular endothelial growth factor gene transcription and tumor angiogenesis. J Natl Cancer Inst 95(6):437–448
Albini A, Marchisone C, Del Grosso F et al (2000) Inhibition of angiogenesis and vascular tumor growth by interferon-producing cells: A gene therapy approach. Am J Pathol 156(4):1381–1393
Indraccolo S, Gola E, Rosato A et al (2002) Differential effects of angiostatin, endostatin and interferon-alpha(1) gene transfer on in vivo growth of human breast cancer cells. Gene Ther 9(13):867–878
Persano L, Moserle L, Esposito G et al (2009) Interferon-alpha counteracts the angiogenic switch and reduces tumor cell proliferation in a spontaneous model of prostatic cancer. Carcinogenesis 30(5):851–860
Indraccolo S, Pfeffer U, Minuzzo S et al (2007) Identification of genes selectively regulated by IFNs in endothelial cells. J Immunol 178(2):1122–1135
Kitaya K, Yasuo T, Yamaguchi T et al (2007) Genes regulated by interferon-gamma in human uterine microvascular endothelial cells. Int J Mol Med 20(5):689–697
Sana TR, Janatpour MJ, Sathe M et al (2005) Microarray analysis of primary endothelial cells challenged with different inflammatory and immune cytokines. Cytokine 29(6):256–269
Taylor KL, Leaman DW, Grane R et al (2008) Identification of interferon-beta-stimulated genes that inhibit angiogenesis in vitro. J Interferon Cytokine Res 28(12):733–740
Guenzi E, Topolt K, Lubeseder-Martellato C et al (2003) The guanylate binding protein-1 GTPase controls the invasive and angiogenic capability of endothelial cells through inhibition of MMP-1 expression. EMBO J 22(15):3772–3782
Lubeseder-Martellato C, Guenzi E, Jorg A et al (2002) Guanylate-binding protein-1 expression is selectively induced by inflammatory cytokines and is an activation marker of endothelial cells during inflammatory diseases. Am J Pathol 161(5):1749–1759
Angiolillo AL, Sgadari C, Taub DD et al (1995) Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J Exp Med 182(1):155–162
Sgadari C, Angiolillo AL, Cherney BW et al (1996) Interferon-inducible protein-10 identified as a mediator of tumor necrosis in vivo. Proc Natl Acad Sci USA 93(24):13791–13796
De Bouard S, Guillamo JS, Christov C et al (2003) Antiangiogenic therapy against experimental glioblastoma using genetically engineered cells producing interferon-alpha, angiostatin, or endostatin. Hum Gene Ther 14(9):883–895
Indraccolo S, Moserle L, Tisato V et al (2006) Gene therapy of ovarian cancer with IFN-alpha-producing fibroblasts: comparison of constitutive and inducible vectors. Gene Ther 13(12):953–965
Indraccolo S, Tisato V, Tosello V et al (2005) Interferon-alpha gene therapy by lentiviral vectors contrasts ovarian cancer growth through angiogenesis inhibition. Hum Gene Ther 16(8):957–970
Rozera C, Carlei D, Lollini PL et al (1999) Interferon (IFN)-beta gene transfer into TS/A adenocarcinoma cells and comparison with IFN-alpha: differential effects on tumorigenicity and host response. Am J Pathol 154(4):1211–1222
Belardelli F, Gresser I, Maury C et al (1983) Antitumor effects of interferon in mice injected with interferon-sensitive and interferon-resistant Friend leukemia cells. III. Inhibition of growth and necrosis of tumors implanted subcutaneously. Int J Cancer 31(5):649–653
Bergers G, Hanahan D (2008) Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8(8):592–603
Curnis F, Gasparri A, Sacchi A et al (2005) Targeted delivery of IFNgamma to tumor vessels uncouples antitumor from counterregulatory mechanisms. Cancer Res 65(7):2906–2913
Tedjarati S, Baker CH, Apte S et al (2002) Synergistic therapy of human ovarian carcinoma implanted orthotopically in nude mice by optimal biological dose of pegylated interferon alpha combined with paclitaxel. Clin Cancer Res 8(7):2413–2422
Samarajiwa SA, Forster S, Auchettl K et al (2009) INTERFEROME: the database of interferon regulated genes. Nucleic Acids Res 37(Database issue):D852–D857
Kerbel RS, Hawley RG (1995) Interleukin 12: newest member of the antiangiogenesis club. J Natl Cancer Inst 87(8):557–559
Sgadari C, Angiolillo AL, Tosato G (1996) Inhibition of angiogenesis by interleukin-12 is mediated by the interferon-inducible protein 10. Blood 87(9):3877–3882
Voest EE, Kenyon BM, O’Reilly MS et al (1995) Inhibition of angiogenesis in vivo by interleukin 12. J Natl Cancer Inst 87(8):581–586
Yao L, Sgadari C, Furuke K et al (1999) Contribution of natural killer cells to inhibition of angiogenesis by interleukin-12. Blood 93(5):1612–1621
Shi X, Cao S, Mitsuhashi M et al (2004) Genome-wide analysis of molecular changes in IL-12-induced control of mammary carcinoma via IFN-gamma-independent mechanisms. J Immunol 172(7):4111–4122
Clark AF, Mellon J, Li XY et al (1999) Inhibition of intraocular tumor growth by topical application of the angiostatic steroid anecortave acetate. Invest Ophthalmol Vis Sci 40(9):2158–2162
Parkins CS, Holder AL, Hill SA et al (2000) Determinants of anti-vascular action by combretastatin A-4 phosphate: role of nitric oxide. Br J Cancer 83(6):811–816
Ingber D, Fujita T, Kishimoto S et al (1990) Synthetic analogues of fumagillin that inhibit angiogenesis and suppress tumour growth. Nature 348(6301):555–557
D’Amato RJ, Loughnan MS, Flynn E et al (1994) Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci USA 91(9):4082–4085
Vacca A, Scavelli C, Montefusco V et al (2005) Thalidomide downregulates angiogenic genes in bone marrow endothelial cells of patients with active multiple myeloma. J Clin Oncol 23(23):5334–5346
