
Abstract
In recent years, the innovation of gene-editing tools such as the CRISPR/Cas9 system improves the translational gap of treatments mediated by gene therapy. The privileges of CRISPR/Cas9 such as working in living cells and organs candidate this technology for using in research and treatment of the central nervous system (CNS) disorders. Parkinson’s disease (PD) is a common, debilitating, neurodegenerative disorder which occurs due to loss of dopaminergic neurons and is associated with progressive motor dysfunction. Knowledge about the pathophysiological basis of PD has altered the classification system of PD, which manifests in familial and sporadic forms. The first genetic linkage studies in PD demonstrated the involvement of Synuclein alpha (SNCA) mutations and SNCA genomic duplications in the pathogenesis of PD familial forms. Subsequent studies have also insinuated mutations in leucine repeat kinase-2 (LRRK2), Parkin, PTEN-induced putative kinase 1 (PINK1), as well as DJ-1 causing familial forms of PD. This review will attempt to discuss the structure, function, and development in genome editing mediated by CRISP/Cas9 system. Further, it describes the genes involved in the pathogenesis of PD and the pertinent alterations to them. We will pursue this line by delineating the PD linkage studies in which CRISPR system was employed. Finally, we will discuss the pros and cons of CRISPR employment vis-à-vis the process of genome editing in PD patients’ iPSCs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genetic screening experiment incorporates a new technology called clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas). Genome editing with CRISPR/Cas9 technology has long been applied to the research and treatment of a range of illnesses such as cancer, various monogenic diseases, AIDS, and central nervous system diseases (Knott and Doudna 2018). CRISPR system as a genome-editing toolbox provides the possibility of addition, deletion, or modification of the genome in the living cells. This system consists of two key components; Cas9, an endonuclease that induces double-strand break (DSB) in DNA at a specific location, and a small guide RNA, which guides Cas9 to the target of interest and guarantees the precision and specificity of genome editing (Safari et al. 2019).
Parkinson’s disease (PD) is a common progressive neurodegenerative disorder characterized by deterioration of dopaminergic (DAergic) neurons in the substantia nigra (SN) (Emamalizadeh et al. 2014; Taghavi et al. 2018). The prevalence of this neurologic disease is 1–2% in the population aged over 65 years and can be seen in people from all over the world and all races (Jamshidi et al. 2014; Schrag 2018). In most cases, PD results in a clinical motor syndrome called degenerative Parkinsonism, which is characterized by resting tremor, bradykinesia, muscle rigidity, and postural instability. The neuropathology of PD is characterized by DAergic neuronal loss with the appearance of Lewy bodies (LB) containing aggregated α-synuclein. However, there are exceptions which deviate from the classical PD pathological morphology. A long line of evidence has shown that gene mutations are responsible for familial forms, but the etiology of sporadic forms yet remains unknown (Lubbe and Morris 2014). The precise etiology of sporadic forms still remains obscure but it is believed that oxidative stress and neuroinflammation may play roles in the mechanisms underlying sporadic PD (Nasrolahi et al. 2019).
Mutations mediate autosomal-dominant PD in α-synuclein (SNCA) and leucine repeat kinase 2 (LRRK2), while mutations trigger autosomal-recessive PD in parkin, PTEN-induced putative kinase 1 (PINK1), and Daisuke-Junko-1 (DJ-1) (Table 1) (Scott et al. 2017). Other genes involved in the pathogenesis of syndromes with PD-like features include mutations in FBX07, ATP13A2, DNAJC1, PLA2G634, and SYNJ1. It is observed that genes such as VPS35, eiF4G1, and CHCHD2 are linked with sporadic forms of PD with varying degrees of penetrance. Also, findings of genome-wide association studies (GWAS) have shown an association of at least 41 risk loci with the pathogenesis of PD (Chang et al. 2017). These results have stated that SNCA is among the most potent risk loci associated with the sporadic form of PD (Devine et al. 2011). However, all genes investigated in monogenic types of PD cannot fully explain the etiology of all PD. The pathways of gene mutations involved in PD pathogenesis are illustrated in Fig. 1.

Illustration of the main molecular pathways involve in PD pathogenesis. Mitochondrial dysfunction and the proteasomal impairment play key roles in contributing to the pathogenic process. PD-associated genes play critical roles in regulation of mitochondrial homeostasis, protein folding and the activity of intracellular clearance systems. The PINK1-parkin axis has significant roles in regulation of the fundamental dynamic properties of mitochondria such as fission–fusion events, mitochondrial arrest, and mitophagy. Furthermore, PINK1 and parkin independently control mitochondrial biogenesis and calcium homeostasis. Mutations in PINK1 and parkin disrupt the autophagy which results in neural cell death. Mutation in α-SYN and LRRK2 also affect mitochondrial function, by impairing membrane potential and oxidative phosphorylation, and inducing reactive oxygen species production and cytochrome-c release. Moreover, PINK1, parkin, and DJ-1 directly promote the degradation of proteasomal substrates, whereas α-SYN is able to impair activity of the ubiquitin-proteasome system (UPS). Finally, SNCA and UCHL-1 mutations impair α-SYN degradation through the chaperone-mediated autophagy (CMA), resulting in α-SYN aggregation and formation of inclusions. Of note, ATP13A2 counteracts the production of α-SYN aggregates. Alpha-SYN and other misfolded proteins accumulate within Lewy bodies, which are the pathologic hallmark of the disease
It has been implicated that neuroinflammation is a main driver of sporadic PD onset and progression (Ghosh et al. 2016). In PD, various physical or chemical insults induce the hyper-activation of both microglia and astrocytes. Environmental neurotoxic agent such as Mn (Luo et al. 2019), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Ghosh et al. 2016) and some of pesticides (Song et al. 2019) cause neuroinflammation through disruption in the signaling pathways between neurons and glial cells. Activation of microglia increases the release of pro-inflammatory cytokines. Gliosis results in the activation and nuclear translocation of NF-κB, and upregulation of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1beta, IL-6, inducible nitric oxide synthase, nitric oxide, cyclooxygenase-2, and prostaglandins E2 (Luo et al. 2019). A long line of evidence demonstrated that in various conditions mitochondrial disorders and neuroinflammation act in line with each other in PD pathogenesis (Sarkar et al. 2017).
Today, a new perspective on CRISPR-related Parkinson’s disease research is emerging, and arguably could cause tectonic shifts in the treatment of PD and other neurological abnormalities.
Today, a tidal wave of CRISPR-related Parkinson’s disease research is ongoing, and it is significant that the Parkinson’s specialist attain the knowledge about how this rigorous technology works. By employing the promising potentials such as gene knocking out/in, gene modification, transcriptional activation/repression, and epigenetic modification, CRISPR/Cas9 technology paves the way to PD treatment and facilitates the genomic investigation of PD pathogenesis.
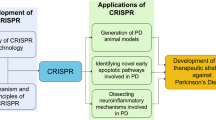
To address the valuable progression in the understanding of the genetic pathogenesis of PD mediated by CRISPR system, in this review we briefly discuss the structure and function of CRISPR system. In continue, the genetic factors involved in PD pathogenesis will be described. In addition, we review the researches that used CRISPR for investigation of PD and also the PD models generated by genome editing technology. At the end, the pros and cons of genome editing and especially induced pluripotent stem cells (iPSCs) generation mediated by CRISPR system are explained.
Genome-Editing Technology: CRISPR/Cas9 System
Traditionally, small interfering RNA (siRNA) and short hair-pin RNA (shRNA) have been employed for genome editing (Safari et al. 2016, 2017). However, one of the most revolutionary hallmarks of the past decade was arguably the discovery of the powerful new gene-editing technology known as CRISPR. This gene-editing technique touched off an explosion of research in 2013, stunning the entire scientific community. It would not be a stretch to claim that it can become the “breakthrough” of our age. CRISPR/Cas9 system could be used to cut any genome at any exciting place, and edit the genome. The nuclease activity of Cas9 protein induces DSB in the intended DNA sequence guided by sgRNA that consequently determines the precision and specificity of the CRISPR system. To repair the DSB, the cellular repair system comes to action, and repairs this site via recruiting two strategies, including non-homologous end joining (NHEJ) and homology-directed repair (HDR). These two strategies permit the random insertion/deletion (in/del) mutation or direct homologous recombination in the presence of donner template (Safari et al. 2019). Also, deactivation of Cas9 (dCas9) nuclease makes a powerful tool for the modification of gene transcription. Combination of activator domains such as V16 and VP64 leads to activation of the gene of interest. In contrast, the fusion of dCas9 with repressor domain such as KRAB allows the repression of intended genes. In state of the art, joining of DNA methyltransferase 3A (DNMT3A), or the DNA demethylase TET or p300 core with dCas9 induces epigenetic modification (Safari et al. 2019).
Even though CRISPR is a promising gene tool kit but it has some limitation such as off-target effect which makes the use of this technology for therapeutic approaches doubtful. Hence to improve its specificity, scientists have invented engineered version of Cas9 such as nickase Cas9 (Cas9n), enhanced specificity Cas9, and high-fidelity Cas9 (Ran et al. 2013). Cas9n as a high-throughput CRISPR system consists of 2 mutated Cas9 nucleases which cause the induction of single strand nicks, two sgRNAs and 2 FOK1 endonuclease enzymes. This complex induces a DSB in the intended gene with high specificity because it needs two sgRNAs for identification of the target site, which increase the precision of this CRISPR system (Safari et al. 2019). The simplicity and efficiency of CRISR provide a hopeful opportunity for using this system in a broad field of research from functional annotation of genes to diseases animal modeling and therapeutic approaches. The versatility of CRISPR extended its application in research and treatment of PD which will be described.
