
Abstract
Chronic and debilitating neurodegenerative diseases, such as Parkinson’s disease (PD), impose an immense medical, emotional, and economic burden on patients and society. Due to a complex interaction between genetic and environmental risk factors, the etiology of PD remains elusive. However, the cumulative evidence emerging from clinical and experimental research over the last several decades has identified mitochondrial dysfunction, oxidative stress, neuroinflammation, and dysregulated protein degradation as the main drivers of PD neurodegeneration. The genome-editing system CRISPR (clustered regularly interspaced short palindromic repeats) has recently transformed the field of biotechnology and biomedical discovery and is poised to accelerate neurodegenerative disease research. It has been leveraged to generate PD animal models, such as Parkin, DJ-1, and PINK1 triple knockout miniature pigs. CRISPR has also allowed the deeper understanding of various PD gene interactions, as well as the identification of novel apoptotic pathways associated with neurodegenerative processes in PD. Furthermore, its application has been used to dissect neuroinflammatory pathways involved in PD pathogenesis, such as the PKCδ signaling pathway, as well as the roles of novel compensatory or protective pathways, such as Prokineticin-2 signaling. This review aims to highlight the historical milestones in the evolution of this technology and attempts to illustrate its transformative potential in unraveling disease mechanisms as well as in the development of innovative treatment strategies for PD.

Graphical Abstract
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Neurodegenerative diseases encompass complex conditions, such as Parkinson’s disease (PD), Alzheimer’s disease (AD), Amyotrophic Lateral Sclerosis (ALS) and Huntington’s disease (HD). The World Health Organization predicts neurodegenerative diseases will overtake cancer as the major cause of death by 2050 (Menken et al. 2000). Yet, due to the complex nature of etiologies and disease progression of these conditions, their cures remain out of reach for modern medicine. Exact causes for many of these conditions, including PD, remain elusive. Common themes have emerged in the progression of neurodegeneration, such as pathways that lead to mitochondrial dysfunction, the accumulation of aggregation-prone proteins, and neuroinflammation. Therefore, it is important to elucidate the neuroinflammatory mechanisms and cell death pathways involved in PD neurodegeneration.
Gene disruption has been a powerful genome-editing approach in uncovering the roles of individual genes in inter- and intra-cellular signaling pathways. However, earlier methods using gene-silencing via RNAi had drawbacks, including difficulty in the delivery and generation of a complete gene knockout. Advances in precise genome-editing using zinc-finger nucleases (ZFN), transcription activator-like effector nucleases (TALENs), and most recently, clustered regularly interspaced short palindromic repeats (CRISPR)-associated nucleases, have completely revolutionized many fields in biological research and are poised to quicken the pace of advances in research for PD, such as causally manipulating early apoptotic pathways and neuroinflammatory mechanisms involved in PD neurodegeneration. Originating from the adaptive bacterial immune system of Streptococcus pyogenes, CRISPR achieves precise genome editing based on the simple Watson-Crick base-pairing rules. Owing to its simplicity in design and synthesis, it has transformed research in many diseases, including cancer and brain diseases. This review will highlight the history of CRISPR and describe its use in studying neuroinflammatory and neuropharmacological mechanisms of PD.
Parkinson’s Disease
PD is the most common age-related movement disorder, affecting 10 million people worldwide. With an aging world population, the estimated prevalence of this neurodegenerative disease is projected to increase by more than 50% by 2030 (Dorsey et al. 2007). Furthermore, the estimated national economic burden for PD is likely to increase to over $13 billion by 2030 and $18 billion by 2050 as the population ages and PD prevalence increases (Kowal et al. 2013). Unfortunately, PD remains incurable and only symptomatic treatments are available to patients. The cardinal motor symptoms of PD include bradykinesia, resting tremor, muscle stiffness, gait abnormalities, postural instability, and fatigue (Barzilai and Melamed 2003; Jarraya et al. 2009; Nassif and Pereira 2018). These motor manifestations can be accompanied or preceded by non-motor symptoms, such as olfactory deficits (hyposmia), sleep disturbances, cognitive deterioration, depression, and pain, some of which can appear up to a decade earlier than motor symptoms. Postmortem examination of PD patient brains reveals an excessive (50–70%) loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), resulting in the dysregulation of extrapyramidal motor circuitry due to the significant de-innervation of nigral dopaminergic input to the striatum.