Majumdar S, Lamothe B, Aggarwal BB (2002) Thalidomide suppresses NF-kappa B activation induced by TNF and H2O2, but not that activated by ceramide, lipopolysaccharides, or phorbol ester. J Immunol 168(6):2644–2651
Ferrari N, Pfeffer U, Dell’Eva R et al (2005) The transforming growth factor-beta family members bone morphogenetic protein-2 and macrophage inhibitory cytokine-1 as mediators of the antiangiogenic activity of N-(4-hydroxyphenyl)retinamide. Clin Cancer Res 11(12):4610–4619
Costa A, Formelli F, Chiesa F et al (1994) Prospects of chemoprevention of human cancers with the synthetic retinoid fenretinide. Cancer Res 54(7 Suppl):2032s–2037s
Blackwell KL, Haroon ZA, Shan S et al (2000) Tamoxifen inhibits angiogenesis in estrogen receptor-negative animal models. Clin Cancer Res 6(11):4359–4364
del Carmen Garcia M, olina Wolgien M, da Silva ID, Villanova FE et al (2005) Differential gene expression assessed by cDNA microarray analysis in breast cancer tissue under tamoxifen treatment. Eur J Gynaecol Oncol 26(5):501–504
Itoh T, Karlsberg K, Kijima I et al (2005) Letrozole-, anastrozole-, and tamoxifen-responsive genes in MCF-7aro cells: a microarray approach. Mol Cancer Res 3(4):203–218
Ferrara N, Hillan KJ, Gerber HP et al (2004) Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 3(5):391–400
Murphy DA, Makonnen S, Lassoued W et al (2006) Inhibition of tumor endothelial ERK activation, angiogenesis, and tumor growth by sorafenib (BAY43–9006). Am J Pathol 169(5):1875–1885
Sun L, Liang C, Shirazian S et al (2003) Discovery of 5-[5-fluoro-2-oxo-1, 2- dihydroindol-(3Z)-ylidenemethyl]-2, 4- dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J Med Chem 46(7):1116–1119
Fischer C, Jonckx B, Mazzone M et al (2007) Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 131(3):463–475
Guba M, von Breitenbuch P, Steinbauer M et al (2002) Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med 8(2):128–135
Jia Z, Zhang J, Wei D et al (2007) Molecular basis of the synergistic antiangiogenic activity of bevacizumab and mithramycin A. Cancer Res 67(10):4878–4885
Yang SX, Steinberg SM, Nguyen D et al (2008) Gene expression profile and angiogenic marker correlates with response to neoadjuvant bevacizumab followed by bevacizumab plus chemotherapy in breast cancer. Clin Cancer Res 14(18):5893–5899
Albini A, Mirisola V, Pfeffer U (2008) Metastasis signatures: genes regulating tumor-microenvironment interactions predict metastatic behavior. Cancer Metastasis Rev 27(1):75–83
Moreno-Vinasco L, Gomberg-Maitland M, Maitland ML et al (2008) Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol Genomics 33(2):278–291
Newell P, Toffanin S, Villanueva A et al (2009) Ras pathway activation in hepatocellular carcinoma and anti-tumoral effect of combined sorafenib and rapamycin in vivo. J Hepatol 51(4):725–733
Kasukabe T, Okabe-Kado J, Kato N et al (2005) Effects of combined treatment with rapamycin and cotylenin A, a novel differentiation-inducing agent, on human breast carcinoma MCF-7 cells and xenografts. Breast Cancer Res 7(6):R1097–R1110
Grolleau A, Bowman J, Pradet-Balade B et al (2002) Global and specific translational control by rapamycin in T cells uncovered by microarrays and proteomics. J Biol Chem 277(25):22175–22184
Park IH, Chen J (2005) Mammalian target of rapamycin (mTOR) signaling is required for a late-stage fusion process during skeletal myotube maturation. J Biol Chem 280(36):32009–32017
Gera JF, Mellinghoff IK, Shi Y et al (2004) AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem 279(4):2737–2746
van de Vijver MJ, He YD, van’t Veer LJ et al (2002) A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 347(25):1999–2009
Ramaswamy S, Ross KN, Lander ES et al (2003) A molecular signature of metastasis in primary solid tumors. Nat Genet 33(1):49–54
Webb T (2003) Microarray studies challenge theories of metastasis. J Natl Cancer Inst 95(5):350–351
Bernards R, Weinberg RA (2002) A progression puzzle. Nature 418(6900):823
Pfeffer U, Noonan D, Albini A (2003) Re: microarray studies challenge theories of metastasis. J Natl Cancer Inst 95(11):829
Albini A, Morini M, D’Agostini F et al (2001) Inhibition of angiogenesis-driven Kaposi’s sarcoma tumor growth in nude mice by oral N-acetylcysteine. Cancer Res 61(22):8171–8178
Garbisa S, Biggin S, Cavallarin N et al (1999) Tumor invasion: molecular shears blunted by green tea. Nat Med 5(11):1216
Yu YM, Wang ZH, Liu CH et al (2007) Ellagic acid inhibits IL-1beta-induced cell adhesion molecule expression in human umbilical vein endothelial cells. Br J Nutr 97(4):692–698
Kumar A, Dhawan S, Hardegen NJ et al (1998) Curcumin (Diferuloylmethane) inhibition of tumor necrosis factor (TNF)-mediated adhesion of monocytes to endothelial cells by suppression of cell surface expression of adhesion molecules and of nuclear factor-kappaB activation. Biochem Pharmacol 55(6):775–783
Lorusso G, Vannini N, Sogno I et al (2009) Mechanisms of Hyperforin as an anti-angiogenic angioprevention agent. Eur J Cancer 45(8):1474–1484
Khachigian LM, Collins T, Fries JW (1997) N-acetyl cysteine blocks mesangial VCAM-1 and NF-kappa B expression in vivo. Am J Pathol 151(5):1225–1229
Sethi G, Ahn KS, Sung B et al (2008) Pinitol targets nuclear factor-kappaB activation pathway leading to inhibition of gene products associated with proliferation, apoptosis, invasion, and angiogenesis. Mol Cancer Ther 7(6):1604–1614
Ahmad R, Raina D, Meyer C et al (2006) Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. J Biol Chem 281(47):35764–35769
Albini A, Dell’Eva R, Vene R et al (2006) Mechanisms of the antiangiogenic activity by the hop flavonoid xanthohumol: NF-kappaB and Akt as targets. FASEB J 20(3):527–529
