
Abstract
The α-synuclein gene (SNCA) plays a major role in the aetiology of Lewy body disease (LBD) including Parkinson’s disease (PD). Point mutations and genetic alterations causing elevated gene expression are causally linked to familial PD. To what extent epigenetic changes play a role in the regulation of α-synuclein expression and may contribute to the aetiology of sporadic LBD is a matter of debate. We analysed the methylation state of the promoter region and a CpG-rich region of intron 1 of α-synuclein in several brain regions in sporadic LBD and controls using 454 GS-FLX-based high-resolution bisulphite sequencing. Our results indicate that there are significant differences in the level of methylation between different brain areas. The overall methylation levels in the promoter and intron 1 of α-synuclein are rather low in controls and—in contrast to previously reported findings—are not significantly different from LBD. However, single CpG analysis revealed significant hyper- and hypomethylation at different positions in various brain regions and LBD stages. A slight overall increase in methylation related to LBD patients’ age was detected.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The term epigenetics has been defined as variability in gene expression underlying characteristic phenotypic traits that are not due to modifications in the genetic sequence. Epigenetic modifications originate from DNA methylation, histone modifications, miRNA expression and nucleosome positioning (Portela and Esteller 2010). It is known that epigenetic mechanisms play an important role in carcinogenesis (Portela and Esteller 2010; Taby and Issa 2010). Over the past years, epigenetic research has also captured the field of neurodegeneration (Marques et al. 2011; Urdinguio et al. 2009).
Epigenetic modifications of SNCA could be the missing link between familial and sporadic forms of LBD such as Parkinson’s disease (PD). Sporadic PD accounts for 90–95% of PD cases and is characterized by the progressive degeneration of dopaminergic nigro-striatal systems and other neuronal networks (Sherer et al. 2001). However, its cause still remains elusive. Nevertheless, one key player in PD, namely α-synuclein, has been identified so far: Lewy bodies and Lewy neurites, histopathological hallmarks of PD, contain mainly α-synuclein (Sherer et al. 2001). Furthermore, missense mutations and amplifications of SNCA are directly related to PD (Ibanez et al. 2004; Singleton et al. 2003; Zarranz et al. 2004; Polymeropoulos et al. 1997), indicating that elevations of α-synuclein expression are directly disease causing. Recent genome-wide association studies (GWAS) have identified SNCA as a major risk locus for PD (Simon-Sanchez et al. 2009; Satake et al. 2009). The expression levels of SNCA could possibly be controlled by changes in methylation levels of SNCA. Latest research has suggested epigenetic regulations by DNA hypomethylation of intron 1 in SNCA in PD, possibly leading to an increased expression of SNCA and contributing to PD aetiology (Jowaed et al. 2010; Matsumoto et al. 2010). These studies only analysed a small number of patients and brain regions, not considering the associated Braak or McKeith stages of LBD. In addition, these publications were based on low resolution and subcloning-based sequencing, so that technically biased evaluation cannot be excluded. As PD pathology evolves in different stages during disease progression (McKeith et al. 2005; Braak et al. 2003), it is mandatory to determine whether epigenetic modifications are stable alterations in different brain regions during the progression of disease.
The aims of the present study were to determine (i) whether methylation changes of SNCA contribute to LBD aetiology, (ii) whether different brain areas affected in LBD (substantia nigra, putamen, cingulate gyrus and temporal cortex) show differences in the methylation of SNCA and (iii) whether the degree of SNCA methylation changes with age or post-mortem (p.m.) time. We used Next-Generation GS-FLX-based bisulphite sequencing to avoid possible technical biases and to obtain high-resolution methylation profiles.
Materials and Methods
Tissue Samples and DNA Extraction
Frozen brain tissue samples of 15 Lewy body cases and 6 controls were obtained from the brain bank (Brain-Net Germany) at the Center of Neuropathology and Prion Research, Ludwig-Maximilians-Universität, Munich according to the approval of the local ethical committee (Table 1). Tissue samples from 4 different brain regions affected in LBD were analysed: substantia nigra (n = 10, controls n = 3), putamen (n = 15, controls n = 6), cingulate gyrus (n = 15, controls n = 6), temporal cortex (n = 15, controls n = 6) as well as tissue from the cerebellum used as an internal control (n = 14, controls n = 6). Histopathological classifications were based on Braak stages (Braak et al. 2003). Braak stages 1 and 2 were taken together as brain-stem predominant stages, Braak stages 3 and 4 as limbic stages and Braak stages 5 and 6 as neocortical stages. All controls were tested for the presence of α-synuclein, abeta, tau etc. using immunohistochemical examinations. Tissue samples were homogenized using 500 μl 25 mM EDTA/10 mM NaCl (Roth, Karlsruhe, Germany and VWR, Darmstadt, Germany), followed by 500 μl 25 mM EDTA/10 mM Tris–HCL/1%SDS (Roth, Karlsruhe, Germany) and treated with RNAse (Sigma-Aldrich, Munich, Germany). Afterwards, samples were digested with Proteinase K (Fermentas, St. Leon-Rot, Germany) followed by phenol/chloroform extraction (VWR, Darmstadt, Germany) and ethanol precipitation of genomic DNA. Finally, DNA was stored in TE buffer (Roth, Karlsruhe, Germany). DNA integrity was checked on 1.2% agarose gels, and DNA was quantified with a Qubit Fluorometer (Invitrogen, Darmstadt, Germany).