Genetic Factors Involved in PD Pathogenesis and PD Prevention
SNCA
Polymeropoulos first discovered the missense mutation in the SNCA gene in 1997. Discovery of this mutation, along with the identification of familial form risk factors, established a strong link between the sporadic and familial forms of PD (Polymeropoulos et al. 1996). It is well established that the lewy bodies (LB) as a common pathological hallmark of PD contains α-synuclein protein. Thus far, five different classes of mutations in SNCA, i.e., three missense mutations and two point mutations, have been reported. These various mutations result in different clinical manifestations and different ages of onset. Missense mutations (A53T, A30P and E46 K) induce severe progressive Parkinsonism with an early start and with an appropriate initial response to L-dopa. Point mutations such as H50Q and G51D trigger distinct clinical manifestations with the different onset and disease severity (Ferreira and Massano 2017).
Tandem repeat in SNCA locus, i.e., duplications and triplications, is classified as the etiological factors for PD. Findings have shown that two excessive copies of the genomic region containing the α-synuclein gene lead to the familial form of PD. Also, the number of SNCA copies determines the severity of PD. Investigation of duplicated SNCA has demonstrated that a 50% enhancement in SNCA expression results in the development of an autosomal-dominant form of PD, which suggests that PD-associated risk variants might cause an enhanced SNCA expression (Kim et al. 2012).
Neuropathological findings of SNCA linked PD showed severe neuronal degeneration in the SN region and locus coeruleus (LC). Furthermore, the extensive spread of LB is observed in the brainstem and cerebral cortex in a patient with SNCA triplication (Farrer et al. 2004).
LRRK2
The autosomal-dominant mutation in LRRK2 has been identified as the underlying genetic cause of PD. The LRRK2 is an abundant multidomain protein that belongs to a ROC–COR (ROCO) superfamily (Marin 2008). However, the precise physiological function of this protein remains unknown, but its involvement in the neurite outgrowth, vesicle trafficking, cytoskeletal maintenance, protein autophagy, and the immune system is well established (Anand and Braithwaite 2009).
It is estimated that four percent of familial forms of PD carry the different types of LRRK2 mutation (Hasegawa et al. 2009). A large number of mutations have been reported to play a role in the etiology of PD. G2019S is the most frequent mutation of LRRK2 in sporadic (1%) and familial (≈ 3–6%) forms of PD (Healy et al. Healy DG 2008).
The clinical manifestation of LRRK2-linked Parkinsonism is not dissimilar from the sporadic cases. The onset age of this PD type is around 60 years. Note worthily, a tremor at presentation and dystonia is more common in the G2019S patients when compared to the non-mutated LRRK2 patients. It is reported that abduction–adduction tremor of the lower limbs could be deemed as a diagnostic marker. Comparison of dyskinesia as an examining scale of PD severity in LRRK2-linked Parkinsonism and idiopathic PD has shown that LRRK2-linked Parkinsonism has a less regular pattern (Healy et al. 2008).
Few pathologic examinations within the same family have shown heterogeneous neuropathological findings LRRK2 associated PD. Most LRRK2 cases demonstrate the presence of LB in the brainstem and loss of neurons in the SN. However, a minority of instances show neurofibrillary tangle pathology, neuronal nigral loss without LB, or glial cytoplasmic inclusions reminiscent of multiple system atrophy (Zimprich et al. 2004).
Parkin
The most common cause of autosomal-recessive early-onset Parkinsonism is mutations in the Parkin (PRKN; also known as PARK2) gene. E3 ubiquitin ligase Parkin encoded by PRKN participates in the mitochondrial quality control and turnover. Primarily, Parkin is a cytosolic protein but is under certain conditions localized to the mitochondria (Panicker et al. 2017). PTEN-induced putative kinase-1 (PINK1) regulates Parkin activity through phosphorylation events (Arkinson and Walden 2018). PINK-1 and PARKIN play critical roles in mitochondrial quality control and mitophagy (Lazarou et al. 2015).
Accumulation of PINK1 on the surface of dysfunctional mitochondria activates the cytosolic PARKIN. Activated PARKIN and S65-phosphorylation mediated via PINK1 extend the preformed ubiquitin chains on multiple mitochondrial surface proteins (Rose et al. 2016). The accretion of S65-phosphorylated ubiquitin (pUb) on mitochondria initiates the signaling mechanism to trigger the autophagy machinery for the selective elimination of dysfunctional mitochondria (Lazarou et al. 2015).
It is proved that 19% of isolated PD cases with an early onset carry the mutations in PARKIN. Furthermore, heterozygous mutations of PARKIN have also been shown in some familial PDs. This fact implies that carriers of a single allele mutated PARKIN might be at risk of developing PD. The carriers of PARKIN mutation shows a clinical profile characterized by a slow progression of the disease and appropriate response to levodopa, with severe early-onset dyskinesias (Lincoln et al. 2003; Lohmann et al. 2003). Mutations in both PINK-1 and PARKIN result in the familial subtype, implying that injured mitophagy is a cardinal feature of PD (Kitada et al. 1998).
Regarding the neuropathology, and the extensive loss of SN dopaminergic neurons, the absence of LB, and the presence of neurofibrillary tangles in the cerebral cortex and brainstem have been reported in PARKIN-linked PD (Mori et al. 1998). Nevertheless, recent reports reveal the presence of LB in the SN and LC, as well as α-synucleinopathy restricted to the mesencephalic reticular formation (Pramstaller et al. 2005).
DJ-1
In 2003, it was reported that a missense mutation, in addition to a homozygous deletion in the DJ-1 gene, caused autosomal-recessive early-onset of PD (Annesi et al. 2005; Bonifati et al. 2003). So far, several novel DJ-1 mutations have been identified in patients with early-onset PD. It is noteworthy that these sort of mutations are quite rare, and can be found in only 1% of early-onset PD patients (Abou-Sleiman et al. 2003). DJ-1 protein also exists in various organs such as the brain. The cellular exposure to oxidative stress prompts the relocation of cytosolic DJ-1 protein into the mitochondria. On the same token, endogenous DJ-1 was expectedly detected in the mitochondrial matrix and intermembrane space. This evidence upholds the hypothesis that DJ-1 may play a role in cellular protection against oxidative stresses. Furthermore, DJ-1 acts as a chaperone molecule to promote proper three-dimensional proteins and refold the damaged ones (Zhu et al. 2017). Neuropathology of DJ-1 is not fully understood, but recent studies have shown the role of LB pathology in DJ-1-associated PD (Taipa et al. 2016).
PINK1
Mutations in PINK1 have been initially observed in a Sicilian family with autosomal-recessive early onset of Parkinsonism. However, the bulk of PINK1 mutations are missense, although few copy number mutations and exonic rearrangements have been reported (Samaranch et al. 2010). These mutations have been identified in both familial and sporadic (2–4%) cases in homozygous and compound heterozygous manifestations. The function of heterozygous mutations in PD pathogenesis supports the theory that carriers of single PINK1 heterozygous mutation are at risk of PD (Nuytemans et al. 2010). It can be argued that the clinical characterization of PINK1-linked PD is on par with Parkin/DJ-1 related PD cases. The morphology of this subtype of PD can be characterized by low progressive disease, which is quite responsive to the application of levodopa. In this light, the functions of PINK-1and Parkin are closely intertwined. It can be fair to note that the PINK-1/Parkin axis controls the fundamental dynamic properties of mitochondria, such as fission–fusion events, mitochondrial arrest, and mitophagy. Furthermore, PINK-1 and Parkin can regulate the calcium homeostasis and mitochondrial biogenesis independent of each other. Also, PINK-1 promotes autophagy by interacting with the protein Beclin-1 (Jian et al. 2018).
A neuropathological study on a PINK-1 linked PD patient has highlighted the neuronal loss as well as LB accumulation within the regions such as substantia nigra (SN) pars compacta, nuclei of the brainstem, and nucleus basalis of Meynert (Valente et al. 2004).
RAB39B
Rab GTPase is low weight proteins that help modulate vesicular trafficking (Cheng et al. 2002). RAB39B is one of the three X-linked RAB genes which are exclusive to the brain (Wilson et al. 2014). Multiple reports in the literature have reported that the mutations in RAB39B result in PD and X-linked intellectual disability. Deletion of ∼ 45-kb of RAB39B, missense mutation, and RAB39B p.Trp186 stop mutation cause RAB39B loss of function and facilitate the shift to Parkinsonism (Lesage et al. 2015). Concerning the neuropathology, Postmortem studies have demonstrated a severe generalized dopaminergic neuronal loss in the SN and widespread classic LB disease in RAB39B related PD (Wilson et al. 2014).
P13
The involvement of mitochondrial defects has been observed in the pathogenesis of sporadic PD patients. Also, many familial PD patients, as well as toxin-induced Parkinsonism, demonstrate mitochondrial dysfunction. Hence, counteracting these mitochondrial defects may be an essential strategy to battle PD. p13 (c7 or f55), as a mitochondrial matrix protein, reduces during the exposure of pancreatic cells to oxidative stress (Higashi et al. 2015). p13 exerts its effects by binding to subunits of the electron transport chain complexes I and V known as NDUFAB1 and ATPAF2 (Floyd et al. 2016). Recently, it has been shown that p13 directly regulates the activity of complex I and increase in p13 expression promotes the assembly of complex I. Such evidence explains the interaction of p13 in the PD models. More recent findings revealed that the reduction in p13 expression suppresses the mitochondrial defects observed in toxin-induced and genetic PD models. Furthermore, the modulatory effect of p13 on mitochondrial function in both in vitro and in vivo PD models has been proved (Inoue et al. 2018).