Lapillonne H, Konopleva M, Tsao T et al (2003) Activation of peroxisome proliferator-activated receptor gamma by a novel synthetic triterpenoid 2-cyano-3, 12-dioxooleana-1, 9-dien-28-oic acid induces growth arrest and apoptosis in breast cancer cells. Cancer Res 63(18):5926–5939
Ahmad R, Raina D, Meyer C et al (2008) Triterpenoid CDDO-methyl ester inhibits the Janus-activated kinase-1 (JAK1)– >signal transducer and activator of transcription-3 (STAT3) pathway by direct inhibition of JAK1 and STAT3. Cancer Res 68(8):2920–2926
Sussan TE, Rangasamy T, Blake DJ et al (2009) Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc Natl Acad Sci USA 106(1):250–255
Chen HH, Zhou HJ, Wu GD et al (2004) Inhibitory effects of artesunate on angiogenesis and on expressions of vascular endothelial growth factor and VEGF receptor KDR/flk-1. Pharmacology 71(1):1–9
Pang X, Yi Z, Zhang X et al (2009) Acetyl-11-keto-beta-boswellic acid inhibits prostate tumor growth by suppressing vascular endothelial growth factor receptor 2-mediated angiogenesis. Cancer Res 69(14):5893–5900
Yi T, Yi Z, Cho SG et al (2008) Gambogic acid inhibits angiogenesis and prostate tumor growth by suppressing vascular endothelial growth factor receptor 2 signaling. Cancer Res 68(6):1843–1850
Larghero P, Vene R, Minghelli S et al (2007) Biological assays and genomic analysis reveal lipoic acid modulation of endothelial cell behavior and gene expression. Carcinogenesis 28(5):1008–1020
Park MJ, Kim EH, Park IC et al (2002) Curcumin inhibits cell cycle progression of immortalized human umbilical vein endothelial (ECV304) cells by up-regulating cyclin-dependent kinase inhibitor, p21WAF1/CIP1, p27KIP1 and p53. Int J Oncol 21(2):379–383
Stierum R, Conesa A, Heijne W et al (2008) Transcriptome analysis provides new insights into liver changes induced in the rat upon dietary administration of the food additives butylated hydroxytoluene, curcumin, propyl gallate and thiabendazole. Food Chem Toxicol 46(8):2616–2628
Meng Q, Velalar CN, Ruan R (2008) Regulating the age-related oxidative damage, mitochondrial integrity, and antioxidative enzyme activity in Fischer 344 rats by supplementation of the antioxidant epigallocatechin-3-gallate. Rejuvenation Res 11(3):649–660
Thangapazham RL, Passi N, Maheshwari RK (2007) Green tea polyphenol and epigallocatechin gallate induce apoptosis and inhibit invasion in human breast cancer cells. Cancer Biol Ther 6(12):1938–1943
Shen G, Xu C, Hu R et al (2005) Comparison of (−)-epigallocatechin-3-gallate elicited liver and small intestine gene expression profiles between C57BL/6 J mice and C57BL/6 J/Nrf2 (−/−) mice. Pharm Res 22(11):1805–1820
Guo S, Yang S, Taylor C et al (2005) Green tea polyphenol epigallocatechin-3 gallate (EGCG) affects gene expression of breast cancer cells transformed by the carcinogen 7, 12-dimethylbenz[a]anthracene. J Nutr 135(12 Suppl):2978S–2986S
Guo S, Lu J, Subramanian A et al (2006) Microarray-assisted pathway analysis identifies mitogen-activated protein kinase signaling as a mediator of resistance to the green tea polyphenol epigallocatechin 3-gallate in her-2/neu-overexpressing breast cancer cells. Cancer Res 66(10):5322–5329
Bae JY, Kanamune J, Han DW et al (2009) Reversible regulation of cell cycle-related genes by epigallocatechin gallate for hibernation of neonatal human tarsal fibroblasts. Cell Transplant 18(4):459–469
Hsu S, Dickinson DP, Qin H et al (2005) Inhibition of autoantigen expression by (−)-epigallocatechin-3-gallate (the major constituent of green tea) in normal human cells. J Pharmacol Exp Ther 315(2):805–811
Wolfram S, Raederstorff D, Preller M et al (2006) Epigallocatechin gallate supplementation alleviates diabetes in rodents. J Nutr 136(10):2512–2518
Nones K, Dommels YE, Martell S et al (2009) The effects of dietary curcumin and rutin on colonic inflammation and gene expression in multidrug resistance gene-deficient (mdr1a−/−) mice, a model of inflammatory bowel diseases. Br J Nutr 101(2):169–181
Sun M, Estrov Z, Ji Y et al (2008) Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Mol Cancer Ther 7(3):464–473
Su CC, Chen GW, Lin JG et al (2006) Curcumin inhibits cell migration of human colon cancer colo 205 cells through the inhibition of nuclear factor kappa B/p65 and down-regulates cyclooxygenase-2 and matrix metalloproteinase-2 expressions. Anticancer Res 26(2A):1281–1288
Ramachandran C, Rodriguez S, Ramachandran R et al (2005) Expression profiles of apoptotic genes induced by curcumin in human breast cancer and mammary epithelial cell lines. Anticancer Res 25(5):3293–3302
Bachmeier BE, Mohrenz IV, Mirisola V et al (2008) Curcumin downregulates the inflammatory cytokines CXCL1 and -2 in breast cancer cells via NFkappaB. Carcinogenesis 29(4):779–789
Bachmeier B, Nerlich AG, Iancu CM et al (2007) The chemopreventive polyphenol Curcumin prevents hematogenous breast cancer metastases in immunodeficient mice. Cell Physiol Biochem 19(1–4):137–152
Arbiser JL, Klauber N, Rohan R et al (1998) Curcumin is an in vivo inhibitor of angiogenesis. Mol Med 4(6):376–383
Dell’Eva R, Pfeffer U, Vene R et al (2004) Inhibition of angiogenesis in vivo and growth of Kaposi’s sarcoma xenograft tumors by the anti-malarial artesunate. Biochem Pharmacol 68(12):2359–2366
Anfosso L, Efferth T, Albini A et al (2006) Microarray expression profiles of angiogenesis-related genes predict tumor cell response to artemisinins. Pharmacogenomics J 6(4):269–278