Bisulphite Treatment
Bisulphite treatment was performed on 500 ng genomic DNA. Briefly, DNA was treated with 2 M sodium bisulphite (Roth, Karlsruhe, Germany) and 0.6 M NaOH (Roth, Karlsruhe, Germany), then denatured for 15 min at 99°C and incubated for 30 min at 50°C. We introduced two thermo-spikes of 99°C for 5 min followed by two incubation steps of 1.5 h at 50°C. Purification was achieved by loading, desulphonation with 0.3 M NaOH and washing with 1× TE on a microcon YM-30 column (Millipore, Schwalbach, Germany). Bisulphite DNA was eluted in 50 μl 1× TE.
Amplicon Generation and 454 GS-FLX Sequencing
Amplicons were generated using region-specific primers (SNCA promoter: 5′ GAAAATTTTGAAGATATTTGAATT 3′ and 5′ AAACTAAAAATAATACCTCTTCCT 3′; SNCA intron 1: 5′ TATAAGGGTTGAGAGATTAGGT 3′ and 5′ CCTCAACTATCTACCCTAAACAAA 3′) with the recommended A and B adaptors (Roche, Mannheim, Germany) at their 5′-end. PCR of bisulphite-treated DNA was performed in a total volume of 30 μl containing 3 μl Hot Start-IT reaction buffer (USB, Cleveland, OH, USA), 2.4 μl dNTPs, 2.5 mM each, 100 nmol of each primer, 1.25 U Hot Start-IT (USB, Cleveland, OH, USA) and 2 μl of bisulphite DNA. After initial denaturation at 95°C for 5 min, 40 cycles were carried out for the promoter region (denaturation 95°C for 1 min, annealing 62°C for 1 min, extension 72°C for 1 min and final extension 72°C for 5 min) and 45 cycles for intron 1 (denaturation 95°C for 1 min, annealing 56.5°C for 1 min, extension 72°C for 1 min and final extension 72°C for 5 min). PCR products were visualized on 1.2% agarose gels, purified using the Gel/PCR DNA Fragments extraction kit (AVEGENE, Taipei, Taiwan) and measured by intercalating fluorescence dye (Qubit HS-Kit, Invitrogen, Darmstadt, Germany) using Qubit Fluorometer (Invitrogen, Darmstadt, Germany). After amplicon pooling, emulsion PCR was performed using Lib-A emPCR protocols. DNA-containing beads were recovered, enriched and sequenced from the A-adaptor on a XLR70 Titanium PicoTiterPlate according to the manufacturer′s protocols (Roche, Mannheim, Germany).
Data Evaluation
From primary sff files, fasta reads were extracted and assigned to the reference sequence. Methylation level and pattern were assessed using multiple sequence alignment with an extended and improved version of BiQ Analyzer (Bock et al. 2005), the Biq Analyzer-HT software (Lutsik et al. 2011). Samples with less than 20 reads (n=6, intron 1 CE sample No. 1, 12, 16, 17; putamen sample No. 20) were excluded from the study to avoid misinterpretations due to low read numbers. Data are shown as mean ± SD. Statistical analysis was performed with SPSS version 18. Results were regarded as significant, if P < 0.05. P values were based on unpaired two-tailed t test. Associations between variables were measured by Pearson’s rank correlation. Co-methylation was analysed with Fisher’s exact test.
Results
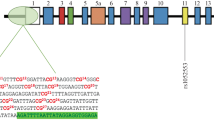
We analysed DNA from 4 different brain regions known to be affected in LBD [substantia nigra (SN), putamen, cingulate gyrus (CG) and temporal cortex (TC)] as well as tissue from the cerebellum (CE) used as an internal control in 187 tissue samples (promoter n = 96, intron 1 n = 91) from 15 LBD cases (brain-stem predominant n = 5, limbic n = 4, neocortical n = 6) and 6 controls. The analysed SNCA regions of interest and the summary of information concerning the patients and controls are shown in Fig. 1 and Table 1.