The manifestation of non-motor dysfunctions before motor dysfunctions indicates that the gradual and progressive loss of dopaminergic neurons began prior to the onset of motor symptoms, with these symptoms not emerging until after various protective, compensatory mechanisms were overwhelmed. Therefore, if an early biomarker were available, the prodromal period preceding massive dopaminergic cell death would afford a critical window of opportunity for a potential therapeutic intervention to alter the course of disease progression (Siderowf and Lang 2012). The exact causes for the destruction of these nigrostriatal dopaminergic neurons are not completely understood, but mounting evidence points to genetic defects and/or exposure to environmental neurotoxicants, such as pesticides and heavy metals, which initiate mitochondrial dysfunction, oxidative stress, neuroinflammation, and dysregulated protein degradation, ultimately contributing to dopaminergic neurodegeneration. A small percentage of PD patients (5–10%) exhibit an inherited form of PD, termed familial PD, attributed to mutations in various PARK genes, especially SNCA (α-synuclein) and LRRK2. Misfolded α-synuclein protein has been linked to the activation of brain microglia and the appearance of α-synuclein-containing Lewy bodies and Lewy neurites, which are correlated with neuroinflammation in PD models (Duffy et al. 2018). This further indicates that most cases of PD result from a complex interplay of human genetics and environmental factors, and that CRISPR-Cas9 gene-editing technology can determine if these proteins and pathways underlie PD.
Nucleases for Precise Genome Editing
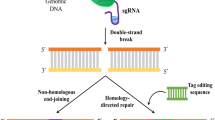
Popular molecular biological techniques in the past decade have been focused on the goal of precise genome editing, with customizable nuclease genome editing systems such as ZFN, meganucleases, TALEN, and most recently, CRISPR. Current methods for precise and versatile genome editing of eukaryotic genomes rely on an essential cell repair pathway to introduce custom-designed alterations into the genome. In a healthy cell, double-stranded breaks (DSBs) in genomic DNA are the most deleterious DNA lesions, which can be generated by both endogenous sources like reactive oxygen species and replication-associated errors, as well as exogenous sources, including ionizing radiation and chemotherapeutic agents. Unrepaired or misrepaired DSBs are generally considered as precursors to chromosome breaks, leading to genomic instability and can ultimately result in carcinogenesis (Nakai et al. 2011). Therefore, eukaryotic cells quickly initiate a number of highly efficient repair mechanisms, including nonhomologous end-joining (NHEJ) and homologous recombination (HR), in attempts to ligate and repair DSBs. Owing to the discovery that targeted DSBs can be used to stimulate genome editing through HR-mediated recombination events, the customizable nuclease proteins, including ZFN, meganuclease, TALEN, and CRISPR, are utilized to produce a site-specific DNA DSB (which is discussed further), where these localized DSBs can induce insertion or deletion mutations (i.e., indels) via the error-prone NHEJ repair pathway, leading to effective genome editing.
While protein-DNA interactions from ZFN can be efficient and precise, adapting the system for genome editing by modifying individual ZFN DNA-binding regions is time-consuming and difficult to validate (Doudna and Charpentier 2014). Furthermore, due to a lack of clear correspondence between meganuclease protein residues and their target DNA sequence specificity, meganucleases have not been widely adopted as a genome-engineering platform. Similarly, TALENs suffer from context-dependent specificity and constructing novel TALEN arrays is labor-intensive and time-consuming. The application of relatively simple-to-generate RNA sequences against known genomic DNA sequences underpins the popularity of CRISPR among researchers. CRISPR is a toolkit with a broad range of uses, including defining gene-driven biological processes and developing translationally relevant animal models of human diseases (Hsu et al. 2014; Lander 2016). Coined as the “CRISPR revolution”, its impact has reverberated through private and public institutions to drive research for development of commercial agricultural products and novel human therapeutics. A recent controversy involving the use of the CRISPR toolkit in germline engineering for the creation of “designer babies” made headlines in the media and raised ethical concerns. With more and more scientists ‘pushing the envelope,’ CRISPR is at the epicenter of this new era in biomedical research (Lander 2016).