Gonzalez-Sarrias A, Espin JC, Tomas-Barberan FA et al (2009) Gene expression, cell cycle arrest and MAPK signalling regulation in Caco-2 cells exposed to ellagic acid and its metabolites, urolithins. Mol Nutr Food Res 53(6):686–698
Gu H, You Q, Liu W et al (2008) Gambogic acid induced tumor cell apoptosis by T lymphocyte activation in H22 transplanted mice. Int Immunopharmacol 8(11):1493–1502
Krusekopf S, Roots I (2005) St. John’s wort and its constituent hyperforin concordantly regulate expression of genes encoding enzymes involved in basic cellular pathways. Pharmacogenet Genomics 15(11):817–829
Vannini N, Lorusso G, Cammarota R et al (2007) The synthetic oleanane triterpenoid, CDDO-methyl ester, is a potent antiangiogenic agent. Mol Cancer Ther 6(12 Pt 1):3139–3146
Yates MS, Kwak MK, Egner PA et al (2006) Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-, 12-dioxooleana-1, 9(11)-dien-28-oyl]imidazole. Cancer Res 66(4):2488–2494
Das A, Mantena SR, Kannan A et al (2009) De novo synthesis of estrogen in pregnant uterus is critical for stromal decidualization and angiogenesis. Proc Natl Acad Sci USA 106(30):12542–12547
Seo KH, Lee HS, Jung B et al (2004) Estrogen enhances angiogenesis through a pathway involving platelet-activating factor-mediated nuclear factor-kappaB activation. Cancer Res 64(18):6482–6488
Johns A, Freay AD, Fraser W et al (1996) Disruption of estrogen receptor gene prevents 17 beta estradiol-induced angiogenesis in transgenic mice. Endocrinology 137(10):4511–4513
Chen Y, Jin X, Zeng Z et al (2009) Estrogen-replacement therapy promotes angiogenesis after acute myocardial infarction by enhancing SDF-1 and estrogen receptor expression. Microvasc Res 77(2):71–77
Hartman J, Lindberg K, Morani A et al (2006) Estrogen receptor beta inhibits angiogenesis and growth of T47D breast cancer xenografts. Cancer Res 66(23):11207–11213
Beral V (1997) Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Collaborative group on hormonal factors in breast cancer. Lancet 350(9084):1047–1059
Fotsis T, Pepper M, Adlercreutz H et al (1993) Genistein, a dietary-derived inhibitor of in vitro angiogenesis. Proc Natl Acad Sci USA 90(7):2690–2694
Ambra R, Rimbach G, de Pascual Teresa S et al (2006) Genistein affects the expression of genes involved in blood pressure regulation and angiogenesis in primary human endothelial cells. Nutr Metab Cardiovasc Dis 16(1):35–43
Piao M, Mori D, Satoh T et al (2006) Inhibition of endothelial cell proliferation, in vitro angiogenesis, and the down-regulation of cell adhesion-related genes by genistein. Combined with a cDNA microarray analysis. Endothelium 13(4):249–266
Li Y, Sarkar FH (2002) Gene expression profiles of genistein-treated PC3 prostate cancer cells. J Nutr 132(12):3623–3631
Suzuki K, Koike H, Matsui H et al (2002) Genistein, a soy isoflavone, induces glutathione peroxidase in the human prostate cancer cell lines LNCaP and PC-3. Int J Cancer 99(6):846–852
Li Y, Kucuk O, Hussain M et al (2006) Antitumor and antimetastatic activities of docetaxel are enhanced by genistein through regulation of osteoprotegerin/receptor activator of nuclear factor-kappaB (RANK)/RANK ligand/MMP-9 signaling in prostate cancer. Cancer Res 66(9):4816–4825
Li Y, Che M, Bhagat S et al (2004) Regulation of gene expression and inhibition of experimental prostate cancer bone metastasis by dietary genistein. Neoplasia 6(4):354–363
Bai J, Sata N, Nagai H et al (2004) Genistein-induced changes in gene expression in Panc 1 cells at physiological concentrations of genistein. Pancreas 29(2):93–98
Takahashi Y, Odbayar TO, Ide T (2009) A comparative analysis of genistein and daidzein in affecting lipid metabolism in rat liver. J Clin Biochem Nutr 44(3):223–230
Zou H, Zhan S, Cao K (2008) Apoptotic activity of genistein on human lung adenocarcinoma SPC-A-1 cells and preliminary exploration of its mechanisms using microarray. Biomed Pharmacother 62(9):583–589
Lee WY, Huang SC, Tzeng CC et al (2007) Alterations of metastasis-related genes identified using an oligonucleotide microarray of genistein-treated HCC1395 breast cancer cells. Nutr Cancer 58(2):239–246
Penza M, Montani C, Romani A et al (2006) Genistein affects adipose tissue deposition in a dose-dependent and gender-specific manner. Endocrinology 147(12):5740–5751
Cooke PS, Selvaraj V, Yellayi S (2006) Genistein, estrogen receptors, and the acquired immune response. J Nutr 136(3):704–708
Wang XJ, Bartolucci-Page E, Fenton SE et al (2006) Altered mammary gland development in male rats exposed to genistein and methoxychlor. Toxicol Sci 91(1):93–103
Lavigne JA, Takahashi Y, Chandramouli GV et al (2008) Concentration-dependent effects of genistein on global gene expression in MCF-7 breast cancer cells: an oligo microarray study. Breast Cancer Res Treat 110(1):85–98
Konstantakopoulos N, Montgomery KG, Chamberlain N et al (2006) Changes in gene expressions elicited by physiological concentrations of genistein on human endometrial cancer cells. Mol Carcinog 45(10):752–763
Igura K, Ohta T, Kuroda Y et al (2001) Resveratrol and quercetin inhibit angiogenesis in vitro. Cancer Lett 171(1):11–16
Gehm BD, McAndrews JM, Chien PY et al (1997) Resveratrol, a polyphenolic compound found in grapes and wine, is an agonist for the estrogen receptor. Proc Natl Acad Sci USA 94(25):14138–14143
Scambia G, Ranelletti FO, Benedetti Panici P et al (1991) Quercetin inhibits the growth of a multidrug-resistant estrogen-receptor-negative MCF-7 human breast-cancer cell line expressing type II estrogen-binding sites. Cancer Chemother Pharmacol 28(4):255–258
