
Abstract
Flow cytometry (FCM) has been widely used in plant science to determine the amount of nuclear DNA, either in absolute units or in relative terms, as an indicator of ploidy. The requirement for fresh material in some applications, however, limits the value of FCM in field research, including plant biosystematics, ecology and population biology. Dried plant samples have proven to be a suitable alternative in some cases (large-scale ploidy screening) although tissue dehydration is often associated with a decrease in the quality of FCM analysis. The present study tested, using time-scale laboratory and in situ field experiments, the applicability of glycerol-treated nuclear suspension for DNA flow cytometry. We demonstrate that plant nuclei preserved in ice-cold buffer + glycerol solution remain intact for at least a few weeks and provide estimates of nuclear DNA content that are highly comparable and of similar quality to those obtained from fresh tissue. The protocol is compatible with both DAPI and propidium iodide staining, and allows not only the determination of ploidy level but also genome size in absolute units. Despite its higher laboriousness, glycerol-preserved nuclei apparently represent the most reliable way of sample preservation for genome size research. We assume that the protocol will provide a vital alternative to other preservation methods, especially when stringent criteria on the quality of FCM analysis are required.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Flow cytometry is a high-throughput, cost-effective and accurate cytogenetic method with broad applications in plant sciences (Doležel et al. 2007a). In plant evolutionary and ecological studies, flow cytometry (FCM) has played a prominent role in the estimation of nuclear DNA content, either in absolute units (picograms of DNA or mega base pairs) or in relative terms as an indicator of ploidy (Kron et al. 2007). By enabling the analysis of large population samples over a short time span, FCM has significantly advanced our knowledge of ploidy and genome size variation in natural systems (Kron et al. 2007; Suda et al. 2007a; Loureiro et al. 2010). The global application of FCM is, however, limited by the need to use fresh plant material for reliable measurements of DNA content (Doležel et al. 2007a; Greilhuber et al. 2007). This prerequisite hampers large-scale population studies in regions without easily accessible FCM facilities (e.g. most of the tropical regions) and may cause difficulties elsewhere (e.g. when the capacity of a laboratory is saturated or during instrument maintenance). Transport of seeds and their direct FCM measurement or analysis of growing seedlings can partly solve the problem (Sliwinska et al. 2005; Suda et al. 2005). Nevertheless, this approach may be hampered by the need to collect plant material during the seed gathering season, difficulties in seed germination ex situ, potential shifts in genome size values estimated from dry seeds (e.g. Sliwinska et al. 2005) and/or by taxonomic complexity (e.g. hybrid origin of the seeds). Tissue preservation using chemical fixatives (ethanol- or formaldehyde-based), although widely used in animal and human FCM studies, have elicited only little interest from the plant community (Kron et al. 2007). The last years have seen several attempts to substitute fresh plant samples with dry or frozen tissue. Suda and Trávníček (2006a, b) introduced a protocol for reliable ploidy estimation (using 4',6-diamidino-2-phenylindole (DAPI)) in desiccated plant material (either silica- or air-dried) and this method has been successfully applied to a number of plant groups (e.g. Šmarda et al. 2005; Suda et al. 2007b; Popp et al. 2008; Hülber et al. 2009; Košnar and Kolář 2009; Volkova et al. 2010). Reasonable FCM histograms can also be achieved by analysing rapidly frozen plant tissues (Dart et al. 2004; Nsabimana and Van Staden 2006; Halverson et al. 2008; Cires et al. 2009).
Despite these promising steps towards the routine use of non-fresh plant samples, the above-mentioned protocols are still considered inadequate for estimating genome size in absolute units (using intercalating fluorochromes). The quality of measurements from dry or frozen tissue samples only rarely reaches the level achievable for fresh material and further decreases with the ageing of the samples. Usual symptoms accompanying the analysis of non-fresh material are: (1) lower reliability of DNA content estimates as evidenced by pronounced shifts in fluorescence intensity compared to that of fresh samples (e.g. Šmarda 2006; Suda and Trávníček 2006b; Cires et al. 2009; Bainard et al. 2011), and (2) decrease in the uniformity of fluorescence, resulting in higher coefficient of variations (CVs) of the peaks and more prominent background (Suda and Trávníček 2006b). These observations are not compatible with the high standards required for some FCM applications, including the determination of absolute genome size (Doležel and Bartoš 2005; Doležel et al. 2007b; Greilhuber et al. 2007). Consequently, the majority of published studies have used non-fresh material solely to determine DNA ploidy levels, which can tolerate some relaxation of the quality criteria (e.g. Eidesen et al. 2007; Schönswetter et al. 2007a; Popp et al. 2008; Bendiksby et al. 2011), or interpret the results as supplementary to fresh tissue analysis (Dušková et al. 2010). Only very recently, Bainard et al. (2011) conducted a careful experimental study to evaluate the potential of silica-dried plant material for genome size research. The authors concluded that sample desiccation introduced comparatively minor variation (<10%), a level of which was species-specific and comparable to other sources of artefactual variation. They considered dehydrated plant samples promising for assessing absolute genome size, yet admitted that relaxed demands should be applied to the quality of analysis and caution must be exercised in interpreting the results.