SNCA regions of interest. Yellow: predicted CpG islands (CpG plot: observed/expected ratio >0.60, percent C + percent G >50.00, length >200 bp), Green: Bis-PCR-Primer
Amplicon-Specific Methylation
The average methylation level over all analysed CpGs at the promoter region was 0.6% for LBD cases and controls (CE excluded, Fig. 2a, Online Resource Table 1). Compared with the promoter region, the intron 1 showed 4.2-fold higher methylation levels (CE excluded, Fig. 2b, Online Resource Table 1). At the intron 1, LBD cases displayed average methylation levels of 2.6% and controls 2.3%.

a, b Methylation levels [%] in different brain areas (SN = substantia nigra, CG = cingulate gyrus, TC = temporal cortex, CE = cerebellum) of LBD samples according to the Braak stages (brain-stem predominant = Braak 1 and 2, limbic = Braak 3 and 4, neocortical = Braak 5 and 6) and controls. P = 0.03 versus controls (unpaired two-tailed t test)
Concerning different LBD stages, the promoter region showed methylation levels of 0.6% for brain-stem predominant, limbic and neocortical stages (CE excluded, Fig. 2a, Online Resource Table 1). At intron 1, brain-stem predominant stages demonstrated methylation levels of 2.8%, limbic ones 2.6% and neocortical ones 2.5% (CE excluded, Fig. 2b, Online Resource Table 1). Additionally, hypomethylation as well as hypermethylation was detected in various LBD stages at both the promoter region and intron 1 compared with controls (Fig. 2a, b, Online Resource Table 1).
In summary, both regions show methylation levels that do not differ markedly between LBD cases and controls. Furthermore, the methylation state is independent on disease state in LBD patients, but one has to bear in mind that individuals and tissues exhibited high variation in methylation state (Fig. 2a, b, Online Resource Table 1). E.g. limbic LBD stages show hypermethylation as well as hypomethylation in the SN compared with controls (Fig. 2a, b, Online Resource Table 1).
Specific Methylation in Different Brain Regions
DNA methylation levels show strong variations in different brain regions (Fig. 2a, b, Online Resource Table 1). Mean methylation levels for CE at the promoter region and intron 1 always showed the lowest methylation levels compared with all other analysed tissues and did not show significant differences between LBD cases and controls. LBD cases demonstrated mean methylation levels of 0.2% at the promoter region and 0.7% at intron 1, whereas controls demonstrated 0.3% at the promoter region and 0.6% at intron 1 (Fig. 2a, b, Online Resource Table 1). A comparison of the methylation levels in CE and the other analysed tissues demonstrated significant hypermethylation for most LBD cases and controls at the intron 1 (Online Resource Table 6). The second lowest methylation levels were found in the putamen (Fig. 2a, b, Online Resource Table 1). There, LBD cases exhibited higher methylation values than controls (Fig. 2a, b, Online Resource Table 1). The highest methylation levels were observed in the cortical areas, but no differences were found between controls and LBD patients (Fig. 2a, b, Online Resource Table 1). Values in the SN must be viewed with caution, since they are based on small numbers of cases and varying admixtures of glial cells in Lewy body cases (see “Discussion”).
Specific Methylation in Different LBD Stages
A comparison of LBD stages revealed hypomethylated as well as hypermethylated alterations at the promoter region and intron 1 in different brain regions (Fig. 2a, b, Online Resource Table 1). Significant changes could only be detected at the intron 1 in the putamen of Lewy body cases with limbic stage compared with controls (Fig. 2b).
In detail, methylation levels at the promoter region in the SN decreased in all three LBD stages (brain-stem predominant and neocortical 14.3%, limbic 28.5%, P = 0.3 each, Fig. 2a and Online Resource Table 1). Concerning the CG, the methylation state also decreased in brain-stem predominant patients (12.5%, P = 0.8), limbic ones (50%, P = 0.2) and neocortical stages (37.5%, P = 0.2, Fig. 2a and Online Resource Table 1). In contrast, promoter methylation analysis of the putamen and TC demonstrated increased but not significantly higher methylation levels in different LBD stages compared with controls; putamen: 1.3-fold in brain-stem predominant stages (P = 0.3), 1.7-fold in limbic stages (P = 0.1), 1.7-fold in neocortical stages (P = 0.2) and TC: 1.2-fold in brain-stem predominant stages (P = 0.5), 1.6-fold in limbic stages (P = 0.1), 1.4-fold in neocortical stages (P = 0.7, Fig. 2a and Online Resource Table 1).