van der Woude H, Ter Veld MG, Jacobs N et al (2005) The stimulation of cell proliferation by quercetin is mediated by the estrogen receptor. Mol Nutr Food Res 49(8):763–771
Woodall BP, Nystrom A, Iozzo RA et al (2008) Integrin alpha2beta1 is the required receptor for endorepellin angiostatic activity. J Biol Chem 283(4):2335–2343
Ambesi A, Klein RM, Pumiglia KM et al (2005) Anastellin, a fragment of the first type III repeat of fibronectin, inhibits extracellular signal-regulated kinase and causes G(1) arrest in human microvessel endothelial cells. Cancer Res 65(1):148–156
Kalluri R (2002) Discovery of type IV collagen non-collagenous domains as novel integrin ligands and endogenous inhibitors of angiogenesis. Cold Spring Harb Symp Quant Biol 67:255–266
Schiemann WP, Blobe GC, Kalume DE et al (2002) Context-specific effects of fibulin-5 (DANCE/EVEC) on cell proliferation, motility, and invasion. Fibulin-5 is induced by transforming growth factor-beta and affects protein kinase cascades. J Biol Chem 277(30):27367–27377
Greenwood JA, Pallero MA, Theibert AB et al (1998) Thrombospondin signaling of focal adhesion disassembly requires activation of phosphoinositide 3-kinase. J Biol Chem 273(3):1755–1763
Orr AW, Pallero MA, Murphy-Ullrich JE (2002) Thrombospondin stimulates focal adhesion disassembly through Gi- and phosphoinositide 3-kinase-dependent ERK activation. J Biol Chem 277(23):20453–20460
Lopes N, Gregg D, Vasudevan S et al (2003) Thrombospondin 2 regulates cell proliferation induced by Rac1 redox-dependent signaling. Mol Cell Biol 23(15):5401–5408
Sudhakar A, Sugimoto H, Yang C et al (2003) Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci USA 100(8):4766–4771
Leali D, Alessi P, Coltrini D et al (2009) Fibroblast growth factor-2 antagonist and antiangiogenic activity of long-pentraxin 3-derived synthetic peptides. Curr Pharm Des 15(30):3577–3589
Cai J, Jiang WG, Grant MB et al (2006) Pigment epithelium-derived factor inhibits angiogenesis via regulated intracellular proteolysis of vascular endothelial growth factor receptor 1. J Biol Chem 281(6):3604–3613
Yamagishi S, Amano S, Inagaki Y et al (2003) Pigment epithelium-derived factor inhibits leptin-induced angiogenesis by suppressing vascular endothelial growth factor gene expression through anti-oxidative properties. Microvasc Res 65(3):186–190
Liu W, Wu Z, Guan M et al (2009) cDNA microarray analysis of pigment epithelium-derived factor-regulated gene expression profile in prostate carcinoma cells. Int J Urol 16(3):323–328
Chen YH, Wu HL, Chen CK et al (2003) Angiostatin antagonizes the action of VEGF-A in human endothelial cells via two distinct pathways. Biochem Biophys Res Commun 310(3):804–810
Bae JS, Rezaie AR (2009) Mutagenesis studies toward understanding the intracellular signaling mechanism of antithrombin. J Thromb Haemost 7(5):803–810
Jouan V, Canron X, Alemany M et al (1999) Inhibition of in vitro angiogenesis by platelet factor-4-derived peptides and mechanism of action. Blood 94(3):984–993
Brooks PC, Silletti S, von Schalscha TL et al (1998) Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell 92(3):391–400
Moses MA, Wiederschain D, Wu I et al (1999) Troponin I is present in human cartilage and inhibits angiogenesis. Proc Natl Acad Sci USA 96(6):2645–2650
Feldman L, Rouleau C (2002) Troponin I inhibits capillary endothelial cell proliferation by interaction with the cell’s bFGF receptor. Microvasc Res 63(1):41–49
Blois A, Srebro B, Mandala M et al (2006) The chromogranin A peptide vasostatin-I inhibits gap formation and signal transduction mediated by inflammatory agents in cultured bovine pulmonary and coronary arterial endothelial cells. Regul Pept 135(1–2):78–84
Yang CR, Hsieh SL, Teng CM et al (2004) Soluble decoy receptor 3 induces angiogenesis by neutralization of TL1A, a cytokine belonging to tumor necrosis factor superfamily and exhibiting angiostatic action. Cancer Res 64(3):1122–1129
Wen L, Zhuang L, Luo X et al (2003) TL1A-induced NF-kappaB activation and c-IAP2 production prevent DR3-mediated apoptosis in TF-1 cells. J Biol Chem 278(40):39251–39258
Hou W, Medynski D, Wu S et al (2005) VEGI-192, a new isoform of TNFSF15, specifically eliminates tumor vascular endothelial cells and suppresses tumor growth. Clin Cancer Res 11(15):5595–5602
Watanabe K, Hasegawa Y, Yamashita H et al (2004) Vasohibin as an endothelium-derived negative feedback regulator of angiogenesis. J Clin Invest 114(7):898–907
Otani A, Slike BM, Dorrell MI et al (2002) A fragment of human TrpRS as a potent antagonist of ocular angiogenesis. Proc Natl Acad Sci USA 99(1):178–183
Tzima E, Reader JS, Irani-Tehrani M et al (2005) VE-cadherin links tRNA synthetase cytokine to anti-angiogenic function. J Biol Chem 280(4):2405–2408
Tong Z, Kunnumakkara AB, Wang H et al (2008) Neutrophil gelatinase-associated lipocalin: a novel suppressor of invasion and angiogenesis in pancreatic cancer. Cancer Res 68(15):6100–6108
Tsuruoka N, Sugiyama M, Tawaragi Y et al (1988) Inhibition of in vitro angiogenesis by lymphotoxin and interferon-gamma. Biochem Biophys Res Commun 155(1):429–435
Strieter RM, Kunkel SL, Arenberg DA et al (1995) Interferon gamma-inducible protein 10 (IP-10), a member of the C-X-C chemokine family, is an inhibitor of angiogenesis. Biochem Biophys Res Commun 210(1):51–57
Koike F, Satoh J, Miyake S et al (2003) Microarray analysis identifies interferon beta-regulated genes in multiple sclerosis. J Neuroimmunol 139(1–2):109–118
Huang T, Tu K, Shyr Y et al (2008) The prediction of interferon treatment effects based on time series microarray gene expression profiles. J Transl Med 6:44