Whereas the effects of physical preservation on FCM estimates of nuclear DNA content have been intensively studied and there are some comparative studies showing advantages and limitations of these approaches (Suda and Trávníček 2006b; Bainard et al. 2011), the potential of chemical fixatives has been largely neglected. This reluctance likely stems from the higher laboriousness of the protocols and potential chemical-induced changes in chromatin condensation, which can affect the stoichiometric staining of DNA using intercalating dyes (Shapiro 2003). The search for alternative modes of preservation is desirable in order to (1) increase the accuracy of non-fresh tissue measurements and (2) extend FCM measurements to species in which other preservation techniques have failed (according to our knowledge, around 15% of plant species do not yield acceptable FCM histograms after dehydration). A promising alternative to physical and chemical preservation of plant tissues is the storage of isolated nuclear suspensions in intact protective solutions such as glycerol (propane-1,2,3-triol). The value of glycerol for the preservation of isolated nuclei for FCM analysis was first mentioned by Chiatante et al. (1990), and a more thorough evaluation of the method was developed by Hopping (1993). This researcher stored isolated nuclei of Actinidia deliciosa in 30% glycerol at approximately −20°C and found that storage for 9 months did not compromise FCM analysis, and the estimated values were highly comparable with those obtained from fresh samples (fluorescence decrease of 5% to 7%). Unfortunately, his results were only based on the analysis of a single plant species and were not subjected to a rigorous statistical evaluation.
This study aimed to investigate the applicability of glycerol-preserved plant nuclei for genome size research and assess the capabilities and limitations of this approach. We conducted two complementary experiments: (1) a time-scale laboratory experiment using six model plant species from different families and covering a range of genome sizes, to systematically compare the glycerol-based protocol with other currently used methods of sample preservation, and (2) a multi-species experiment using a set of tropical species collected and preserved in the field, to test the feasibility of the methodology in situ. The effects of fluorescent dyes with different modes of DNA binding (AT-selective DAPI vs. intercalating propidium iodide (PI)) were also investigated.
Methods
Plant material
Six plant species from five plant families and spanning nearly 18-fold range of genome sizes (from 1.52 pg/2C to 26.9 pg/2C) were selected for a time-scale laboratory experiment. They included both frequently used FCM reference standards (Bellis perennis—Asteraceae, Pisum sativum ‘Ctirad’—Fabaceae and Vicia faba ‘Inovec’—Fabaceae) and representatives of the major tropical families analysed in the second experiment (Euphorbia milii—Euphorbiaceae, Ficus elastica—Moraceae and Galium album—Rubiaceae). This species selection comprised both plants with soft and rapidly decaying leaves (B. perennis, G. album and P. sativum) and plants with rather tough or even leathery leaves (E. milii, F. elastica and V. faba). Plants of P. sativum and V. faba were grown from seeds (kindly provided by J. Doležel, Institute of Experimental Botany, Olomouc, the Czech Republic) while the remaining species were available from the living collection of the Botanical Garden, Faculty of Science, Charles University in Prague, Czech Republic.
The in situ experiment involved 21 species from 12 angiosperm families (both monocots and dicots) that were collected in primary and secondary rainforests around the Wannang village (approximately 60 km west of Madang) in northern Papua New Guinea in August 2006 (see Table S1). Representatives of species-rich tropical genera (e.g. Ficus and Macaranga) as well as economically important crops (e.g. Musa, Strychnos and Syzigium) were included. Herbarium vouchers are kept in CBFS.
Sample preservation
Four different methods of sample preservation were tested in a time-scale experiment: (1) young healthy leaves stored in a moist plastic bag at 4°C in a refrigerator (further referred to as ‘plastic bag’), (2) leaf tissue rapidly dehydrated using silica gel (‘silica gel’), (3) isolated nuclei suspended in Otto I buffer + glycerol (see below) and kept at −18°C in a freezer (‘ice-cold glycerol’; note that the solution remained liquid at this temperature), and (4) isolated nuclei kept in the same solution at room temperature (23 ± 2°C; ‘RT glycerol’). In addition, fresh leaves picked from the cultivated plants were used as ‘control’.