CRISPR: Historical Perspective
Like many scientific breakthroughs, the discovery of CRISPR was serendipitous. As outlined in Fig. 1, the pioneers were not initially driven by the need to address human diseases or to edit the human genome, but were developing tools to identify resistant bacteria plaguing the dairy industry as an effort to counteract the threat of bio-terrorism or understand salt-resistant microbes – issues that were costly and pressing in their field (Mojica et al. 1993, 1995, 2000, 2005; Pourcel et al. 2005; Horvath et al. 2008; Lander 2016). In the early 1990’s, the Spanish microbiologist Francisco Mojica was studying the genomes of Haloferax mediterranei and H. volcanii, extreme salt-tolerant Archaean microbes from the salt marshes of Spain’s Costa Blanca. In their genomes, Mojica had discovered multiple copies of 30 bases of near-perfect palindromic repeat sequences each separated by spacers of roughly 36-base pair sequences. Around the same time, a team led by Japanese scientist Dr. Ishino witnessed a similar pattern of repeat sequences in the bacterial genome of Escherichia coli (Ishino et al. 1987). Mojica coined these repeats found in prokaryotes as short regularly spaced repeats (SRSRs), later renamed CRISPR (Jansen et al. 2002). By 2003, Mojica had shown sequence similarity between the spacer sequences and the P1 bacteriophage in E. coli, and concluded that the CRISPR loci encoding the instructions for an adaptive immune system protected microbes against specific infections (Mojica et al. 2005; Lander 2016).

Evolution of the CRISPR-Cas9 system
In another region of the world, the biological weapons threat posed by Saddam Hussein pushed the French Defense Ministry to recruit Gilles Vergnaud to study the tandem-repeat polymorphism in strains of anthrax bacteria (Pourcel et al. 2005). Unaware of Mojica’s work on E. coli, he identified identical tandem-repeat sequences at the CRISPR locus with Yersinia pestis strains differing in spacer sequences, which corresponded to the dominant prophage present in its genome. With this work, Vergnaud further validated Mojica’s theory that the CRISPR loci were involved in the immune defense mechanism of prokaryotes (Mojica et al. 2005; Pourcel et al. 2005).
After Mojica and Vergnaud’s discoveries, another breakthrough in the search for the function of these spacer sequences came from Phillippe Horvath’s studies in the 2000s on S. thermophilus phage infections, a recurrent issue in the dairy industry. To better understand this critical issue, Horvath was recruited by the Danish firm Danisco to develop a DNA-based identification method for different strains. He genotyped the strains and noticed a positive correlation between spacers and resistance to infection. With the collaborative effort of Rodolphe Barrangou and Sylvain Moineau, they verified that phage-resistant strains had acquired sequences at the CRISPR loci, with the insertion of multiple spacers correlating with increased resistance. Furthermore, they identified and characterized the roles of two cas (CRISPR-associated) genes, cas7 and cas9, where cas7 was shown to be involved in the generation of spacers and repeats, whereas cas9 contained two nuclease motifs, HNH and RuvC, and thus was presumably necessary for cutting nucleic acids. The group then demonstrated that a single-base change in the genome altered the immunity of the bacteria, suggesting that spacer sequences must match exactly the target DNA sequences (Barrangou et al. 2007; Horvath et al. 2008; Barrangou 2012; Barrangou and Marraffini 2014).
The Dutch microbiologist John van der Oost was focusing on cyanobacteria and its use in the production of biofuels and further characterized the CRISPR system while collaborating with a microbial evolution expert, Eugene Koonin. By systemically knocking-out each component of a five-Cas protein complex known as Cascade, they demonstrated that Cascade was required to make a 61-nucleotide CRISPR RNA (crRNA). The crRNA sequence formed a secondary structure within its palindromic repeats, implying a significant role for crRNA in CRISPR-based resistance. To test CRISPR-based resistance, the group designed a CRISPR-based array and reprogrammed E. coli strains carrying genes for phage lambda. Oost and Koonin’s group demonstrated that the new CRISPR sequence conferred resistant to phage lambda. Through their work, Oost and Koonin hypothesized that crRNA targets DNA and not RNA as previously suggested by other groups (Brouns et al. 2008; van der Oost et al. 2014).