Chen Y, Antoniou E, Liu Z et al (2007) A microarray analysis for genes regulated by interferon-tau in ovine luminal epithelial cells. Reproduction 134(1):123–135
Zou W, Kim JH, Handidu A et al (2007) Microarray analysis reveals that Type I interferon strongly increases the expression of immune-response related genes in Ubp43 (Usp18) deficient macrophages. Biochem Biophys Res Commun 356(1):193–199
Crow MK, Kirou KA, Wohlgemuth J (2003) Microarray analysis of interferon-regulated genes in SLE. Autoimmunity 36(8):481–490
Cozzolino F, Torcia M, Aldinucci D et al (1990) Interleukin 1 is an autocrine regulator of human endothelial cell growth. Proc Natl Acad Sci USA 87(17):6487–6491
Tamura T, Nakanishi T, Kimura Y et al (1996) Nitric oxide mediates interleukin-1-induced matrix degradation and basic fibroblast growth factor release in cultured rabbit articular chondrocytes: a possible mechanism of pathological neovascularization in arthritis. Endocrinology 137(9):3729–3737
Williams MR, Kataoka N, Sakurai Y et al (2008) Gene expression of endothelial cells due to interleukin-1 beta stimulation and neutrophil transmigration. Endothelium 15(1):73–165
Zhao B, Stavchansky SA, Bowden RA et al (2003) Effect of interleukin-1beta and tumor necrosis factor-alpha on gene expression in human endothelial cells. Am J Physiol Cell Physiol 284(6):C1577–C1583
Elaraj DM, Weinreich DM, Varghese S et al (2006) The role of interleukin 1 in growth and metastasis of human cancer xenografts. Clin Cancer Res 12(4):1088–1096
Shi J, Schmitt-Talbot E, DiMattia DA et al (2004) The differential effects of IL-1 and TNF-alpha on proinflammatory cytokine and matrix metalloproteinase expression in human chondrosarcoma cells. Inflamm Res 53(8):377–389
Berchtold LA, Larsen CM, Vaag A et al (2009) IL-1 receptor antagonism and muscle gene expression in patients with type 2 diabetes. Eur Cytokine Netw 20(2):81–87
Ching S, Zhang H, Chen Q et al (2007) Differential expression of extracellular matrix and adhesion molecule genes in the brain of juvenile versus adult mice in responses to intracerebroventricular administration of IL-1. Neuroimmunomodulation 14(1):46–56
Volpert OV, Fong T, Koch AE et al (1998) Inhibition of angiogenesis by interleukin 4. J Exp Med 188(6):1039–1046
Schnyder B, Lugli S, Feng N et al (1996) Interleukin-4 (IL-4) and IL-13 bind to a shared heterodimeric complex on endothelial cells mediating vascular cell adhesion molecule-1 induction in the absence of the common gamma chain. Blood 87(10):4286–4295
Matsumoto K, Ohi H, Kanmatsuse K (1999) Interleukin-4 cooperates with interleukin-10 to inhibit vascular permeability factor release by peripheral blood mononuclear cells from patients with minimal-change nephrotic syndrome. Am J Nephrol 19(1):21–27
Chaitidis P, O’Donnell V, Kuban RJ et al (2005) Gene expression alterations of human peripheral blood monocytes induced by medium-term treatment with the TH2-cytokines interleukin-4 and -13. Cytokine 30(6):366–377
Lee YW, Eum SY, Chen KC et al (2004) Gene expression profile in interleukin-4-stimulated human vascular endothelial cells. Mol Med 10(1–6):19–27
Coughlin CM, Salhany KE, Gee MS et al (1998) Tumor cell responses to IFNgamma affect tumorigenicity and response to IL-12 therapy and antiangiogenesis. Immunity 9(1):25–34
Dias S, Boyd R, Balkwill F (1998) IL-12 regulates VEGF and MMPs in a murine breast cancer model. Int J Cancer 78(3):361–365
Renneson J, Dutta B, Goriely S et al (2009) IL-12 and type I IFN response of neonatal myeloid DC to human cytomegalovirus infection. Eur J Immunol 39(10):2789–2799
Shi Y, Zou M, Baitei EY et al (2008) Cannabinoid 2 receptor induction by IL-12 and its potential as a therapeutic target for the treatment of anaplastic thyroid carcinoma. Cancer Gene Ther 15(2):101–107
Hoey T, Zhang S, Schmidt N et al (2003) Distinct requirements for the naturally occurring splice forms Stat4alpha and Stat4beta in IL-12 responses. EMBO J 22(16):4237–4248
Hodge DL, Schill WB, Wang JM et al (2002) IL-2 and IL-12 alter NK cell responsiveness to IFN-gamma-inducible protein 10 by down-regulating CXCR3 expression. J Immunol 168(12):6090–6098
Cao R, Farnebo J, Kurimoto M et al (1999) Interleukin-18 acts as an angiogenesis and tumor suppressor. FASEB J 13(15):2195–2202
Kim J, Kim C, Kim TS et al (2006) IL-18 enhances thrombospondin-1 production in human gastric cancer via JNK pathway. Biochem Biophys Res Commun 344(4):1284–1289
Coma G, Pena R, Blanco J et al (2006) Treatment of monocytes with interleukin (IL)-12 plus IL-18 stimulates survival, differentiation and the production of CXC chemokine ligands (CXCL)8, CXCL9 and CXCL10. Clin Exp Immunol 145(3):535–544
Wiener Z, Pocza P, Racz M et al (2008) IL-18 induces a marked gene expression profile change and increased Ccl1 (I-309) production in mouse mucosal mast cell homologs. Int Immunol 20(12):1565–1573
Seo M, Park M, Yook Y et al (2008) IL-18 gene expression pattern in exogenously treated AML cells. BMB Rep 41(6):461–465
Saha S, Gonzalez J, Rosenfeld G et al (2009) Prolactin alters the mechanisms of B cell tolerance induction. Arthritis Rheum 60(6):1743–1752
Charoenphandhu N, Wongdee K, Teerapornpuntakit J et al (2008) Transcriptome responses of duodenal epithelial cells to prolactin in pituitary-grafted rats. Mol Cell Endocrinol 296(1–2):41–52
Tomblyn S, Langenheim JF, Jacquemart IC et al (2005) The role of human prolactin and its antagonist, G129R, in mammary gland development and DMBA-initiated tumorigenesis in transgenic mice. Int J Oncol 27(5):1381–1389