The in situ experiment involved the same preservation methods except for the silica gel treatment. For all the used treatments, there was an approximately 24-h interruption in low-temperature storage due to sample transportation from Papua New Guinea to the Czech Republic.
FCM analysis
Sample preparation generally followed the simplified two-step procedure using Otto’s buffers (Doležel et al. 2007b). Briefly, ~50 mg of sample leaf tissue and the same amount of the fresh internal reference standard were chopped with a sharp razor blade in a Petri dish containing 0.5 mL of ice-cold Otto I buffer (0.1 M citric acid, 0.5% Tween-20) (Otto 1990). The suspension was filtered through a 42-μm nylon mesh and incubated for approximately 15 min at room temperature. Samples were then stained for 10 min at room temperature. The staining solution consisted of 1 mL of Otto II buffer (0.4 M Na2HPO4·12 H2O), β-mercaptoethanol (final concentration of 2 μL mL−1) and a fluorochrome. Two DNA-binding fluorochromes were employed: (1) intercalating PI plus RNase IIA (both at final concentrations of 50 μg mL−1) and (2) AT-selective DAPI at a final concentration of 4 μg mL−1. Stained nuclei were run on a flow cytometer and excited either with (1) a green diode-pumped solid-state laser (Cobolt Samba, 532 nm, 100 mW; Cobolt, Stockholm, Sweden) embedded in a Partec CyFlow SL instrument (Partec GmbH., Münster, Germany) (for PI staining) or (2) a UV mercury arc lamp embedded in a Partec PA II instrument (for DAPI staining). In the time-scale experiment, fluorescence intensity (measured in linear scale) and forward and side scatter (both in logarithmic scale) were recorded in laser-based measurements while only the first parameter was recorded in lamp-based measurements; in both cases, 5,000 particles were analysed. The following instrument settings were kept constant throughout the experiment: (1) the position of the first G 0/G 1 peak on channel 100 (using a 1,024-channel scale), (2) the discriminator for fluorescence (i.e. the lowest recorded value) on channels 30 and 50 in the time-scale laboratory experiment and in the in situ experiment, respectively and (3) discriminators for forward and side scatter on channels 30 and 10, respectively. In the in situ experiment, only fluorescence intensity was recorded for both laser- and lamp-based instruments and the setting was adjusted independently for each sample to achieve optimal FCM results.
The following modifications were adopted for the analysis of glycerol-preserved samples (see Supplementary file S4 for a summarised procedure): approximately 300 mg of intact fresh leaf tissue of the sample was chopped together with the same amount of the internal reference standard in a Petri dish containing 6 mL of Otto I buffer. The suspension was filtered through a 42-μm nylon mesh, divided into twelve 0.5-mL aliquots and 0.5 mL of 60% glycerol solution was added to each aliquot. Six aliquots were kept in a freezer (−18°C) until FCM analysis (‘ice-cold glycerol’), while the other six were left at room temperature (‘RT glycerol’). Before FCM analysis, the suspension was centrifuged for 3 min at 3,200 rpm, the supernatant was discarded and 100 μL of ice-cold Otto I buffer was added to resuspend the nuclei. The sample tubes were gently shaken and the nuclear suspension was incubated for 15 min at room temperature. Finally, 1 mL of staining solution (Otto II buffer supplemented with β-mercaptoethanol and a fluorochrome) was added, and after 10 min of incubation, the samples were run on a flow cytometer.
B. perennis (2C = 3.38 pg; Schönswetter et al. 2007b) served as the internal reference standard for E. milii, F. elastica and P. sativum, while P. sativum (2C = 8.84 pg; Greilhuber et al. 2007) was used as a reference point for B. perennis, G. album and V. faba. Zea mays from a local field in Madang was used as a reference standard for the tropical species included in the in situ experiment.
FCM histograms were evaluated using Partec Flomax 2.4d software. ‘Fit Gauss Peaks’ function was used to calculate basic descriptive statistics (mean position, CV and number of particles) of G 0/G 1 peaks. Because non-fresh material was measured, we adopted more relaxed quality standards, and as successful considered analyses with CVs of sample G 0/G 1 peaks up to 10%. The proportion of background noise was determined as a ratio between the number of particles outside and inside the area of G 0/G 1 peaks defined by the ‘Fit Gauss Peaks’ function.
In the time-scale experiment, all except ‘silica gel’ samples were analysed after 1, 7 and 15 days of storage; the ‘silica gel’ samples were only analysed after 15 days of storage. Each measurement (including sample preparation) was repeated on three subsequent days to minimise potential artefactual instrumental drift. As a result of this experimental design, each species was analysed 18 times per treatment (three times of storage, three replicates and two fluorochromes), except ‘silica gel’ for which each species was analysed only six times (three replicates and two fluorochromes). In the in situ experiment, samples were analysed once after 15 days of storage.