Ghrelin
Greline is a hormone that activated its receptor, the G-protein coupled receptor the growth hormone secretagogue receptor (GHS-R). This protein regulates the growth hormone secretion, food intake, memory performance and reward-seeking behavior (Abizaid et al. 2006). Ghrelin directly regulates the neuronal activity, and the expression of GHS-R has been observed in different brain areas such as the SNc, ventral tegmental area, hypothalamus, and hippocampus (Osterstock et al. 2010). It is reported that the electrical activity of ghrelin triggers the dopaminergic neurons in SNc and enhances the level of dopamine in the striatum (Shi et al. 2013). Also, the neuroprotective effects of ghrelin are commonly known; as it is seen preventing the loss of dopaminergic neurons within SNc in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of PD (Andrews et al. 2009).
Prokineticin 2
Prokineticin belongs to the AVIT protein family and consists of Prokineticin 1 and Prokineticin 2 (PK2). These two chemokines signal via the Prokineticin receptors PKR1 and PKR2. Because of their ability to contract gastrointestinal smooth muscle from isolated ileal segments, they were named Prokineticin (Pitteloud et al. 2007). PK2 regulates wide-ranging processes, including neurogenesis, hematopoiesis, pain reception, reproductive functions, and circadian rhythm. In the brain, PK2 regulates neurogenesis during olfactory bulb biogenesis by guiding the migration of progenitor cells from the subventricular zone. In addition, it regulates circadian rhythms by playing role as an output molecule from the suprachiasmatic nucleus (SCN) (Cheng et al. 2005). Recently, the involvement of PK2 in thermoregulation and energy expenditure through the hypothalamus has been reported 8. The result of recent study showed that PK2 mRNA overexpressed during dopaminergic (DAergic) cell death triggered by tumor necrosis factor alpha (TNFa) (Gordon et al. 2012). Hence, it is hypothesized that PK2 is a promising signaling mediator secreted during dopaminergic degeneration.
Protein Kinase Cδ (PKCδ)
PKCδ belongs to a novel PKC isoform family and involves in numerous signal transduction pathways include proliferation, cell cycle progression, differentiation, as well as apoptosis. Findings showed that the expression of Kinase is high in SNpc dopaminergic neurons, dopaminergic cell lines primary, and dopaminergic cultures (Zhang et al. 2007). During neurotoxic stress, PKCδ is proteolytically cleaved by caspase-3 in dopaminergic cells which causes apoptotic neuronal cell death (Hanrott et al. 2008). Furthermore, it is reported that stimulation of TNFα receptors in dopaminergic neurons proteolytically activates PKCδ and triggers apoptosis (Gordon et al. 2012). Along with the role in apoptosis, PKCδ regulates inflammatory and immune responses in peripheral immune cells such as macrophages, neutrophils, and B cells. Findings provide evidence that activated microglia induced by various inflammatory agents include LPS, TNFα, as well as aggregated α-synuclein, results in the activation of PKCδ. This activation is accompanied by upregulation of PKCδ gene and kinase activity which supports the hypothesis that microglial activation is integrated with enhanced PKCδ protein expression and associated kinase activity (Gordon et al. 2016b).
Findings provide evidence that activated microglia induced by various inflammatory agents include TNFα, LPS, as well as aggregated α-synuclein, results in the activation of PKCδ. This activation is accompanied by upregulation of PKCδ gene and kinase activity which supports the hypothesis that microglial activation is integrated with enhanced PKCδ protein expression and associated kinase activity. Furthermore, the exposure of N27 dopaminergic neuronal cells to pesticide such as an endosulfan rapidly triggers the autophagy. However, prolonged endosulfan exposure activates apoptotic signaling and triggers caspase-2 and -3 activation and PKCδ proteolytic activation. Eventually, the activation of this signal pathway leads to the cell death. Therefore, it concludes that apoptosis following autophagy during endosulfan neurotoxicity (Song et al. 2019).
Multiple studies have scrutinized the relation between mentioned genes and the pathogenesis of PD. The development of genetic engineering technologies could render this category of investigations more feasible. The recently emerged CRISPR-Cas9 genome editing technologies have enabled efficient and precise genetic and epigenetic manipulations of genomes and revolutionized the study of cellular function in health and disease. PD as a neurodegenerative disease predicts to affect a numerous people in a century ahead. Hence, efforts for more understanding of PD underlying mechanisms and finding an efficient therapeutic approaches are ongoing. In the following paragraphs, we review the researches in which CRISPR system has been used for investigation and treatment of PD.
Gene Editing Mediated by CRISPR/Cas9 in PD Linkage Researches
SNCA Gene Editing Mediated by CRISPR
According to data derived from the GWAS, SNCA is one of the common risk loci associated with sporadic PD patients (Schrag 2018). It is worth mentioning that none of the sporadic PD-associated single-nucleotide polymorphisms (SNPs) affect the protein coding sequence of a-synuclein (Lubbe and Morris 2014). It has been proposed that these variants may enhance the gene expression levels (Scott et al. 2017); therefore, the aggregation of abnormal protein and formation of LB is a classic pathological feature of PD. To validate this hypothesis, Soldner et al. have employed iPS cells, GWAS technology, and CRISPR/Cas9 system to detect different SNCA variants containing SNP, and accordingly manipulate them. They utilized these data to analyze the link between SNCA variants and the pathogenesis of PD. In this way, they introduced a common variant of SNCA embedded in a non-coding distal enhancer element. It is hypothesized that this variant upregulates the expression of SNCA via enhancing the affinity of transcription factors to this enhancer element (Soldner et al. 2016).
To assess this hypothesis, scientists used CRISPR/Cas9 to remove the enhancer region in sequence that contains two risk-associated SNPs, rs356168 and rs3756054, in human embryonic stem cells. After that, the insertion of one of the two enhancer-deleted alleles with one possible variant leads to the generation of four heterozygous cells. Importantly, the homozygous enhancer-deleted cell was used as the control. Following this, the cells were differentiated into neural precursors or neurons. Besides, they utilized the TaqMan SNP genotyping assays to measure the relative levels of the SNCA transcriptions in the genetically edited cells. Their findings showed that a common risk-associated variant could alter the expression of the a-syn via altering the affinity of the transcription factors (TFs) to the enhancer. It is the first report in which investigators elucidated the influences of genetic variants on PD risk at the molecular level by using CRISPR/Cas9 technology according to GWAS data. Therefore, the empirical evidence shed light on the involvement of non-coding genetic variants in the elevation of gene expression levels. However, in this study, innovative use of GWAS data, CRISPR/Cas9 and TaqMan qRT–PCR analysis facilitates the quantification of the allele-specific expression levels in a single multiplex reaction, but it represents some limitations (Soldner et al. 2016).
For example, in this study, only transcriptional regulation was assessed, but other possible regulatory mechanisms of the disease-associated variants have not been investigated. Another limitation is the generalization of the in vitro data presented in this study to the in vivo situation. To address these limitations, researchers have considered using techniques that enable direct observation in brain slices, and even in animal brains (Piper et al. 2018).
In another effort to investigate the transcriptional dysregulation of SNCA, researches have progressed to epigenetic studies. These studies indicated that hypomethylation of SNCA regulatory region has a significant influence on increasing the expression of SNCA in sporadic PD (de Boni et al. 2011; Jowaed et al. 2010; Matsumoto et al. 2010). Lack of complete understanding of SNCA expression in pathologic conditions prompted the researchers to use a molecular tool/system to detect the alterations in transcription and also investigate the effect of endogenous epigenetic modulation of the gene. Luciferase reporter fused to the promotor of a gene of interest is the most common tool used for the gene transcriptional activity studies (Solberg and Krauss 2013).
In this respect, the plasmid-based exogenous reporter systems provide low efficiency for investigating a whole aspect of gene expression regulation. Therefore, Basu and his colleagues developed a novel reporter tagged at the 3′end of SNCA endogenously (Basu et al. 2017). This promising tool allowed the analysis of gene transcriptional activity without perturbation in epigenetic architecture. The advantages of this NanoLuc luciferase reporter include being 150-fold brighter and remarkably smaller in size as compared to another luciferase such as Firefly or Renilla. These properties make NanoLuc luciferase reporter an ideal tag even for genes with a low level of expression. To endogenously insert this tag in SNCA, CRISPR/Cas9 has been applied (Basu et al. 2017). SNCA NanoLuc tagging provided a sensitive and real-time approach to measure the changes in transcriptional activity in response to various conditions of stimuli. As the SNCA gene has been demonstrated to get extensively regulated by epigenetic structure, this tagging system may help the study of SNCA expression (de Boni et al. 2011). Furthermore, α -SYN is a molecular marker whose level of expression is directly associated with the PD pathogenesis and severity (Grundemann et al. 2008). In this context, NanoLuc can be deemed as a strong candidate for the treatment for PD.