Gass S, Harris J, Ormandy C et al (2003) Using gene expression arrays to elucidate transcriptional profiles underlying prolactin function. J Mammary Gland Biol Neoplasia 8(3):269–285
Dillner K, Kindblom J, Flores-Morales A et al (2003) Gene expression analysis of prostate hyperplasia in mice overexpressing the prolactin gene specifically in the prostate. Endocrinology 144(11):4955–4966
Dillner K, Kindblom J, Flores-Morales A et al (2002) Molecular characterization of prostate hyperplasia in prolactin-transgenic mice by using cDNA representational difference analysis. Prostate 52(2):139–149
Hou Z, Bailey JP, Vomachka AJ et al (2000) Glycosylation-dependent cell adhesion molecule 1 (GlyCAM 1) is induced by prolactin and suppressed by progesterone in mammary epithelium. Endocrinology 141(11):4278–4283
Quan H, Xu Y, Lou L (2008) p38 MAPK, but not ERK1/2, is critically involved in the cytotoxicity of the novel vascular disrupting agent combretastatin A4. Int J Cancer 122(8):1730–1737
Wang YQ, Luk JM, Chu AC et al (2006) TNP-470 blockage of VEGF synthesis is dependent on MAPK/COX-2 signaling pathway in PDGF-BB-activated hepatic stellate cells. Biochem Biophys Res Commun 341(1):239–244
Chen GJ, Weylie B, Hu C et al (2007) FGFR1/PI3 K/AKT signaling pathway is a novel target for antiangiogenic effects of the cancer drug fumagillin (TNP-470). J Cell Biochem 101(6):1492–1504
Koseki Y, Zava DT, Chamness GC et al (1977) Estrogen receptor translocation and replenishment by the antiestrogen tamoxifen. Endocrinology 101(4):1104–1110
Inai T, Mancuso M, Hashizume H et al (2004) Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol 165(1):35–52
Nghiemphu PL, Liu W, Lee Y et al (2009) Bevacizumab and chemotherapy for recurrent glioblastoma: a single-institution experience. Neurology 72(14):1217–1222
Maier AK, Kociok N, Zahn G et al (2007) Modulation of hypoxia-induced neovascularization by JSM6427, an integrin alpha5beta1 inhibiting molecule. Curr Eye Res 32(9):801–812
Strumberg D (2005) Preclinical and clinical development of the oral multikinase inhibitor sorafenib in cancer treatment. Drugs Today (Barc) 41(12):773–784
Gragoudas ES, Adamis AP, Cunningham ET Jr et al (2004) Pegaptanib for neovascular age-related macular degeneration. N Engl J Med 351(27):2805–2816
Banerjee S, Zvelebil M, Furet P et al (2009) The vascular endothelial growth factor receptor inhibitor PTK787/ZK222584 inhibits aromatase. Cancer Res 69(11):4716–4723
Rosenfeld PJ, Brown DM, Heier JS et al (2006) Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 355(14):1419–1431
Hosoi H, Dilling MB, Shikata T et al (1999) Rapamycin causes poorly reversible inhibition of mTOR and induces p53-independent apoptosis in human rhabdomyosarcoma cells. Cancer Res 59(4):886–894
Preiss T, Baron-Benhamou J, Ansorge W et al (2003) Homodirectional changes in transcriptome composition and mRNA translation induced by rapamycin and heat shock. Nat Struct Biol 10(12):1039–1047
Palanki MS, Akiyama H, Campochiaro P et al (2008) Development of prodrug 4-chloro-3-(5-methyl-3-{[4-(2-pyrrolidin-1-ylethoxy)phenyl]amino}-1, 2, 4-be nzotriazin-7-yl)phenyl benzoate (TG100801): a topically administered therapeutic candidate in clinical trials for the treatment of age-related macular degeneration. J Med Chem 51(6):1546–1559
Fabbrini M, Trachsel E, Soldani P et al (2006) Selective occlusion of tumor blood vessels by targeted delivery of an antibody-photosensitizer conjugate. Int J Cancer 118(7):1805–1813
Billerey-Larmonier C, Uno JK, Larmonier N et al (2008) Protective effects of dietary curcumin in mouse model of chemically induced colitis are strain dependent. Inflamm Bowel Dis 14(6):780–793
Nguyen KT, Shaikh N, Shukla KP et al (2004) Molecular responses of vascular smooth muscle cells and phagocytes to curcumin-eluting bioresorbable stent materials. Biomaterials 25(23):5333–5346
Chen HW, Yu SL, Chen JJ et al (2004) Anti-invasive gene expression profile of curcumin in lung adenocarcinoma based on a high throughput microarray analysis. Mol Pharmacol 65(1):99–110
Mariadason JM, Corner GA, Augenlicht LH (2000) Genetic reprogramming in pathways of colonic cell maturation induced by short chain fatty acids: comparison with trichostatin A, sulindac, and curcumin and implications for chemoprevention of colon cancer. Cancer Res 60(16):4561–4572
Papoutsi Z, Kassi E, Chinou I et al (2008) Walnut extract (Juglans regia L.) and its component ellagic acid exhibit anti-inflammatory activity in human aorta endothelial cells and osteoblastic activity in the cell line KS483. Br J Nutr 99(4):715–722
Fassina G, Vene R, Morini M et al (2004) Mechanisms of inhibition of tumor angiogenesis and vascular tumor growth by epigallocatechin-3-gallate. Clin Cancer Res 10(14):4865–4873
Garbisa S, Sartor L, Biggin S et al (2001) Tumor gelatinases and invasion inhibited by the green tea flavanol epigallocatechin-3-gallate. Cancer 91(4):822–832
Vittal R, Selvanayagam ZE, Sun Y et al (2004) Gene expression changes induced by green tea polyphenol (-)-epigallocatechin-3-gallate in human bronchial epithelial 21BES cells analyzed by DNA microarray. Mol Cancer Ther 3(9):1091–1099
Weinreb O, Mandel S, Youdim MB (2003) Gene and protein expression profiles of anti- and pro-apoptotic actions of dopamine, R-apomorphine, green tea polyphenol (-)-epigallocatechine-3-gallate, and melatonin. Ann NY Acad Sci 993:351–61 (discussion 87–93)
Wang SI, Mukhtar H (2002) Gene expression profile in human prostate LNCaP cancer cells by (–) epigallocatechin-3-gallate. Cancer Lett 182(1):43–51