Statistical analyses
Parameters describing the quality of FCM histograms (CVs and proportions of background noise) were analysed using a mixed-effect analysis of variance (ANOVA) with species identity as a random factor. Separate ANOVAs were conducted for (1) all treatments (i.e. ‘control’, ‘plastic bag’, ‘ice-cold glycerol’, ‘RT glycerol’ and ‘silica gel’), testing the effects of stain and preservation method, and (2) all except ‘silica gel’ treatments, testing the effect of storage time. Differences among individual treatments were further analysed by a Fisher’s LSD test.
The stability of sample/standard fluorescence ratio across treatments was tested separately for each fluorochrome using a linear mixed-effect model with species identity as a random factor. The effects of the preservation method and storage time (including their interactions) were tested after the exclusion of the ‘silica gel’ treatment, while the effect of the preservation method alone was tested on the data from the 15th day of storage (i.e. with the ‘silica gel’ treatment included). The probability of success of FCM analysis for individual treatments was analysed using a binomial generalised linear mixed-effect model fit with the Laplace approximation (Bates and Maechler 2009).
ANOVAs with post hoc comparisons were calculated in Statistica version 8 (StatSoft, Inc. 2008), while R package version 2.9.2 (R Development Core Team 2009) was used to calculate mixed-effect and generalised mixed-effect linear models.
Results
Time-scale experiment
FCM acquisitions resulted in histograms with sample peak CVs ranging from 0.98% to 9.58% (mean 2.4%; Figs. 1 and 2); thus, all analyses were considered successful. The mixed-effect ANOVA on the entire dataset revealed significant effect of the preservation method on the quality of analysis (F 4, 20 = 67.62, p < 0.001): the ‘silica gel’ samples generally exhibited the highest CVs (mean 4.2%) while the ‘plastic bag’ and ‘ice-cold glycerol’ samples (means for both treatments 2.0%) exhibited CV values comparable to those of fresh ‘control’ (mean 1.7%). PI-stained samples had generally slightly higher CVs than their DAPI-stained counterparts (means 2.67% and 2.19%, respectively; F 1, 5 = 29.28, p = 0.003).

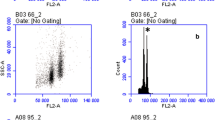
Illustrative histograms of fluorescence intensities and side scatter/fluorescence scattergraphs of ‘control’ (A, D), 15-day-old samples of B. perennis stored in ‘ice-cold glycerol’ (B, E) and ‘silica gel’ (C, F), analysed together with the internal reference standard, P. sativum and stained with propidium iodide. CVs (%) of G 0/G 1 peaks of Bellis/Pisum are 1.92/1.68, 1.84/1.82 and 9.04/1.92 for the ‘control’, ‘ice-cold glycerol’ and ‘silica gel’ samples, respectively

The effects of preservation method and the time of storage on the quality of FCM analysis of six plant species expressed as the CV (mean ± SD) of sample G 0/G 1 peak. Treatments significantly (at α = 0.05) different from the control in a particular time point are highlighted in grey. Freshly collected leaves from the cultivated plants were used as control at each time point. Silica gel samples were only analysed after 15 days of storage and not tested for the temporal variation
There was a significant interaction between the preservation method and the time of storage (F 6, 30 = 42.43, p < 0.001). After 15 days of storage, ‘plastic bag’ and ‘RT glycerol’ samples generally showed higher CVs than freshly collected tissue. By contrast, the quality of ‘ice-cold glycerol’ samples was highly comparable to that of fresh control (Fig. 2). The effect of fluorochrome remained significant (F 1, 5 = 11.43, p = 0.020); however, no significant interaction with the time of storage or preservation method was detected.
The proportion of background noise significantly differed among the preservation methods (F 4, 20 = 19.3, p < 0.001); the highest background levels were generally observed in ‘silica gel’ samples. However, if the storage time was considered (i.e. with ‘silica gel’ samples excluded), no significant differences were detected either among the preservation methods (F 3, 15 = 2.85, p = 0.072) or their interactions with the time of storage (F 6, 30 = 0.97, p = 0.462).