SNCA gene encoding a-synuclein may be the most viable target in the context of gene therapy for PD (Flierl et al. 2014; McCormack et al. 2010; Takahashi et al. 2015). It is postulated that alterations in the levels of SNCA expression have beneficial impacts. However, results of RNAi mediated gene silencing have shown a robust reduction in SNCA levels, which has prompted incidents neurotoxicity (Gorbatyuk et al. 2010; Khodr et al. 2011). Therefore, identification of a target tightly regulating the SNCA transcription may maintain baseline physiological levels of a-syn. Several genetic and epigenetic regulatory mechanisms are involved in the regulation of SNCA expression levels (Guhathakurta et al. 2017; Tagliafierro and Chiba-Falek 2016). Epigenetic mechanisms such as DNA methylation are essential mechanisms in the transcriptional regulation. It is known that amplified SNCA expression is directly related to the demethylation of CpGs at SNCA intron-1 (Jowaed et al. 2010; Wang et al. 2013). Furthermore, several studies have reported disease-related differential DNA methylation of SNCA intron-1. Investigation of blood samples and postmortem brain tissues of PD patients have shown that the methylation levels at SNCA intron-1 were lower in PD patients when compared with the control donors (Ai et al. 2014). Hence, DNA methylation studies may add to our knowledge about the PD pathogenesis and could also act an attractive target for therapeutic manipulation of SNCA gene expression. Recently, new engineered variants of the CRISPR-Cas9 system, including dCas9, provide us with a unique opportunity to modify the gene expression with high precision (Liao et al. 2017; Liu et al. 2018). Epigenome editing is a novel and promising branch of gene therapy which could pave the way for the development of new drugs via targeting specific genes. In a recent study, an innovative epigenome-editing approach was developed to manipulate the endogenous SNCA levels. In this study, the SNCA locus of human induced pluripotent stem cell (hiPSC)-derived neurons was triplicated, which resulted in the upregulation of SNCA expression (Kantor et al. 2018).
The characterization of these hiPSC-derived neurons has demonstrated that spontaneous a-syn aggregation and an increase in oxidative stress markers propagates alterations to the cell viability. These changes cause a higher vulnerability to cell death mediated by oxidative stress (Byers et al. 2012; Flierl et al. 2014). The production of hiPSCs derived from the neural tissue of PD individuals containing a genomic triplication of the SNCA gene opens a new therapeutic avenue for targeting SNCA expression levels.
In reversing the overexpression of SNCA, a novel all-in-one lentiviral vector—which has undergone modifications of gRNA, dCas9 nuclease, and the catalytic domain of the DNMT3A—is developed. This vector was designed to target specific hypomethylated CpG islands in the SNCA intron one region. With this novel technique, transfection of dopaminergic neural progenitor cells derived from hiPSCs of a PD patient resulted in the reduction of SNCA mRNA and protein expression. Consequentially, this reversed phenotypic perturbations related to the PD. In this context, Kantor et al. showed that the modification of gene expression by epigenome editing provides a valuable therapeutic strategy for neurological disorders, such as PD (Kantor et al. 2018).
Cell therapy by transplantation of fetal ventral mesencephalic cells into the striatum has been acknowledged as an experimental treatment for PD (Lindvall et al. 1990). However, some patients exhibited long-term alleviation of motor symptoms (Li et al. 2016), but numerous patients have suffered from graft-induced dyskinesias (Piccini et al. 2005). It is reported that the graft-induced dyskinesia may be associated with serotonergic hyperinnervation from the graft (Politis et al. 2010). Furthermore, observation of acquired LB pathology in most grafts older than ten years proposed that host-to-graft spread of disease might take place (Li et al. 2008). The aggregation of LBs in the graft has resulted in a low therapeutic benefit for patients (Chu and Kordower 2010). These clinical findings highlight the need for engineered cells which are resistant to the formation of Lewy bodies (Spillantini et al. 1997). The utilization of disease-resistant cells will be particularly useful for PD patients with early-onset or familial form of the disease. These forms of PD burden a substantial α-synuclein such as GBA mutation carriers and SNCA multiplications (Neumann et al. 2009).
This phenomenon can be triggered by the injection of preformed fibrils (PFFs) of recombinant α-syn protein modeling of synucleinopathy in cell culture has shown. Results of molecular pathology have demonstrated that the phosphorylation of α-syn at position serine-129 as a prominent post-translational event has taken place during the first stage of α-synuclein aggregation (Fujiwara et al. 2002). From these observations, it can be argued that in the absence of α-syn expression, neurons are less susceptible to α-syn PFF seeding (Volpicelli-Daley et al. 2011). It is reported that neurons with low endogenous expression of α-syn are left uninjured in the PD (Braak et al. 2004). Also, α-synuclein PFFs receiving Snca −/− mice showed no signs of synucleinopathy or neurodegeneration (Luk et al. 2012).
To this end, Chen and his colleagues established an engineered pluripotent stem cell with reduced or removed SNCA alleles that encoded for α-syn. Employing Cas9n technology with four gRNAs, they were able to successfully remove the first coding exon of the SNCA gene in human embryonic stem cells (hESCs) to produce several SNCA ± and SNCA −/− clonal cell lines. Furthermore, they engineered an iPSC line which harbored three α-synuclein genes and generated triple knockout cell lines.
Successful differentiation of these all genotypes of the cell line to mDA neurons suggested that knockout of the α-synuclein protein did not have any influence on the differentiation tendency of pluripotent stem cells into this neuronal subtype. Functional studies and analysis of transcriptome revealed that although the numbers of dopaminergic neurons in the substantia nigra of Snca −/− mice were normal, dopamine content of striatum was reduced (Abeliovich et al. 2000). Furthermore, findings of this study proposed that α-synuclein-free grafts may have some protective effect against neurotoxins such as MPTP.
In this context, the CRISPR system can be seen as an efficient technology for the generation of SNCA ± and SNCA −/− mDA neurons from hESCs or iPSCs. Moreover, various engineered variants of Cas9 protein such as Cas9n may pave the way of production of the next generation of Parkinson-résistant cells (Chen et al. 2018).
Parkin Gene Editing Mediated by CRISPR
PINK1 and PARKIN are critical in mitochondrial quality control and mitophagy. In previous studies, RNAi screens have been employed to identify the regulators of PINK1/PARKIN-mediated mitophagy (McCoy et al. 2014). To elucidate on the set mitophagy threshold of the cells, researchers have used CRISPR/Cas9 gene-editing technology as a screening tool which appears to outdo RNAi for phenotypic screening (DeJesus et al. 2016).
To detect the regulators of PARKIN, a phenotypic genome-wide CRISPR/Cas9 pooled screen has been performed. In this effort, scientist used cells which express a PARKIN reporter protein from the endogenous PARK2 promoter. The level of PARKIN determined the kinetics of pUb accumulation in this study. The results of this project showed that 53 positive and negative factors were involved in the regulation of PARKIN.
In line with bioinformatics results, it is asserted that the transcriptional repression negatively regulates the level of endogenous PARKIN. Remarkably, the reduction of THAP11 affects both pUb accumulation and PARKIN protein levels in multiple cell types. Moreover, THAP11 targeting by CRISPR/Cas9 in iPSC-derived inducible Neurogenin 2 (iNGN2) neurons resulted in de-repression of PARK2 transcription and enhanced pUb accumulation (Potting et al. 2018).
Even though the exact mechanism by which PARKIN mediates PD-like syndromes yet remains unknown, researchers have generated a monogenic form of PD-specific iPSCs-derived DA neurons which harbor PARKIN mutations. Accordingly, CRISPR/Cas9 can generate PARKIN-knockin knockout (KIKO) iPSCs by insertion of a donor sequence in the exon 2 of gene silencing the PARKIN function. Investigation of this isogenic cell line has shown that the significant reduction in the mRNA expression of both GHSR1a and GHSR1b. Also, their data have elucidated the effect of PARK2 mutation effect on the expression of GHSR1a. PARK2 mutation may also alter the transfer of ubiquitin onto substrate proteins, which could change the transcriptional level of GHSR1a (Suda et al. 2018).
P13 Gene Editing Mediated by CRISPR
As previously discussed, p13 is involved in the modulation of mitochondrial function, so it is proposed as a therapeutic target for PD. To assess this hypothesis, Inoue et al. investigated the p13 levels in both toxin-induced and genetic models of PD. They demonstrated that p13 levels are downregulated in vitro as well as in vivo toxin-induced models of PD (human and mice neuroblastoma cells). Although similar low levels were not observed in the PD, a difference between acute and chronic models can be an underlying mechanism beneath this finding.
Also, they discovered that a further decrease in p13 levels of expression induced neuroprotective effects, and rescued the toxin-induced as well as the genetic PD models. Downregulation of p13 levels by using shRNA inhibited the disruption of complex I assembly induced by the neurotoxin. On the other hand, overexpression of p13 accelerated the generation of the toxin-induced PD models phenotypes. In further probing, they found that the p13 reduction in the toxin-induced PD models took place as a response to the mitochondrial dysfunction and cellular injury. Therefore, reduction in the p13 level might “hyperstabilize” the Tne complex 1 and protect the mitochondria before the accumulation of toxins (Fig. 2).

Downregulation of p 13 triggers the assembly of mitochondrial complex I and inhibits cell death mediated by mitochondrial dysfunction in Parkinson’s disease
In the same context, Inoue et al. created a knockout mouse which lacked p13 expression via CRISPR/Cas9. Intriguingly, these p13 knockout mice represented no motor deficits or dopaminergic neuron loss following treatment of MPTP. Their results implied that in p13 knockout mice, an assemblage of complex I occurred more readily with toxin exposure. Hence, the mitochondrial function was preserved at a heightened level, which maintained the normalcy neural cells (Inoue et al. Inoue N 2018).