In intron 1, the SN and the putamen showed increased methylation levels: SN 1.6-fold in brain-stem predominant stages (P = 0.6), 1.3-fold in neocortical stages (P = 0.6); putamen 1.4-fold in brain-stem predominant stages (P = 0.4), 1.9-fold in limbic stages (P = 0.03), 1.4-fold in neocortical stages compared with controls (P = 0.2, Fig. 2b and Online Resource Table 1). The TC also displayed increased methylation levels in brain-stem predominant (1.1-fold, P = 0.9) and limbic stages (1.1-fold, P = 0.8) but decreased methylation levels in neocortical stages (6.9%, P = 0.8) compared with controls (Fig. 2b and Online Resource Table 1). Decreased methylation levels were also detected in the CG for limbic and neocortical stages (10.7%, P = 0.5 each, Fig. 2b and Online Resource Table 1).
Methylation State of Single CpGs
Other studies carried out so far suggested methylation changes analysing single CpGs in intron 1 (Table 2). Our analysis of single CpGs in the SNCA promoter region and intron 1 (promoter: 17 CpG positions, intron 1 19 CpG positions; Fig. 1) revealed no significant differences between various LBD stages and controls (Fig. 3a–d, Online Resource Tables 2–5). With regard to intron 1, methylation levels of different CpGs only varied marginally. Differences in methylation levels only accounted for 10%. The highest methylation levels always peaked at positions 7 and 19 (up to 10.1%), the lowest methylation levels (0.1%) mainly at positions 3, 4 and 14 (Fig. 3a–d, Online Resource Tables 3, 5).

a–d Methylation levels [%] of analysed CpGs (intron 1) in different brain areas of LBD samples according to the Braak stages (brain–stem predominant = Braak 1 and 2, limbic = Braak 3 and 4, neocortical = Braak 5 and 6) and controls. P < 0.05 versus controls (unpaired two-tailed t test)
Concerning different tissue samples, only sparse CpG positions demonstrated significant hypo- as well as hypermethylation in the promoter region and intron 1. Compared with the other studies, only significant increased, but not decreased methylation levels in LBD affected brain regions could be detected in intron 1 (putamen CpG no. 5 limbic stage (4.8-fold, P = 0.03), CpG no. 7 neocortical stage (2.0-fold, P = 0.02), CG CpG no. 18 neocortical stage (2.9-fold, P = 0.04), Fig. 3a–d, Online Resource Tables 3, 5). Since we analysed brain regions consisting of different cell types, looking at co-methylation of CpG positions within the amplicons might shed light on cell type–specific methylation profiles. For example, Fisher’s exact test revealed no co-methylation of the hypermethylated CpGs No. 5 and 7 in the putamen of LBD cases (P < 0.0001, Online Resource Fig. 2).
Regarding different LBD stages, significant methylation changes were detected in early and late LBD stages in intron 1 as well as the promoter region, indicating that methylation changes do not antecede or parallel disease progression. (Fig. 3a–d, Online Resource Tables 2–5).
Correlations Between Methylation State and Age and p.m. Time
Matsumoto et al. suggested correlations between methylation state and disease duration, but not age (Matsumoto et al. 2010). Our results show that the averaged methylation state was unaffected by age (r = −0.03; P=0.9) or p.m. time (r = −0.02; P = 0.9) for controls and p.m. time for LBD cases (r = −0.2; P = 0.2; Online Resource Fig. 1 A, B). A slightly significant positive correlation was found between methylation state and age in LBD cases (r = 0.3, P = 0.01, Online Resource Fig. 1 A).
Discussion
In the present study, we have investigated whether (i) differences in the methylation state of the promoter region and intron 1 of SNCA appear in LBD cases versus controls, (ii) different brain areas show variable DNA methylation of SNCA and (iii) the degree of SNCA methylation changes with age or p.m. time. We analysed the methylation state in sporadic cases of LBD of different stages according to the Braak classification (Braak et al. 2003) and compared them with controls. Our results indicate that the overall methylation levels in both regions of SNCA are rather low in controls and various LBD stages compared with reported methylation levels of other genes (most of them ranging from 40 to 70%) in the brain (Xin et al. 2010; Ladd-Acosta et al. 2007). Furthermore, methylation levels varied markedly between different brain areas in our study, the CE consistently showed the lowest methylation levels compared to the other analysed regions of the brain, and the cortex always showed the highest ones. The consistent differences between different brain regions can be interpreted as an indirect methodological control in relation to the absence of measurable differences between LBD brains and control brains.