Schempp CM, Kiss J, Kirkin V et al (2005) Hyperforin acts as an angiogenesis inhibitor. Planta Med 71(11):999–1004
Martinez-Poveda B, Quesada AR, Medina MA (2005) Hyperforin, a bio-active compound of St. John’s Wort, is a new inhibitor of angiogenesis targeting several key steps of the process. Int J Cancer 117(5):775–780
Quiney C, Billard C, Mirshahi P et al (2006) Hyperforin inhibits MMP-9 secretion by B-CLL cells and microtubule formation by endothelial cells. Leukemia 20(4):583–589
Rakshit S, Bagchi J, Mandal L et al (2009) N-acetyl cysteine enhances imatinib-induced apoptosis of Bcr-Abl + cells by endothelial nitric oxide synthase-mediated production of nitric oxide. Apoptosis 14(3):298–308
Yi T, Cho SG, Yi Z et al (2008) Thymoquinone inhibits tumor angiogenesis and tumor growth through suppressing AKT and extracellular signal-regulated kinase signaling pathways. Mol Cancer Ther 7(7):1789–1796
Su SJ, Yeh TM, Chuang WJ et al (2005) The novel targets for anti-angiogenesis of genistein on human cancer cells. Biochem Pharmacol 69(2):307–318
Wang TT, Sathyamoorthy N, Phang JM (1996) Molecular effects of genistein on estrogen receptor mediated pathways. Carcinogenesis 17(2):271–275
Li Y, Sarkar FH (2002) Down-regulation of invasion and angiogenesis-related genes identified by cDNA microarray analysis of PC3 prostate cancer cells treated with genistein. Cancer Lett 186(2):157–164
Regenbrecht CR, Jung M, Lehrach H et al (2008) The molecular basis of genistein-induced mitotic arrest, exit of self-renewal in embryonal carcinoma, primary cancer cell lines. BMC Med Genomics 1:49
Pie JE, Park JH, Park YH et al (2006) Effect of genistein on the expression of bone metabolism genes in ovariectomized mice using a cDNA microarray. J Nutr Biochem 17(3):157–164
Takahashi Y, Lavigne JA, Hursting SD et al (2004) Using DNA microarray analyses to elucidate the effects of genistein in androgen-responsive prostate cancer cells: identification of novel targets. Mol Carcinog 41(2):108–119
Adachi T, Ono Y, Koh KB et al (2004) Long-term alteration of gene expression without morphological change in testis after neonatal exposure to genistein in mice: toxicogenomic analysis using cDNA microarray. Food Chem Toxicol 42(3):445–452
Chen WF, Huang MH, Tzang CH et al (2003) Inhibitory actions of genistein in human breast cancer (MCF-7) cells. Biochim Biophys Acta 1638(2):187–196
Naciff JM, Jump ML, Torontali SM et al (2002) Gene expression profile induced by 17alpha-ethynyl estradiol, bisphenol A, and genistein in the developing female reproductive system of the rat. Toxicol Sci 68(1):184–199
Chen CC, Shieh B, Jin YT et al (2001) Microarray profiling of gene expression patterns in bladder tumor cells treated with genistein. J Biomed Sci 8(2):214–222
Kobori M, Masumoto S, Akimoto Y et al (2009) Dietary quercetin alleviates diabetic symptoms and reduces streptozotocin-induced disturbance of hepatic gene expression in mice. Mol Nutr Food Res 53(7):859–868
Natsume Y, Kadota K, Satsu H et al (2009) Effect of quercetin on the gene expression profile of the mouse intestine. Biosci Biotechnol Biochem 73(3):722–725
Odbayar TO, Kimura T, Tsushida T et al (2009) Isoenzyme-specific up-regulation of glutathione transferase and aldo-keto reductase mRNA expression by dietary quercetin in rat liver. Mol Cell Biochem 325(1–2):121–130
Soundararajan R, Wishart AD, Rupasinghe HP et al (2008) Quercetin 3-glucoside protects neuroblastoma (SH-SY5Y) cells in vitro against oxidative damage by inducing sterol regulatory element-binding protein-2-mediated cholesterol biosynthesis. J Biol Chem 283(4):2231–2245
Murtaza I, Marra G, Schlapbach R et al (2006) A preliminary investigation demonstrating the effect of quercetin on the expression of genes related to cell-cycle arrest, apoptosis and xenobiotic metabolism in human CO115 colon-adenocarcinoma cells using DNA microarray. Biotechnol Appl Biochem 45(Pt 1):29–36
Whyte L, Huang YY, Torres K et al (2007) Molecular mechanisms of resveratrol action in lung cancer cells using dual protein and microarray analyses. Cancer Res 67(24):12007–12017
Golkar L, Ding XZ, Ujiki MB et al (2007) Resveratrol inhibits pancreatic cancer cell proliferation through transcriptional induction of macrophage inhibitory cytokine-1. J Surg Res 138(2):163–169
Jones SB, DePrimo SE, Whitfield ML et al (2005) Resveratrol-induced gene expression profiles in human prostate cancer cells. Cancer Epidemiol Biomarkers Prev 14(3):596–604
Yang SH, Kim JS, Oh TJ et al (2003) Genome-scale analysis of resveratrol-induced gene expression profile in human ovarian cancer cells using a cDNA microarray. Int J Oncol 22(4):741–750
Narayanan BA, Narayanan NK, Re GG et al (2003) Differential expression of genes induced by resveratrol in LNCaP cells: P53-mediated molecular targets. Int J Cancer 104(2):204–212
Acknowledgments
This work has been made possible by contributions from the Italian Health Ministry to UP, the Regione Liguria to UP, the Compagnia San Paolo di Torino to UP and AA, the AIRC (Associazione Italiana per la Ricerca sul Cancro; to DN and AA), the Ministero della Salute (to AA and DN), and the Università degli Studi dell’Insubria (to DN). AA is the recipient of a Berlucchi Award for Scientific Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Albini, A., Indraccolo, S., Noonan, D.M. et al. Functional genomics of endothelial cells treated with anti-angiogenic or angiopreventive drugs. Clin Exp Metastasis 27, 419–439 (2010). https://doi.org/10.1007/s10585-010-9312-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-010-9312-5