PK2 Gene Editing Mediated by CRISPR
Recent studies have demonstrated a major role of PK2 in mitochondrial biogenesis. It is reported that PK2 signaling is enhanced in PD postmortem brains. Facing with neurotoxic stress results in overexpression of PK2 which preserves mitochondrial biogenesis through increasing the levels of PGC-1α and BCL2 and retrieves loss in ATP production mediated by neurotoxic agents (Gordon et al. 2016a). In MPTP-induced gliosis model, overexpression of PK2 decreased the processes of reactive astrocytes and increased the process of neuroprotective A2 astrocyte phenotype production (Neal et al. 2018). This compensatory effect may neutralize the neuroinflammation induced by reactive A1 astrocytes (Liddelow et al. 2017).
To confirm the neuroprotective effect of PK2, CRISPR/Cas9 system has been used to block this gene and its receptor. The results of these in vitro studies showed that a PK2 knockout increased the neuronal susceptibility to cell death induced by neurotoxic agent (Gordon et al. 2016a). Furthermore, CRISPR-Cas9-mediated knockout of PKR1, put an end to the effects of PK2 on the activation of alternative A2 astrocyte (Neal et al. 2018). Together with, these evidence shed light on the association of mitochondrial dysfunction with neuroinflammation and display the utility of CRISPR-Cas9 in clarifying these mechanisms (Fig. 3).

Knocked out PK2 mediated by CRISPR/Cas9 illustrates the protective role of PK2 in anti-apoptotic responses against neurotoxic stress in both neurons and astrocytes. Neurotoxic stress can activate a variety of transcription factors, including HIF1α. This transcription factor binds to the promoter of the PK2 gene and upregulates the expression of this protein. PK2 enhances the mitochondrial biogenesis and upregulates the BCL-2 and PGC1α which result in the cell survival. In addition, PK2 in astrocytes upregulates the expression of genes associated with the anti-inflammatory alternative activation (A2) phenotype. PK2 gene ablation by CRISPR-Cas9 produces unintended effects and increases the cell susceptibility to a neurotoxic stress
PKCδ Gene Editing Mediated by CRISPR
Prolonged endosulfan treatment triggers the apoptotic cascade include activation of caspases, release of cytochrome c, proteolytic activation of PKCδ, and fragmentation of DNA. To elucidate the role of PKCδ in cell death mediated by endosulfan, PKCδ was knock-downed by utilization of CRISPR/Cas9 in N27 dopaminergic cells. This stable knock-downed cells revealed the reduction in the levels of endosulfan-induced caspase-3 which suggests that a PKCδ plays regulatory role in apoptosis induced by endosulfan. Further investigation by using autophagy and caspase-2 or -3 inhibitors showed that autophagy is an upstream process than apoptosis and plays protective roles in exposure with endosulfan. These results imply that autophagy dysfunction plays a critical role in the etiology of PD; hence, findings of an efficient autophagy enhancer may be taken into consideration as a potential PD therapies (Song et al. 2019).
Parkinson Disease Modeling
Animal models are viable tools for investigating a different range of neurological abnormalities such as PD. Since PD has a heterogenic etiology, various approaches have been used to generate the disease in the animal models. These approaches include using pharmaceutical products and neurotoxins, which induce dysfunctions attributed to the midbrain dopaminergic signaling. It also includes genetic engineering strategies in modeling the genetic forms of PD. The 6-hydroxydopamine (6-OHDA) and MPTP have been extensively used to generate pharmacologically based models of PD (Dawson et al. 2018). Although introduced models were appropriate in symptomatic therapies of the motor symptoms in PD, they were not applicable in therapeutic approaches (Athauda and Foltynie 2016). Therefore, efforts are directed toward generating genetics-based models of PD. Unsurprisingly, CRISPR has emerged as a de facto technique in the domain of genetic animal modeling with respect to the PD tissues.
In vivo Disease Modeling Mediated by CRISPR System
In previous decades, scientists have been employing transgenic mouse models expressing the wild-type or mutant α-synuclein to study the molecular pathophysiology of PD (Blesa and Przedborski 2014). Until now, few transgenic models have been generated with the ability to mirror the unique and cardinal pathologic characteristics of human PD, such as an actual α-synuclein-immunopositive pathology or progressive dopaminergic neuron loss in the SN (Blesa and Przedborski 2014; Chesselet and Richter 2011). Since the pathogenesis of PD lasts longer than the lifespan of a mouse, development of long-lifespan, large animal models of PD is deemed as necessary (Zhu et al. 2018).
Pigs and minipigs in specific can be considered as a proper large animal model for human diseases (Yao et al. 2016). There are substantial advantages to the minipigs, which include a decade-long lifespan, body size similar to humans when compared to rodents. They also have short gestation periods (about 4 months). There are also large numbers of offspring with one pregnancy (more than 10 piglets), in addition to the existence of an adolescence-onset (5 to 6 months). Also, the similarities in the context of neurophysiology and neuroanatomy of minipigs with humans make them useful tool vis-a-vis neurodegenerative maladies (Dolezalova et al. 2014).
In this context, CRISPR/Cas9 has been used for the generation of Guangxi Bama minipigs harboring PD-causing mutations in the SCNA including E46 K, H50Q and G51D. Gene editing of somatic cells was followed by the somatic cell nuclear transfer (SCNT). The combination of the CRISPR/Cas9 system with SCNT made minipigs that represented PD-specific pathological changes, i.e., α-synuclein-immunopositive pathology along with the loss of dopaminergic neurons in SN (Zhu et al. 2018).
As pointed out before, recessively inherited mutations in Parkin, DJ-1, and PINK1 genes are tightly linked to the familial forms of early-onset PD (Bonifati et al. 2002). TThe lack of a proper model is a significant barrier to the development of practical therapeutic approaches to the PD in humans. Albeit, this lack of appropriate models has been exclusive to human PD, as recently a triple knockout was generated to ameliorate our understanding of the PD pathophysiology in the pig models and to improve the symptomatic management of DJ-1, Parkin, and PINK1 in them. In this effort, the CRISPR/Cas9 system was, used to modify the genes biallelically. In this study, dual sgRNAs were employed for specific gene targeting, which remarkably enhanced the CRISPR/Cas9-mediated genome editing (Wang et al. 2016).
In vitro PD Modeling Mediated by CRISPR/Cas9
IPSC technology provides us with a promising opportunity to develop a wide range of PD disease modeling. These models facilitate the analysis of pathological effects of LRRK2 mutation (LRRK2-G2019S) such as raising the stress sensitivity and reducing the neurite complexity (Nguyen et al. 2011; Sanchez-Danes et al. 2012). As the genetic background is a critical element in comparing the patients and the control group, isogenic cell lines with identical genetic backgrounds only differ in mutations which are introduced. Various LRRK2-G2019S isogeneic cell lines were generated with technologies such as Zinc finger and Cre/LoxP systems (Reinhardt et al. 2013).
The major limitation within the Cre/LoxP system is the deletion of a 34-bp-long LoxP of the chromatin, usually in an intron juxtaposed to the position where mutations are introduced or corrected. It is reported that the deletion of this LoxP site in the intron exerts influence on the expression of the targeted allele (Zou et al. 2011). Also, in heterozygous mutations, the random insertion of the LoxP site in an unknown regulatory element in the intron may also affect the regulation of other genes (Meier et al. 2010). These drawbacks have prompted the scientific community to generate footprint-free isogenic cell lines.
Simplicity and efficiency of CRISPR/Cas9 compared with zinc finger nucleases and TALENs underlines the superiority of this system for gene editing (Gaj et al. 2013). In recent studies, the CRISPR/Cas9 system was employed efficiently to introduce mutations by reporter insertion in the LRRK2 gene (Qing et al. 2017). Beside the CRISPR system, piggyback transposon has also been used for complete removal of the selected cassette to ensure the generation of a footprint-free isogeneic cell line (Ye et al. 2014). This LRRK2-G2019S isogenic cell line summarizes PD-specific phenotypes such as neurite complexity and makes new insights into the role of S129P-αS (Qing et al. 2017).
In other in vitro PD models, isogenic hESC lines with a deletion of RAB39B are generated. These cell models will pave the way for a better understanding of PD pathogenesis. Further, they can deepen our understanding of the RAB39B function. In this study, the parental male hESC line was transfected by CRISPR guides targeting either exon 1 or 2 of RAB39B. These hESC lines represent typical morphological characters like their unmodified parental lines such as small and tightly packed cells, prominent nucleoli and high nucleus to cytoplasm ratio. By using Yamanaka’s factors, RAB39B knocked out hESC lines were converted to iPSC, which verified by flow cytometry. This iPSC cell line can differentiate into neuronal models to study the pathogenic mechanisms underlying PD (Gao et al. 2018).
Pros and Cons of CRISPR for iPSCs Genome Editing
CRISPR, as an emergent genome editing system, improves the biomedical research and therapeutic approach toward PD [108]. This technology provides us with the opportunities for all types of genome modifications such as in/del point mutations, homologous recombination, or deletion of long-nucleotide sequences as well as transcription modification of specific genetic elements. Practical nature, as well as resistance against epigenetic changes, makes CRISPR/Cas9 the most popular technique of genome editing technology in modeling human disease in vitro and in vivo [109]. Most recently, genome editing of stem or somatic cells mediated by the CRISPR/Cas9 system has become ever relevant. In the same context, iPSCs have become useful models for understanding the patient-specific pathology. These cells lines as patient in a tube can be surely used for establishing different therapeutic substances (Vasil’eva et al. 2015). Target modifications mediated via CRISPR/Cas9 in the iPSC genome provide a useful cell line to identify the genes involved in pathogenesis at various stages of the disease in both in vivo and in vitro models. However, there have been problems with the cell reprogramming using these technologies for therapeutic approaches such as observation of genomic abnormalities during long-term culture (Hussein et al. 2011). Findings of molecular and cytogenetic studies revealed the emergence of unintended silent point mutations, chromosome aberrations, aneuploidies, and polyploidies (Dekel-Naftali et al. 2012).