Single CpG analysis revealed significant hyper- and hypomethylation at different positions in various brain regions and LBD stages. Interestingly, we detected a slight overall increase in methylation related to the LBD patients’ age. A statistically significant difference was seen in intron 1 in the putamen, when control cases were compared with LBD patients in the limbic stage. The biological meaning of this difference is questionable, in particular, since no differences were found when controls were compared with brain-stem predominant LBD cases, neocortical cases, or all LBD cases together as one cohort.
Regional differences of methylation in the brain (Ghosh et al. 2010) with a distinct cerebellar methylation profile have been described (Xin et al. 2010). Other studies have shown that the cerebellar methylation levels are strikingly lower than in other brain regions (Xin et al. 2010; Ladd-Acosta et al. 2007). On the one hand, these studies demonstrated regional methylation patterns that correlated strongly within a brain region across individuals (Ladd-Acosta et al. 2007). On the other hand, interindividual methylation variability of different brain regions was discovered (Xin et al. 2010). It is discussed that these differences are due to experience-based plasticity and may explain functional differences of the brain regions.
Other studies (Table 2) have shown that the methylation state in the SNCA promoter region is quite low and not altered in PD (Matsumoto et al. 2010; Jowaed et al. 2010). However, the age was associated with an increase in methylation levels of LBD cases in our study, but not in others (Matsumoto et al. 2010). Furthermore, two studies indicated that sporadic PD is associated with hypomethylation of SNCA intron 1 (Jowaed et al. 2010; Matsumoto et al. 2010; Table 2). Jowaed et al. showed methylation differences in LBD cases concerning group comparisons and single CpG position in the SN (position 8 and 17) and putamen (position 8 and 12). No methylation changes were detected in the cortex (2010).
Transcription factor binding sites (TFBS) within SNCA intron 1 have previously been shown to be implicated in transcriptional activation (Scherzer et al. 2008), and TF misregulation was proposed to play a role in neurodegeneration (Jowaed et al. 2010). Interestingly, the above-mentioned hypermethylated CpGs within intron 1 overlap with a binding region for the TFBS p107/E2F. Both are potentially involved in cell cycle regulation (Zhu et al. 1993; Nevins 1992). p107 associates with Sp1 and E2F (Datta et al. 1995), and the phosphorylated retinoblastoma protein (pRB)/E2F pathway is activated in dopaminergic neurons of PD patients mediating cell death (Hoglinger et al. 2007). Other studies also detected an increase in E2F-1 levels in the CG of PD patients compared with controls strengthening the potential implication of E2F in neuronal apoptosis (Alvira et al. 2008). Sp1 is a commonly found transcription factor of CG-rich promoters (Briggs et al. 1986), and E2F binds to CpGs at promoter regions with sensitivity towards DNA methylation (Jenal et al. 2009). A potential role of intron 1-specific CpG methylation on transcription factor-mediated regulation of SNCA remains an open question.
Matsumoto et al. 2010) only found alterations of the methylation state in the SN, but not CG or putamen. Moreover, a third study described reduced methylation levels using methylation-specific PCR (MSP) and an antibody against 5-methylcytosine (5-mC) in samples of the frontal cortex (FC) together with decreased expressions of DNA (cytosine-5)-methyltransferase 1 (DNMT1) and an inverse correlation with oligomerization of SNCA (Desplats et al. 2011).
In detail, regarding the percentage of methylation state in the SN, Jowaed et al. 2010 found <5% methylation in PD at most CpG positions and up to ~40% in controls, whereas Matsumoto et al. 2010 found almost 100% methylation in controls and ~0% in LBD cases. Concerning our data, methylation levels of LBD cases were comparable with those of Jowaed et al., but controls did not display methylation levels above 16%.
In the presented study, we chose the 454 GS-FLX platform, which is pyrosequencing-based, thus avoiding subcloning that was performed in previous studies (Jowaed et al. 2010; Matsumoto et al. 2010). It is important to note that during cloning, the ratio of methylated to unmethylated PCR products is not faithfully maintained (Warnecke et al. 2002) and can result in bias against AT-rich clones (Chhibber and Schroeder 2008). High-throughput parallel bisulphite sequencing can scan DNA methylation in genomic regions of interest at single methylcytosine resolution in a more direct and robust fashion, making it superior to traditional bisulphite sequencing technologies (Taylor et al. 2007).