With the exception of ‘RT glycerol’ samples, tissue preservation using any of the methods caused only a negligible shift in fluorescence intensity relative to the standard over the time span tested. Although the effect of both preservation method and its interaction with the time of storage was significant in both DAPI and PI datasets (Table 1), this was largely caused by ‘RT glycerol’ samples, which showed a significant decrease in sample/standard fluorescence ratio compared to the fresh ‘control’ (t 203 = −2.65, p = 0.008 and t 203 = −2.14, p = 0.033 for DAPI and PI data, respectively). The most stable were ‘ice-cold glycerol’ samples (absolute difference between preserved/fresh tissue below 2%), followed by ‘plastic bag’ (difference 0.2% to 3.1% and 0.3% to 5.7% for DAPI and PI staining, respectively) and ‘silica gel’ (difference 0.2% to 2.6% and 1.6% to 12.9% for DAPI and PI staining, respectively) treatments. The difference in ‘RT glycerol’ samples usually exceeded 2% and reached up to ~18% in some cases (Table 2).
In situ experiment
Eighty-one out of 126 samples transported from Papua New Guinea and stored ~15 days had been successfully analysed (i.e. yielded histograms with distinct peaks and CVs <10%, for illustrative histograms see Fig. S2). Acceptable histograms using at least one preservation technique were obtained in all but one species (Macaranga fallacina, Euphorbiaceae). The probability of success was significantly influenced by the preservation method (binomial generalised linear mixed-effect model χ 2 = 79.99, p < 10−6; see also Fig. 3) and marginally also by the DNA fluorochrome (χ 2 = 4.5054, p = 0.034; DAPI slightly less successful). Regardless of the fluorochrome, the most successful was the ‘ice-cold glycerol’ treatment whereas the ‘RT glycerol’ one was the least successful.

The probability of achieving successful FCM analysis (i.e. sample CV below 10%) using three different preservation methods in the multi-species in situ experiment. Open circles samples, black squares mean probability success for a particular treatment. To visualise all samples, a small error variance was added to each value representing individual measurements
In successful analyses, the type of preservation significantly influenced the sample CV (F 2, 24 = 19.51, p < 0.001). The highest CVs were recorded in ‘RT glycerol’ samples (mean 8.20%) while CVs for ‘ice-cold glycerol’ (mean 4.51%) and ‘plastic bag’ (mean 5.02%) samples did not differ significantly from each other at α = 0.05. Nuclei preserved in ‘ice-cold glycerol’ yielded the best FCM results (in terms of the lowest CVs) in more than two-thirds of the analysed species (14 and 13 species in DAPI and PI analyses, respectively), while the remaining species gave the lowest CVs when stored in a plastic bag in a refrigerator. ‘Ice-cold glycerol’ was the only mode of preservation that allowed successful FCM analysis in four species (Endospermum labios, Macaranga aleuritoides, Osmoxylon novo-guineense and Versteegia sp.; Table S1).
Discussion
The value of glycerol-preserved nuclei
Unlike animal and human biology, fresh samples still dominate plant FCM research, especially when absolute genome size values are required (Doležel et al. 2007b; Kron et al. 2007). Nonetheless, the ever-increasing number of applications in biosystematics, ecology and evolutionary biology has accelerated the search for methods of sample preservation applicable in field conditions and allowing longer-term sample storage. An ideal protocol should be simple and rapid (to be easily performed outside the laboratory), universal (applicable to a wide range of plant species) and reliable (introducing no artefactual shift in fluorescence intensity). The present study adds to previous methodological attempts and describes the advantages, limitations and potential use of glycerol-treated nuclear suspensions.
The suitability of glycerol solution for preserving nuclear suspension was first documented by Hopping (1993), who achieved promising results (i.e. distinct peaks and only a small shift in fluorescence intensity) using nuclei of A. deliciosa stored up to 9 months in the frost and stained by PI. We tested the value of his protocol on a set of plants from different families and covering a range of genome sizes (including popular plant reference standards), using two the most important DNA-selective fluorochromes with different binding modes (DAPI and PI). In addition, we applied the methodology in situ in the tropics to estimate the amount of nuclear DNA in 21 native species.
The preservation of nuclear suspensions in a 30% glycerol–Otto I buffer solution (Otto 1990; Doležel et al. 2007b) and sample storage at −18°C was found to be a reliable method for the FCM estimation of genome size in plants in both absolute and relative units. The quality of analysis (expressed as CVs of G 0/G 1 sample peaks and the proportion of background noise) as well as their reliability (i.e. the stability of fluorescence intensity) appeared to be unaffected by the glycerol treatment, at least during the time period studied (15 days). After 2 weeks of storage in frost, both the quality measures of resulting FCM histograms and genome size estimates were fully comparable to those of fresh control samples (Fig. 2; Table 2). Furthermore, light scattering properties of the nuclei showed no signs of the so-called tannic acid effect, indicating that the analyses were not negatively affected by interfering secondary metabolites and/or nuclei aggregation (Loureiro et al. 2006; see Fig. 1). In general, the analyses of glycerol-preserved nuclei mostly fulfilled the stringent criteria applied for the genome size estimation in absolute units in freshly collected samples, including the requirements of stable sample/standard fluorescence ratio and CVs below 5% (e.g. Doležel et al. 2007a). A crucial step of the proposed protocol seems to be the storage of glycerol-preserved nuclei at −18°C, as samples kept at room temperature deteriorated quickly (Fig. 2).