The mutant genes’ products may cause the instability of either nuclear or mitochondrial genomes (Sanders et al. 2014). This is an important phenomenon and must be taken into consideration for genome editing. The other limitation of iPSC genome editing is the sensitivity of the cells to DNA damages, which results in the low frequency of homological insertion. To scrutinize this, researches performed cytogenetic analysis of neural cells carrying corrected G2019S mutation in LRRK2 gene mediated by CRISPR/Cas9. Findings of this analysis have shown a mosaic variant of tetraploidy 92 XXYY/46, XY in 24-43 percent of cells derived from various clones in neuronal precursors differentiated from the iPSC. Also, single cases of translocations and chromosome breaks were reported. These findings imply the significance of new techniques ensuring genome stability in CRISPR/Cas9-edited cell cultures. This data may insinuate that each gene-edited culture should be thoroughly assessed for the presence of probable mutations before autological transplantation (Vetchinova et al. 2018). It is postulated here that genome aberrations occur independently with the use of integration/non-integration methods of transcription factors delivered for reprogramming (Gore et al. 2011).
A frequency of off-target effect is a major challenge of the genome editing through CRISPR-Cas9. Because, Cas9 that expressed in plasmid platform could tolerate single- and double-base mismatches which results in unintended mutagenesis (Fu et al. 2013). Developing of precise web base tools for designing of specific gRNA and utilization of recombinant Cas9 protein which degrades immediately after exerting its intended gene-editing effect may reduce the off-target effects. Moreover, efficient delivery approaches for targeting CNS system is another challenge of CRISPR utility. Electroporation, microinjection, lentivirus, AAV, and nanoparticle or liposomal are common delivery methods but a few of them are practical for CNS delivery of CRISPR-Cas9-based therapy (Arruda et al. 2017). To address this limitation, a neuron-preferring chimeric AAV-based CRISPR-Cas9 system has been developed for inducing brain-specific gene deletion (Murlidharan et al. 2016).
Conclusion
Neurodegenerative abnormalities such as Parkinson are debilitating for patients and their families and also impose a growing economic burden on health systems. The gap in the research and the clinic is has remained wide open. To address this gap, platforms for CRISPR-based genome editing in human neurons have been developed. These platforms facilitate the identification of cellular factors in controlling the vulnerability of cellular processes underlying neurodegeneration in PD. Combinations of CRISPR/Cas9, GWAS, and iPSCs technology shed light on the critical elements behind the PD pathogenesis. This may pave the way to introduce possible therapeutic targets. To improve the limitations of CRISPR/Cas9, we recommend using other types of CRISPR system like Cas12a (Cpf1) with less unforeseen consequences. The advantages of Cas12a include the ability to target T-rich motifs-without trans-activating crRNA- and induction of a staggered double-strand break as well as the potential for both RNA processing and DNA nuclease activity. This programmable nuclease may become our de facto compass in navigating among different therapeutic approaches to PD, as well as other neurodegenerative maladies.
References
Abeliovich A et al (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25:239–252
Abizaid A et al (2006) Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Investig 116:3229–3239. https://doi.org/10.1172/jci29867
Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW (2003) The role of pathogenic DJ-1 mutations in Parkinson’s disease. Ann Neurol 54(3):283–286. https://doi.org/10.1002/ana.10675
Ai SX et al (2014) Hypomethylation of SNCA in blood of patients with sporadic Parkinson’s disease. J Neurol Sci 337:123–128. https://doi.org/10.1016/j.jns.2013.11.033
Anand VS, Braithwaite SP (2009) LRRK2 in Parkinson’s disease: biochemical functions. FEBS J 276(22):6428–6435. https://doi.org/10.1111/j.1742-4658.2009.07341.x
Andrews ZB et al (2009) Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J Neurosci 29(45):14057–14065. https://doi.org/10.1523/jneurosci.3890-09.2009
Annesi G et al (2005) DJ-1 mutations and parkinsonism-dementia-amyotrophic lateral sclerosis complex. Ann Neurol 58(5):803–807. https://doi.org/10.1002/ana.20666
Arkinson C, Walden H (2018) Parkin function in Parkinson’s disease. Science 360:267–268. https://doi.org/10.1126/science.aar6606
Arruda VR, Doshi BS, Samelson-Jones BJ (2017) Novel approaches to hemophilia therapy: successes and challenges. Blood 130:2251–2256. https://doi.org/10.1182/blood-2017-08-742312
Athauda D, Foltynie T (2016) Challenges in detecting disease modification in Parkinson’s disease clinical trials. Parkinsonism Relat Disord 32:1–11. https://doi.org/10.1016/j.parkreldis.2016.07.019
Basu S, Adams L, Guhathakurta S, Kim YS (2017) A novel tool for monitoring endogenous alpha-synuclein transcription by NanoLuciferase tag insertion at the 3′end using CRISPR-Cas9 genome editing technique. Sci Rep 7:45883. https://doi.org/10.1038/srep45883
Blesa J, Przedborski S (2014) Parkinson’s disease: animal models and dopaminergic cell vulnerability. Front Neuroanat 8:155. https://doi.org/10.3389/fnana.2014.00155
Bonifati V et al (2002) Localization of autosomal recessive early-onset parkinsonism to chromosome 1p36 (PARK7) in an independent dataset. Ann Neurol 51(2):253–256
Bonifati V et al (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299(5604):256–259. https://doi.org/10.1126/science.1077209
Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318:121–134. https://doi.org/10.1007/s00441-004-0956-9
Byers B, Lee HL, Reijo Pera R (2012) Modeling Parkinson’s disease using induced pluripotent stem cells. Curr Neurol Neurosci Rep 12:237–242. https://doi.org/10.1007/s11910-012-0270-y
Chang D et al (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49(10):1511–1516. https://doi.org/10.1038/ng.3955
Chen Y et al (2018) Engineering synucleinopathy-resistant human dopaminergic neurons by CRISPR-mediated deletion of the SNCA gene. Eur J Neurosci. https://doi.org/10.1111/ejn.14286
Cheng H et al (2002) Isolation and characterization of a human novel RAB (RAB39B) gene. Cytogenet Genome Res 97(1–2):72–75. https://doi.org/10.1159/000064047
Cheng MY, Bittman EL, Hattar S, Zhou QY (2005) Regulation of prokineticin 2 expression by light and the circadian clock. BMC Neurosci 6(1):17. https://doi.org/10.1186/1471-2202-6-17
Chesselet MF, Richter F (2011) Modelling of Parkinson’s disease in mice. Lancet Neurol 10:1108–1118. https://doi.org/10.1016/s1474-4422(11)70227-7
Chu Y, Kordower JH (2010) Lewy body pathology in fetal grafts. Ann N Y Acad Sci 1184:55–67. https://doi.org/10.1111/j.1749-6632.2009.05229.x
Dawson TM, Golde TE, Lagier-Tourenne C (2018) Animal models of neurodegenerative diseases. Nat Neurosci 21:1370–1379. https://doi.org/10.1038/s41593-018-0236-8
de Boni L, Tierling S, Roeber S, Walter J, Giese A, Kretzschmar HA (2011) Next-generation sequencing reveals regional differences of the alpha-synuclein methylation state independent of Lewy body disease. NeuroMol Med 13:310–320. https://doi.org/10.1007/s12017-011-8163-9
DeJesus R et al (2016) Functional CRISPR screening identifies the ufmylation pathway as a regulator of SQSTM1/p62. ELife 5:e17290. https://doi.org/10.7554/elife.17290
Dekel-Naftali M et al (2012) Screening of human pluripotent stem cells using CGH and FISH reveals low-grade mosaic aneuploidy and a recurrent amplification of chromosome 1q. Eur J Hum Genet 20(12):1248–1255. https://doi.org/10.1038/ejhg.2012.128
Devine MJ, Gwinn K, Singleton A, Hardy J (2011) Parkinson’s disease and alpha-synuclein expression. Mov Disord 26:2160–2168. https://doi.org/10.1002/mds.23948
Dolezalova D et al (2014) Pig models of neurodegenerative disorders: utilization in cell replacement-based preclinical safety and efficacy studies. J Comp Neurol 522:2784–2801. https://doi.org/10.1002/cne.23575
Emamalizadeh B et al (2014) RIT2, a susceptibility gene for Parkinson’s disease in Iranian population. Neurobiol Aging 35(12):e27–e28. https://doi.org/10.1016/j.neurobiolaging.2014.07.013
Farrer M et al (2004) Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol 55:174–179. https://doi.org/10.1002/ana.10846
Ferreira M, Massano J (2017) An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol Scand 135:273–284. https://doi.org/10.1111/ane.12616
Flierl A et al (2014) Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an alpha-synuclein gene triplication. PLoS ONE 9:e112413. https://doi.org/10.1371/journal.pone.0112413
Floyd BJ et al (2016) Mitochondrial protein interaction mapping identifies regulators of respiratory chain function. Mol Cell 63:621–632. https://doi.org/10.1016/j.molcel.2016.06.033