Whilst examining genetic alterations in SNCA, we came across annotated polymorphisms within the underlying PCR primer sequences in the studies of Jowaed and Matsumoto, which may have resulted in specific and preferential amplifications of alleles differing in methylation state (Warnecke et al. 2002; Wojdacz et al. 2008; Shen et al. 2007).
Moreover, the limited number of fresh frozen brain samples in previous studies might have led to over-interpretations. We also want to stress the point that, so far, only tissue homogenates containing multiple cell types were analysed. Consequently, detected methylation changes might not represent alterations in affected neurons only as cell numbers diverge during disease progression. The administration of L-Dopa or other drugs could possibly alter methylation levels. From our study, we cannot conclude any influence of medication, but we cannot argue with certainty that methylation levels are not changed by L-Dopa because we do not have sufficient information on the medication of all patients and controls.
Overall, our data raise the question whether alterations of methylation levels play a role in the aetiology of LBD. We have investigated DNA methylation in the promoter and intron 1 of SNCA. But DNA methylation can also occur at CpG shores (Portela and Esteller 2010), and methylated stretches of DNA have also been found in the promoter and coding regions of actively transcribed genes (Graff et al. 2011). Highly expressed genes were found to have hypomethylated promoters and hypermethylated gene bodies (Ball et al. 2009; Esteller 2007), but the influence of methylated CpGs within genomic regions is still poorly understood. Recent evidence also suggests the involvement of histone modifications and miRNAs in the aetiology of LBD (Doxakis 2010; Du et al. 2010; Jin et al. 2011a; Junn et al. 2009; Kontopoulos et al. 2006; Marques et al. 2011; Asikainen et al. 2010). Furthermore, 5-hydroxymethylcytosine (5-hmC), which is widely expressed in the human and murine brain, may significantly alter the biological functions of promoter and gene methylation (Robertson et al. 2011; Kinney et al. 2011; Jin et al. 2011b). The investigation of its function awaits further technical development, as it is indistinguishable from 5-mC by presently available sequencing techniques.
References
Alvira, D., Ferrer, I., Gutierrez-Cuesta, J., Garcia-Castro, B., Pallas, M., & Camins, A. (2008). Activation of the calpain/cdk5/p25 pathway in the girus cinguli in Parkinson’s disease. Parkinsonism Related Disorder, 14(4), 309–313. doi:10.1016/j.parkreldis.2007.09.005.
Asikainen, S., Rudgalvyte, M., Heikkinen, L., Louhiranta, K., Lakso, M., Wong, G., et al. (2010). Global microRNA expression profiling of Caenorhabditis elegans Parkinson’s disease models. Journal of Molecular Neuroscience, 41(1), 210–218. doi:10.1007/s12031-009-9325-1.
Ball, M. P., Li, J. B., Gao, Y., Lee, J. H., LeProust, E. M., Park, I. H., et al. (2009). Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Natural Biotechnology, 27(4), 361–368. doi:10.1038/nbt.1533.
Bock, C., Reither, S., Mikeska, T., Paulsen, M., Walter, J., & Lengauer, T. (2005). BiQ analyzer: Visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics, 21(21), 4067–4068. doi:10.1093/bioinformatics/bti652.
Braak, H., Del Tredici, K., Rub, U., de Vos, R. A., Jansen Steur, E. N., & Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of Aging, 24(2), 197–211.
Briggs, M. R., Kadonaga, J. T., Bell, S. P., & Tjian, R. (1986). Purification and biochemical characterization of the promoter-specific transcription factor, Sp1. Science, 234(4772), 47–52.
Chhibber, A., & Schroeder, B. G. (2008). Single-molecule polymerase chain reaction reduces bias: Application to DNA methylation analysis by bisulfite sequencing. Analytical Biochemistry, 377(1), 46–54. doi:10.1016/j.ab.2008.02.026.
Datta, P. K., Raychaudhuri, P., & Bagchi, S. (1995). Association of p107 with Sp1: Genetically separable regions of p107 are involved in regulation of E2F- and Sp1-dependent transcription. Molecular and Cellular Biology, 15(10), 5444–5452.
Desplats, P., Spencer, B., Coffee, E., Patel, P., Michael, S., Patrick, C., et al. (2011). Alpha-synuclein sequesters DNMT1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. Journal of Biological Chemistry, 286(11), 9031–9037. doi:10.1074/jbc.C110.212589.
Doxakis, E. (2010). Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. Journal of Biological Chemistry, 285(17), 12726–12734. doi:10.1074/jbc.M109.086827.