Comparison with other modes of sample preservation
Despite the ongoing debate about the use of non-fresh (preserved) plant material for FCM analysis (Doležel and Bartoš 2005; Doležel et al. 2007a; Bainard et al. 2011), only a handful of studies have ventured to explore the value of fixed plant nuclei, cells and/or tissues to estimate genome size. Rapid tissue desiccation (most conveniently done using silica gel) is the only way of sample preservation that has received wider attention in ecological and evolutionary plant FCM studies. However, the dehydrated samples were mostly stained with AT-selective fluorochrome DAPI, which has favourable staining properties (e.g. comparatively low sensitivity to chromatin condensation and high increase in quantum efficiency after binding to the DNA molecule; Shapiro 2003), but precludes genome size estimation in absolute units. The artefactual shift in fluorescence intensity often observed after tissue dehydration (e.g. Šmarda 2006; Suda and Trávníček 2006b; Cires et al. 2009) and lower quality of FCM analysis also work against its use for absolute genome size estimation. Only recently, the potential of dried plant material for genome size research was thoroughly evaluated (Bainard et al. 2011). The authors considered the fluorescence shift introduced by drying (<10%) to be acceptable, as it fell within the limits introduced by other methodological factors (e.g. seasonal variation, instrument and buffer used, among others), and concluded that PI-stained samples can represent a promising option.
Despite its higher laboriousness in comparison with tissue dehydration, glycerol-preserved nuclei apparently represent the most reliable way of sample preservation for genome size research, at least in a short-term time frame. In the present study, estimates of nuclear DNA in glycerol-treated samples after 2 weeks of storage in frost were highly comparable to those obtained using fresh material. Whereas the ‘silica gel’ samples experienced up to a 12.9% shift in fluorescence intensity and CVs of 4.2%, on average, the ‘ice-cold glycerol’ samples showed very stable fluorescence (maximum difference <1.7%) and much lower CVs (Table 2). Interestingly, 2-week-old glycerol-preserved nuclei yielded better results than living plant tissues kept for the same time in a humid environment in the cold (Fig. 2), which is usually the first choice for short-term sample storage (Suda et al. 2007a). It should be noted that the period for which fresh tissues can be stored in a refrigerator before FCM analysis is considerably influenced by leaf characteristics. Whereas species with small and soft leaves (in our set, for instance, B. perennis and P. sativum) deteriorate quickly, plants with tough and leathery leaves (e.g. E. milii and F. elastica) seem to be generally less sensitive (Fig. S3). In parallel, glycerol preservation represented the only way to analyse several soft-leaved tropical plants in the in situ experiment (Table S1). Although the fluorescence properties of glycerol-preserved nuclei after long-term storage are largely unknown and in need of further study (but note that Hopping (1993) suggests that nuclei are likely to remain intact for at least several months), the presented methodology appears to be a very promising way of sample preservation for genome size studies.
Applicability of the protocol
Of particular importance is the fact that plant nuclei stored in ice-cold glycerol remain intact for at least a few weeks and provide estimates of nuclear DNA content that are highly comparable and of similar quality to those obtained from fresh tissue. Furthermore, the protocol is compatible with both DAPI and PI staining, allowing the determination of not only ploidy level but, more importantly, genome size in absolute units. Moreover, high resolution of resulting FCM histograms opens the possibility of detecting small differences in nuclear DNA content. Finally, glycerol-preserved nuclei also offer opportunity to cytotype plant species with soft and rapidly decaying leaves, in which other modes of storage usually provide less satisfactory results or completely fail.
The major limitations of the proposed protocol stem from higher demands on sample preparation, i.e. the need for basic laboratory facilities, including a freezer, and the necessity for suitable reference standard(s), a selection of which should meet several criteria (see Suda and Leitch 2010). In addition, the reliability of the protocol after longer-term storage (several months) needs to be assessed.
A detailed comparison of current methodologies of sample preservation for plant FCM, including their pros and cons, is provided in Table 3. Considering these facts, we assume that glycerol-treated nuclei will provide a vital alternative to other preservation methods, especially when stringent criteria on FCM analysis are required (e.g. in genome size studies) and/or if a detailed investigation of a single or a few plant species is intended. The storage of fresh tissue in cold will likely be the method of choice for short-term field trips while large-scale ploidy studies in remote areas will probably still be dominated by silica-dried samples.