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD (2013) High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31:822–826. https://doi.org/10.1038/nbt.2623
Fujiwara H et al (2002) α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4(2):160–164. https://doi.org/10.1038/ncb748
Gaj T, Gersbach CA, Barbas CF III (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31(7):397–405. https://doi.org/10.1016/j.tibtech.2013.04.004
Gao Y, Wilson GR, Bozaoglu K, Elefanty AG, Stanley EG, Dottori M, Lockhart PJ (2018) Generation of RAB39B knockout isogenic human embryonic stem cell lines to model RAB39B-mediated Parkinson’s disease. Stem Cell Res 28:161–164. https://doi.org/10.1016/j.scr.2018.02.015
Ghosh A et al (2016) Mitoapocynin treatment protects against neuroinflammation and dopaminergic neurodegeneration in a preclinical animal model of parkinson’s disease. J Neuroimmune Pharmacol 11:259–278. https://doi.org/10.1007/s11481-016-9650-4
Gorbatyuk OS et al (2010) In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol Ther 18:1450–1457. https://doi.org/10.1038/mt.2010.115
Gordon R, Anantharam V, Kanthasamy AG, Kanthasamy A (2012) Proteolytic activation of proapoptotic kinase protein kinase Cdelta by tumor necrosis factor alpha death receptor signaling in dopaminergic neurons during neuroinflammation. J Neuroinflammation 9:82. https://doi.org/10.1186/1742-2094-9-82
Gordon R et al (2016a) Prokineticin-2 upregulation during neuronal injury mediates a compensatory protective response against dopaminergic neuronal degeneration. Nat Commun 7:12932. https://doi.org/10.1038/ncomms12932
Gordon R et al (2016b) Protein kinase Cdelta upregulation in microglia drives neuroinflammatory responses and dopaminergic neurodegeneration in experimental models of Parkinson’s disease. Neurobiol Dis 93:96–114. https://doi.org/10.1016/j.nbd.2016.04.008
Gore A et al (2011) Somatic coding mutations in human induced pluripotent stem cells. Nature 471:63–67. https://doi.org/10.1038/nature09805
Grundemann J, Schlaudraff F, Haeckel O, Liss B (2008) Elevated alpha-synuclein mRNA levels in individual UV-laser-microdissected dopaminergic substantia nigra neurons in idiopathic Parkinson’s disease. Nucleic Acids Res 36:e38. https://doi.org/10.1093/nar/gkn084
Guhathakurta S, Bok E, Evangelista BA, Kim YS (2017) Deregulation of alpha-synuclein in Parkinson’s disease: insight from epigenetic structure and transcriptional regulation of SNCA. Prog Neurobiol 154:21–36. https://doi.org/10.1016/j.pneurobio.2017.04.004
Hanrott K, Murray TK, Orfali Z, Ward M, Finlay C, O’Neill MJ, Wonnacott S (2008) Differential activation of PKC delta in the substantia nigra of rats following striatal or nigral 6-hydroxydopamine lesions. Eur J Neurosci 27(5):1086–1096. https://doi.org/10.1111/j.1460-9568.2008.06097.x
Hasegawa K, Stoessl AJ, Yokoyama T, Kowa H, Wszolek ZK, Yagishita S (2009) Familial parkinsonism: study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat Disord 15(4):300–306. https://doi.org/10.1016/j.parkreldis.2008.07.010
Healy DG et al (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol 7(7):583–590. https://doi.org/10.1016/s1474-4422(08)70117-0
Higashi S et al (2015) p13 overexpression in pancreatic beta-cells ameliorates type 2 diabetes in high-fat-fed mice. Biochem Biophys Res Commun 461(4):612–617. https://doi.org/10.1016/j.bbrc.2015.04.074
Hussein SM et al (2011) Copy number variation and selection during reprogramming to pluripotency. Nature 471:58–62. https://doi.org/10.1038/nature09871
Inoue N et al (2018) Knockdown of the mitochondria-localized protein p13 protects against experimental parkinsonism. EMBO Rep. https://doi.org/10.15252/embr.201744860
Jamshidi J et al (2014) HLA-DRA is associated with Parkinson’s disease in Iranian population. Int J Immunogenet 41:508–511. https://doi.org/10.1111/iji.12151
Jian F et al (2018) Sam50 Regulates PINK1-Parkin-mediated mitophagy by controlling PINK1 stability and mitochondrial morphology. Cell Rep 23(10):2989–3005. https://doi.org/10.1016/j.celrep.2018.05.015
Jowaed A, Schmitt I, Kaut O, Wullner U (2010) Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J Neurosci 30:6355–6359. https://doi.org/10.1523/jneurosci.6119-09.2010
Kantor B et al (2018) Downregulation of SNCA expression by targeted editing of DNA methylation: a potential strategy for precision therapy in PD. Mol Ther 26:2638–2649. https://doi.org/10.1016/j.ymthe.2018.08.019
Khodr CE et al (2011) An alpha-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson’s disease, but displays toxicity in dopamine neurons. Brain Res 1395:94–107. https://doi.org/10.1016/j.brainres.2011.04.036
Kim HJ, Jeon BS, Yoon MY, Park SS, Lee KW (2012) Increased expression of alpha-synuclein by SNCA duplication is associated with resistance to toxic stimuli. J Mol Neurosci 47:249–255. https://doi.org/10.1007/s12031-012-9732-6
Kitada T et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608. https://doi.org/10.1038/33416
Knott GJ, Doudna JA (2018) CRISPR-Cas guides the future of genetic engineering. Science 361(6405):866–869. https://doi.org/10.1126/science.aat5011
Lazarou M et al (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524(7565):309–314. https://doi.org/10.1038/nature14893
Lesage S et al (2015) Loss-of-function mutations in RAB39B are associated with typical early-onset Parkinson disease. Neurol Genet 1(1):e9. https://doi.org/10.1212/nxg.0000000000000009
Li JY et al (2008) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 14:501–503. https://doi.org/10.1038/nm1746
Li W et al (2016) Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc Natl Acad Sci USA 113(23):6544–6549. https://doi.org/10.1073/pnas.1605245113
Liao HK et al (2017) In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell 171(7):1495–1507. https://doi.org/10.1016/j.cell.2017.10.025
Liddelow SA et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. https://doi.org/10.1038/nature21029
Lincoln SJ et al (2003) Parkin variants in North American Parkinson’s disease: cases and controls. Mov Disord 18:1306–1311. https://doi.org/10.1002/mds.10601
Lindvall O et al (1990) Grafts of fetal dopamine neurons survive and improve motor function in Parkinson’s disease. Science 247(4942):574–577
Liu XS et al (2018) Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell 172(5):979–992. https://doi.org/10.1016/j.cell.2018.01.012
Lohmann E et al (2003) How much phenotypic variation can be attributed to parkin genotype? Ann Neurol 54:176–185. https://doi.org/10.1002/ana.10613
Lubbe S, Morris HR (2014) Recent advances in Parkinson’s disease genetics. J Neurol 261:259–266. https://doi.org/10.1007/s00415-013-7003-2
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338(6109):949–953. https://doi.org/10.1126/science.1227157
Luo J et al (2019) Utilization of the CRISPR-Cas9 gene editing system to dissect neuroinflammatory and neuropharmacological mechanisms in parkinson’s disease. J Neuroimmune Pharmacol. https://doi.org/10.1007/s11481-019-09844-3
Marin I (2008) Ancient origin of the Parkinson disease gene LRRK2. J Mol Evol 67(1):41–50. https://doi.org/10.1007/s00239-008-9122-4
Matsumoto L, Takuma H, Tamaoka A, Kurisaki H, Date H, Tsuji S, Iwata A (2010) CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS ONE 5:e15522. https://doi.org/10.1371/journal.pone.0015522
McCormack AL, Mak SK, Henderson JM, Bumcrot D, Farrer MJ, Di Monte DA (2010) Alpha-synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PLoS ONE 5:e12122. https://doi.org/10.1371/journal.pone.0012122
McCoy MK, Kaganovich A, Rudenko IN, Ding J, Cookson MR (2014) Hexokinase activity is required for recruitment of parkin to depolarized mitochondria. Hum Mol Genet 23:145–156. https://doi.org/10.1093/hmg/ddt407
Meier ID et al (2010) Short DNA sequences inserted for gene targeting can accidentally interfere with off-target gene expression. FASEB J 24:1714–1724. https://doi.org/10.1096/fj.09-140749
Mori H et al (1998) Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 51(3):890–892
Murlidharan G et al (2016) CNS-restricted transduction and CRISPR/Cas9-mediated gene deletion with an engineered AAV vector. Mol Ther 5:e338. https://doi.org/10.1038/mtna.2016.49
Nasrolahi A et al (2019) Immune system and new avenues in Parkinson’s disease research and treatment. Rev Neurosci. https://doi.org/10.1515/revneuro-2018-0105
Neal M et al (2018) Prokineticin-2 promotes chemotaxis and alternative A2 reactivity of astrocytes. Glia 66(10):2137–2157. https://doi.org/10.1002/glia.23467
Neumann J et al (2009) Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132:1783–1794. https://doi.org/10.1093/brain/awp044
Nguyen HN et al (2011) LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell stem cell 8(3):267–280. https://doi.org/10.1016/j.stem.2011.01.013
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C (2010) Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 31(7):763–780. https://doi.org/10.1002/humu.21277