Du, G., Liu, X., Chen, X., Song, M., Yan, Y., Jiao, R., et al. (2010). Drosophila histone deacetylase 6 protects dopaminergic neurons against {alpha}-synuclein toxicity by promoting inclusion formation. Molecular Biology Cell, 21(13), 2128–2137. doi:10.1091/mbc.E10-03-0200.
Esteller, M. (2007). Cancer epigenomics: DNA methylomes and histone-modification maps. Natural Review of Genetics, 8(4), 286–298. doi:10.1038/nrg2005.
Ghosh, S., Yates, A. J., Fruhwald, M. C., Miecznikowski, J. C., Plass, C., & Smiraglia, D. (2010). Tissue specific DNA methylation of CpG islands in normal human adult somatic tissues distinguishes neural from non-neural tissues. Epigenetics, 5(6), 527–538.
Graff, J., Kim, D., Dobbin, M. M., & Tsai, L. H. (2011). Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiological Reviews, 91(2), 603–649. doi:10.1152/physrev.00012.2010.
Hoglinger, G. U., Breunig, J. J., Depboylu, C., Rouaux, C., Michel, P. P., Alvarez-Fischer, D., et al. (2007). The pRb/E2F cell-cycle pathway mediates cell death in Parkinson’s disease. Proceedings of the Natural Academy of Science U S A, 104(9), 3585–3590. doi:10.1073/pnas.0611671104.
Ibanez, P., Bonnet, A. M., Debarges, B., Lohmann, E., Tison, F., Pollak, P., et al. (2004). Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet, 364(9440), 1169–1171. doi:10.1016/S0140-6736(04)17104-3.
Jenal, M., Trinh, E., Britschgi, C., Britschgi, A., Roh, V., Vorburger, S. A., et al. (2009). The tumor suppressor gene hypermethylated in cancer 1 is transcriptionally regulated by E2F1. Molecular Cancer Research, 7(6), 916–922. doi:10.1158/1541-7786.mcr-08-0359.
Jin, H., Kanthasamy, A., Ghosh, A., Yang, Y., Anantharam, V., & Kanthasamy, A. G. (2011). Alpha-synuclein negatively regulates protein kinase C delta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. Journal of Neuroscience, 31(6), 2035–2051. doi:10.1523/JNEUROSCI.5634-10.2011.
Jin, S. G., Wu, X., Li, A. X., & Pfeifer, G. P. (2011). Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Research, 39(12), 5015–5024, doi:10.1093/nar/gkr120.
Jowaed, A., Schmitt, I., Kaut, O., & Wullner, U. (2010). Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. Journal of Neuroscience, 30(18), 6355–6359. doi:10.1523/JNEUROSCI.6119-09.2010.
Junn, E., Lee, K. W., Jeong, B. S., Chan, T. W., Im, J. Y., & Mouradian, M. M. (2009). Repression of alpha-synuclein expression and toxicity by microRNA-7. Proceedings of the Natural Academy of Science U S A, 106(31), 13052–13057. doi:10.1073/pnas.0906277106.
Kinney, S. M., Chin, H. G., Vaisvila, R., Bitinaite, J., Zheng, Y., Esteve, P. O., Feng, S., Stroud, H., Jacobsen, S. E., & Pradhan, S. (2011). Tissue-specific distribution and dynamic changes of 5-hydroxymethylcytosine in mammalian genomes. Journal of Biological Chemistry, 286(28), 24685–24693. doi:10.1074/jbc.M110.217083.
Kontopoulos, E., Parvin, J. D., & Feany, M. B. (2006). Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Human Molecular Genetics, 15(20), 3012–3023. doi:10.1093/hmg/ddl243.
Ladd-Acosta, C., Pevsner, J., Sabunciyan, S., Yolken, R. H., Webster, M. J., Dinkins, T., et al. (2007). DNA methylation signatures within the human brain. American Journal of Human Genetics, 81(6), 1304–1315. doi:10.1086/524110.
Lutsik, P., Feuerbach, L., Arand, J., Lengauer, T., Walter, J., & Bock, C. (2011). BiQ analyzer HT: Locus-specific analysis of DNA methylation by high-throughput bisulfite sequencing. Nucleic Acids Research, 39(suppl 2), W551–W556. doi:10.1093/nar/gkr312.
Marques, S. C., Oliveira, C. R., Pereira, C. M., & Outeiro, T. F. (2011). Epigenetics in neurodegeneration: A new layer of complexity. Progress in Neuropsychopharmacology and Biological Psychiatry, 35(2), 348–355. doi:10.1016/j.pnpbp.2010.08.008.