Abbreviations
- CV:
-
Coefficient of variation
- DAPI:
-
4',6-diamidino-2-phenylindole
- FCM:
-
Flow cytometry/flow cytometric
- PI:
-
Propidium iodide
- SD:
-
Standard deviation
References
Bainard JD, Husband BC, Baldwin SJ, Fazekas AJ, Gregory TR, Newmaster SG, Kron P (2011) The effects of rapid desiccation on estimates of plant genome size. Chromosome Res 19:825–842
Bates D, Maechler M (2009) Package ‘lme4’—linear mixed-effects models using S4 classes. R Foundation for Statistical Computing, Vienna
Bendiksby M, Tribsch A, Borgen L, Trávníček P, Brysting AK (2011) Allopolyploid origins of the Galeopsis tetraploids—revisiting Müntzing’s classical textbook example using molecular tools. New Phytol 191:1150–1167
Chiatante D, Brusa P, Levi M, Sgorbati S, Sparvoli E (1990) A simple protocol to purify fresh nuclei from milligram amounts of meristematic pea root tissue for biochemical and flow cytometry applications. Physiol Plantarum 78:501–506
Cires E, Cuesta C, Peredo EL, Revilla MA, Prieto JAF (2009) Genome size variation and morphological differentiation within Ranunculus parnassifolius group (Ranunculaceae) from calcareous screes in the northwest of Spain. Plant Syst Evol 281:193–208
Dart S, Kron P, Mable BK (2004) Characterizing polyploidy in Arabidopsis lyrata using chromosome counts and flow cytometry. Can J Bot 82:185–197
Doležel J, Bartoš J (2005) Plant DNA flow cytometry and estimation of nuclear genome size. Ann Bot 95:99–110
Doležel J, Greilhuber J, Suda J (2007a) Flow cytometry with plant cells: analysis of genes, chromosomes and genomes. Wiley-VCH, Weinheim
Doležel J, Greilhuber J, Suda J (2007b) Estimation of nuclear DNA content in plants using flow cytometry. Nat Protoc 2:2233–2244
Dušková E, Kolář F, Sklenář P, Rauchová J, Kubešová M, Fér T, Suda J, Marhold K (2010) Genome size correlates with growth form, habitat and phylogeny in the Andean genus Lasiocephalus (Asteraceae). Preslia 82:127–148
Eidesen PB, Alsos IG, Popp M, Stensrud Ø, Suda J, Brochmann C (2007) Nuclear vs. plastid data: complex Pleistocene history of a circumpolar key species. Mol Ecol 16:3902–3925
Greilhuber J, Temsch EM, Loureiro J (2007) Nuclear DNA content measurement. In: Doležel J, Greilhuber J, Suda J (eds) Flow cytometry with plant cells: analysis of genes, chromosomes and genomes. Wiley-VCH Verlag, Weinheim, pp 67–101
Halverson K, Heard SB, Nason JD, Stireman JO (2008) Origins, distribution, and local co-occurrence of polyploid cytotypes in Solidago altissima (Asteraceae). Am J Bot 95:50–58
Hopping M (1993) Preparation and preservation of nuclei from plant tissues for quantitative DNA analysis by flow cytometry. N Z J Bot 31:391–401
Hülber K, Sonnleitner M, Flatscher R, Berger A, Dobrovsky R, Niessner S, Nigl T, Schneeweiss GM, Kubešová M, Rauchová J, Suda J, Schönswetter P (2009) Ecological segregation drives fine-scale cytotype distribution of Senecio carniolicus in the Eastern Alps. Preslia 81:309–319
Košnar J, Kolář F (2009) A taxonomic study of selected European taxa of the Tortula muralis (Pottiaceae, Musci) complex: variation in morphology and ploidy level. Preslia 81:399–421
Kron P, Suda J, Husband BC (2007) Applications of flow cytometry to evolutionary and population biology. Annu Rev Ecol Evol Syst 38:847–876
Loureiro J, Rodriguez E, Doležel J, Santos C (2006) Flow cytometric and microscopic analysis of the effect of tannic acid on plant nuclei and estimation of DNA content. Ann Bot 98:515–527
Loureiro J, Trávníček P, Rauchová J, Urfus T, Vít P, Štech M, Castro S, Suda J (2010) The use of flow cytometry in the biosystematics, ecology and population biology of homoploid plants. Preslia 82:3–21
Nsabimana A, Van Staden J (2006) Ploidy investigation of bananas (Musa spp.) from the National Banana Germplasm Collection at Rubona-Rwanda by flow cytometry. S Afr J Bot 72:302–305
Otto F (1990) DAPI staining of fixed cells for high-resolution flow cytometry of nuclear DNA. In: Crissman HA, Darzynkiewicz Z (eds) Methods in cell biology 33. Academic, San Diego, pp 105–110