Osterstock G et al (2010) Ghrelin stimulation of growth hormone-releasing hormone neurons is direct in the arcuate nucleus. PLoS ONE 5:e9159. https://doi.org/10.1371/journal.pone.0009159
Panicker N, Dawson VL, Dawson TM (2017) Activation mechanisms of the E3 ubiquitin ligase parkin. Biochem J 474(18):3075–3086. https://doi.org/10.1042/bcj20170476
Piccini P et al (2005) Factors affecting the clinical outcome after neural transplantation in Parkinson’s disease. Brain 128:2977–2986. https://doi.org/10.1093/brain/awh649
Piper DA, Sastre D, Schule B (2018) Advancing stem cell models of alpha-synuclein gene regulation in neurodegenerative disease. Front Neurosci 12:199. https://doi.org/10.3389/fnins.2018.00199
Pitteloud N et al (2007) Loss-of-function mutation in the prokineticin 2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA 104(44):17447–17452. https://doi.org/10.1073/pnas.0707173104
Politis M et al (2010) Serotonergic neurons mediate dyskinesia side effects in Parkinson’s patients with neural transplants. Sci Transl Med 2(38):38ra46. https://doi.org/10.1126/scitranslmed.3000976
Polymeropoulos MH et al (1996) Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science 274(5290):1197–1199
Potting C et al (2018) Genome-wide CRISPR screen for PARKIN regulators reveals transcriptional repression as a determinant of mitophagy. Proc Natl Acad Sci USA 115:E180–E189. https://doi.org/10.1073/pnas.1711023115
Pramstaller PP et al (2005) Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol 58(3):411–422. https://doi.org/10.1002/ana.20587
Qing X, Walter J, Jarazo J, Arias-Fuenzalida J, Hillje AL, Schwamborn JC (2017) CRISPR/Cas9 and piggyBac-mediated footprint-free LRRK2-G2019S knock-in reveals neuronal complexity phenotypes and alpha-Synuclein modulation in dopaminergic neurons. Stem Cell Res 24:44–50. https://doi.org/10.1016/j.scr.2017.08.013
Ran FA et al (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154(6):1380–1389. https://doi.org/10.1016/j.cell.2013.08.021
Reinhardt P et al (2013) Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS ONE 8:e59252. https://doi.org/10.1371/journal.pone.0059252
Rose CM et al (2016) Highly multiplexed quantitative mass spectrometry analysis of ubiquitylomes. Cell Syst 3(395–403):e394. https://doi.org/10.1016/j.cels.2016.08.009
Safari F, Tamaddon AM, Zarghami N, Abolmali S, Akbarzadeh A (2016) Polyelectrolyte complexes of hTERT siRNA and polyethyleneimine: effect of degree of PEG grafting on biological and cellular activity. Artif Cells Nanomed Biotechnol 44(6):1561–1568. https://doi.org/10.3109/21691401.2015.1064936
Safari F, Rahmani Barouji S, Tamaddon AM (2017) Strategies for improving siRNA-induced gene silencing efficiency. Adv Pharm Bull 7:603–609. https://doi.org/10.15171/apb.2017.072
Safari F, Zare K, Negahdaripour M, Barekati-Mowahed M, Ghasemi Y (2019) CRISPR Cpf1 proteins: structure, function and implications for genome editing. Cell Biosci 9:36. https://doi.org/10.1186/s13578-019-0298-7
Samaranch L et al (2010) PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133(4):1128–1142. https://doi.org/10.1093/brain/awq051
Sanchez-Danes A et al (2012) Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med 4:380–395. https://doi.org/10.1002/emmm.201200215
Sanders LH et al (2014) LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiol Dis 62:381–386. https://doi.org/10.1016/j.nbd.2013.10.013
Sarkar S et al (2017) Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ 3(1):30. https://doi.org/10.1038/s41531-017-0032-2
Schrag A (2018) Testing the MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 33:1518–1520. https://doi.org/10.1002/mds.27543
Scott L, Dawson VL, Dawson TM (2017) Trumping neurodegeneration: targeting common pathways regulated by autosomal recessive Parkinson’s disease genes. Exp Neurol 298:191–201. https://doi.org/10.1016/j.expneurol.2017.04.008
Shi L et al (2013) Peptide hormone ghrelin enhances neuronal excitability by inhibition of Kv7/KCNQ channels. Nat Commun 4:1435. https://doi.org/10.1038/ncomms2439
Solberg N, Krauss S (2013) Luciferase assay to study the activity of a cloned promoter DNA fragment. Methods Mol Biol 977:65–78. https://doi.org/10.1007/978-1-62703-284-1_6
Soldner F et al (2016) Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 533:95–99. https://doi.org/10.1038/nature17939
Song C et al (2019) Mechanistic interplay between autophagy and apoptotic signaling in endosulfan-induced dopaminergic neurotoxicity: relevance to the adverse outcome pathway in pesticide neurotoxicity. Toxicol Sci. https://doi.org/10.1093/toxsci/kfz049
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840. https://doi.org/10.1038/42166
Suda Y et al (2018) Down-regulation of ghrelin receptors on dopaminergic neurons in the substantia nigra contributes to Parkinson’s disease-like motor dysfunction. Mol Brain 11:6. https://doi.org/10.1186/s13041-018-0349-8
Taghavi S et al (2018) A clinical and molecular genetic study of 50 families with autosomal recessive parkinsonism revealed known and novel gene mutations. Mol Neurobiol 55(4):3477–3489. https://doi.org/10.1007/s12035-017-0535-1
Tagliafierro L, Chiba-Falek O (2016) Up-regulation of SNCA gene expression: implications to synucleinopathies. Neurogenetics 17:145–157. https://doi.org/10.1007/s10048-016-0478-0
Taipa R, Pereira C, Reis I, Alonso I, Bastos-Lima A, Melo-Pires M, Magalhaes M (2016) DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 139(6):1680–1687. https://doi.org/10.1093/brain/aww080
Takahashi M et al (2015) Normalization of overexpressed alpha-synuclein causing parkinson’s disease by a moderate gene silencing with rna interference. Mol Ther 4:e241. https://doi.org/10.1038/mtna.2015.14
Valente EM et al (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304(5674):1158–1160. https://doi.org/10.1126/science.1096284
Vasil’eva EA, Melino D, Barlev NA (2015) CRISPR/Cas system for genome editing in pluripotent stem cells. Tsitologiia 57(1):19–30
Vetchinova AS et al (2018) Cytogenetic analysis of the results of genome editing on the cell model of parkinson’s disease. Bull Exp Biol Med. https://doi.org/10.1007/s10517-018-4174-y
Volpicelli-Daley LA et al (2011) Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72:57–71. https://doi.org/10.1016/j.neuron.2011.08.033
Wang Y, Wang X, Li R, Yang ZF, Wang YZ, Gong XL, Wang XM (2013) A DNA methyltransferase inhibitor, 5-aza-2′-deoxycytidine, exacerbates neurotoxicity and upregulates Parkinson’s disease-related genes in dopaminergic neurons. CNS Neurosci Ther 19(3):183–190. https://doi.org/10.1111/cns.12059
Wang X et al (2016) One-step generation of triple gene-targeted pigs using CRISPR/Cas9 system. Sci Rep 6:20620. https://doi.org/10.1038/srep20620
Wilson GR et al (2014) Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with alpha-synuclein pathology. Am J Hum Genet 95(6):729–735. https://doi.org/10.1016/j.ajhg.2014.10.015
Yao J, Huang J, Zhao J (2016) Genome editing revolutionize the creation of genetically modified pigs for modeling human diseases. Hum Genet 135:1093–1105. https://doi.org/10.1007/s00439-016-1710-6
Ye L et al (2014) Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5Delta32 mutation confers resistance to HIV infection. Proc Natl Acad Sci USA 111(26):9591–9596. https://doi.org/10.1073/pnas.1407473111
Zhang D, Kanthasamy A, Yang Y, Anantharam V, Kanthasamy A (2007) Protein kinase C delta negatively regulates tyrosine hydroxylase activity and dopamine synthesis by enhancing protein phosphatase-2A activity in dopaminergic neurons. J Neurosci 27(20):5349–5362. https://doi.org/10.1523/jneurosci.4107-06.2007
Zhu M (1861) S HP, Han S (2017) DJ-1, a Parkinson’s disease related protein, aggregates under denaturing conditions and co-aggregates with alpha-synuclein through hydrophobic interaction. Biochim et Biophys Acta 7:1759–1769. https://doi.org/10.1016/j.bbagen.2017.03.013
Zhu XX, Zhong YZ, Ge YW, Lu KH, Lu SS (2018) CRISPR/Cas9-mediated generation of guangxi bama minipigs harboring three mutations in alpha-synuclein causing parkinson’s disease. Sci Rep 8(1):12420. https://doi.org/10.1038/s41598-018-30436-3
Zimprich A et al (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44(4):601–607. https://doi.org/10.1016/j.neuron.2004.11.005
Zou J, Mali P, Huang X, Dowey SN, Cheng L (2011) Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood 118:4599–4608. https://doi.org/10.1182/blood-2011-02-335554
Funding
It is certified that no funding has been received for the preparation of this manuscript
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
As no human participants are involved in this study no informed consent had been obtained.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Safari, F., Hatam, G., Behbahani, A.B. et al. CRISPR System: A High-throughput Toolbox for Research and Treatment of Parkinson’s Disease. Cell Mol Neurobiol 40, 477–493 (2020). https://doi.org/10.1007/s10571-019-00761-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-019-00761-w