Matsumoto, L., Takuma, H., Tamaoka, A., Kurisaki, H., Date, H., Tsuji, S., et al. (2010). CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS One, 5(11), e15522, doi:10.1371/journal.pone.0015522.
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, et al. (2005). Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology, 65(12), 1863–1872. doi:10.1212/01.wnl.0000187889.17253.b1.
Nevins, J. R. (1992). Transcriptional regulation. A closer look at E2F. Nature, 358(6385), 375–376. doi:10.1038/358375a0.
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science, 276(5321), 2045–2047.
Portela, A., & Esteller, M. (2010). Epigenetic modifications and human disease. Natural Biotechnology, 28(10), 1057–1068. doi:10.1038/nbt.1685.
Robertson, J., Robertson, A. B., & Klungland, A. (2011). The presence of 5-hydroxymethylcytosine at the gene promoter and not in the gene body negatively regulates gene expression. Biochemistry and Biophysics Research Community, 411(1), 40–43. doi:10.1016/j.bbrc.2011.06.077.
Satake, W., Nakabayashi, Y., Mizuta, I., Hirota, Y., Ito, C., Kubo, M., et al. (2009). Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Natural Genetics, 41(12), 1303–1307. doi:10.1038/ng.485.
Scherzer, C. R., Grass, J. A., Liao, Z., Pepivani, I., Zheng, B., Eklund, A. C., et al. (2008). GATA transcription factors directly regulate the Parkinson’s disease-linked gene alpha-synuclein. Proceedings of Natural Academy of Science U S A, 105(31), 10907–10912, doi:10.1073/pnas.0802437105.
Shen, L., Guo, Y., Chen, X., Ahmed, S., & Issa, J. P. (2007). Optimizing annealing temperature overcomes bias in bisulfite PCR methylation analysis. Biotechniques, 42(1), 48, 50, 52 passim.
Sherer, T. B., Betarbet, R., & Greenamyre, J. T. (2001). Pathogenesis of Parkinson’s disease. Current Opinion in Investigation Drugs, 2(5), 657–662.
Simon-Sanchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., et al. (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Natural Genetics, 41(12), 1308–1312. doi:10.1038/ng.487.
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). Alpha-synuclein locus triplication causes Parkinson’s disease. Science, 302(5646), 841. doi:10.1126/science.1090278.
Taby, R., & Issa, J. P. (2010). Cancer epigenetics. CA: A Cancer Journal for Clinicians, 60(6), 376–392. doi:10.3322/caac.20085.
Taylor, K. H., Kramer, R. S., Davis, J. W., Guo, J., Duff, D. J., Xu, D., et al. (2007). Ultradeep bisulfite sequencing analysis of DNA methylation patterns in multiple gene promoters by 454 sequencing. Cancer Research, 67(18), 8511–8518. doi:10.1158/0008-5472.can-07-1016.
Urdinguio, R. G., Sanchez-Mut, J. V., & Esteller, M. (2009). Epigenetic mechanisms in neurological diseases: Genes, syndromes, and therapies. The Lancet Neurology, 8(11), 1056–1072. doi:10.1016/S1474-4422(09)70262-5.
Warnecke, P. M., Stirzaker, C., Song, J., Grunau, C., Melki, J. R., & Clark, S. J. (2002). Identification and resolution of artifacts in bisulfite sequencing. Methods, 27(2), 101–107.
Wojdacz, T. K., Hansen, L. L., & Dobrovic, A. (2008). A new approach to primer design for the control of PCR bias in methylation studies. BMC Research Notes, 1, 54. doi:10.1186/1756-0500-1-54.
Xin, Y., Chanrion, B., Liu, M. M., Galfalvy, H., Costa, R., Ilievski, B., et al. (2010). Genome-wide divergence of DNA methylation marks in cerebral and cerebellar cortices. PLoS One, 5(6), e11357. doi:10.1371/journal.pone.0011357.
Zarranz, J. J., Alegre, J., Gomez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Annals of Neurology, 55(2), 164–173, doi:10.1002/ana.10795.
Zhu, L., van den Heuvel, S., Helin, K., Fattaey, A., Ewen, M., Livingston, D., et al. (1993). Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes & Development, 7(7A), 1111–1125.
Acknowledgments
This work was supported by the Friedrich-Baur-Stiftung (Grant Number 13/10) and Brain Net.
Conflicts of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
de Boni, L., Tierling, S., Roeber, S. et al. Next-Generation Sequencing Reveals Regional Differences of the α-Synuclein Methylation State Independent of Lewy Body Disease. Neuromol Med 13, 310–320 (2011). https://doi.org/10.1007/s12017-011-8163-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-011-8163-9