Popp M, Gizaw A, Nemomissa S, Suda J, Brochmann C (2008) Colonization and diversification in the African ‘sky islands’ by Eurasian lychnis L. (Caryophyllaceae). J Biogeogr 35:1016–1029
R Development Core Team (2009) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Schönswetter P, Lachmayer M, Lettner C, Prehsler D, Rechnitzer S, Reich DS, Sonnleitner M, Wagner I, Hülber I, Schneeweiss GM, Trávníček P, Suda J (2007a) Sympatric diploid and hexaploid cytotypes of Senecio carniolicus (Asteraceae) in the Eastern Alps are separated along an altitudinal gradient. J Plant Res 120:721–725
Schönswetter P, Suda J, Popp M, Weiss-Schneeweiss H, Brochmann C (2007b) Circumpolar phylogeography of Juncus biglumis (Juncaceae) inferred from AFLP fingerprints, cpDNA sequences, nuclear DNA content and chromosome numbers. Mol Phyl Evol 42:92–103
Shapiro H (2003) Practical flow cytometry, 4th edn. Wiley-Liss, New York
Sliwinska E, Zielinska E, Jedrzejczyk I (2005) Are seeds suitable for flow cytometric estimation of plant genome size? Cytom Part A 64A:72–79
Šmarda P (2006) DNA ploidy levels and intraspecific DNA content variability in Romanian fescues (Festuca, Poaceae) measured in fresh and herbarium material. Folia Geobot 41:417–432
Šmarda P, Müller J, Vrána J, Kočí K (2005) Ploidy level variability of some Central European fescues (Festuca subg. Festuca, Poaceae). Biologia (Bratislava) 60:25–36
StatSoft, Inc. (2008) STATISTICA (data analysis software system), version 8.0
Suda J, Leitch IJ (2010) The quest for suitable reference standards in genome size research. Cytom Part A 77A:717–720
Suda J, Trávníček P (2006a) Estimation of relative nuclear DNA content in dehydrated plant tissues by flow cytometry. Curr Protoc Cytom 38:7.30.1–7.30.14
Suda J, Trávníček P (2006b) Reliable DNA ploidy determination in dehydrated tissues of vascular plants by DAPI flow cytometry—new prospects for plant research. Cytom Part A 69A:273–280
Suda J, Kyncl T, Jarolímová V (2005) Nuclear DNA amounts in Macaronesian angiosperms: forty percent of Canarian endemic flora completed. Plant Syst Evol 252:215–238
Suda J, Kron P, Husband BC, Trávníček P (2007a) Flow cytometry and ploidy: applications in plant systematics, ecology and evolutionary biology. In: Doležel J, Greilhuber J, Suda J (eds) Flow cytometry with plant cells: analysis of genes, chromosomes and genomes. Wiley-VCH, Weinheim, pp 103–130
Suda J, Weiss-Schneeweiss H, Tribsch A, Schneeweiss G, Trávníček P, Schönswetter P (2007b) Complex distribution patterns of di-, tetra-, and hexaploid cytotypes in the European high mountain plant Senecio carniolicus (Asteraceae). Am J Bot 94:1391–1401
Volkova PA, Trávníček P, Brochmann C (2010) Evolutionary dynamics across discontinuous freshwater systems: rapid expansions and repeated allopolyploid origins in the Palearctic white water-lilies (Nymphaea). Taxon 59:483–494
Acknowledgements
Jana Krejčíková and Pavel Trávníček helped us with the flow cytometric analyses and Jan Lepš, Kateřina Štajerová and Kenneth Molem kindly assisted with the collection and determination of the tropical species. The project was supported by projects 206/08/H049 and P506/10/0704 from the Czech Science Foundation. JT was supported by project 138/2010/P. Additional support was provided by the Academy of Science of the Czech Republic within the Institutional Research Programme AV0Z60050516 and the Ministry of Education, Youth and Sports of the Czech Republic (projects MSM0021620828 and MSM6007665801).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Hans de Jong.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
(PDF 15.7 kb)
Fig. S2
(PDF 28 kb)
Fig. S3
(PDF 31 kb)
Supplementary file S4
(PDF 99.1 kb)
Rights and permissions
About this article
Cite this article
Kolář, F., Lučanová, M., Těšitel, J. et al. Glycerol-treated nuclear suspensions—an efficient preservation method for flow cytometric analysis of plant samples. Chromosome Res 20, 303–315 (2012). https://doi.org/10.1007/s10577-012-9277-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-012-9277-0