Around the same time, American scientists Luciano Marraffini and Erik Sontheimer also sought to answer whether DNA or RNA was the target for crRNA. During his graduate years, Marraffini recognized that CRISPR could interfere with plasmid conjugation along with viruses in Staphylococcus. The two scientists sought to identify whether DNA was indeed the target of CRISPR in the S. epidermidis model, which included a spacer sequence found in S. aureus for the antibiotic-resistant plasmid gene nes that encodes nickase. Since the S. epidermidis CRISPR system was complicated and still poorly characterized, they genetically modified the nes gene by inserting a self-splicing intronic sequence in the middle as a foreign spacer. They hypothesized that if mRNA was indeed the target, then inserting a self-splicing intron would not interfere with the CRISPR system. However, if DNA was the target, the intronically generated mismatched sequence would prevent cleavage. Their results showed that crRNA could not interfere with the expression of the target gene, suggesting that a DNA-matched sequence is required for CRISPR (Marraffini and Sontheimer 2008; Barrangou and Marraffini 2014).
With limited knowledge of the cas genes and crRNA, much of the function and mechanism of the CRISPR loci was still unknown. Sylvain Moineau, with support from Danisco, studied the mechanism by which CRISPR cleaved DNA. Using S. thermophilus as their model, his group recognized that the Cas9 nuclease was responsible for a single, precise, blunt-end cleavage of three nucleotides upstream of the proto-spacer adjacent motif (PAM) sequence. They further revealed that the number of spacers matching a target corresponded to the number of cuts, thus concluding that Cas9 functions as the primary nuclease involved in CRISPR immunity (Deveau et al. 2008; Marraffini and Sontheimer 2008; Garneau et al. 2010). A few years later, Virginijus Siksnys pushed the limits of CRISPR technology, where he reconstituted a CRISPR system, transferred across genera, from S. thermophilus into the distant microbe E. coli in vitro. He confirmed that Cas9 was the only nuclease required for interference, and that the HNH and RuvC nuclease domains were essential for interference. He further demonstrated that Cas9 could be re-programmed in vitro with custom-designed spacers to cut any target site of their choosing (Sapranauskas et al. 2011; Gasiunas et al. 2012).
The third important component of the CRISPR system is the trans-activating CRISPR RNA (tracrRNA), discovered serendipitously by Emmanuelle Charpentier, who was characterizing intergenic regions of the sequences that coded for regulatory non-coding RNA in S. pyogenes. Charpentier collaborated with Jorg Vogel, whose focus was using next-generation sequencing to build a microbial transcriptome catalog. Their combined expertise led to the identification of a novel small RNA with 25 base pairs of near-perfect complementarity to the CRISPR repeats. Through their extensive study, they determined tracrRNA to not only be essential in the formation of crRNA, but also was critical for Cas9 nuclease activity (Mangold et al. 2004; Deltcheva et al. 2011).
Recognizing CRISPR’s potential as a powerful biological toolkit for genetic engineering, Charpentier went on to collaborate with Jennifer Doudna, an RNA expert and world-renowned structural biologist. In exploring the potential of CRISPR in vitro, they discovered that Cas9 could be reprogrammed with custom crRNA, that the function of Cas9 was dependent on the two nuclease domains, and that both crRNA and tracrRNA could be genetically engineered as single-guide RNA (gRNA) while still maintaining its functionality (Brouns et al. 2008; Deltcheva et al. 2011; Jinek et al. 2012; Sternberg and Doudna 2015). Their publication in Science revealed CRISPR’s microbiological potential, but for the broader scientific community its use in mammalian cell lines presented new challenges.
Finally, in mid-2012, the Chinese American scientist Feng Zhang repurposed CRISPR in a robust three-component system which included Cas9 from S. pyogenes and S. thermophilus, tracrRNA and crRNA. By specifically targeting sites in humans and mice, genes of interest could be disrupted with high efficiency and accuracy via the NHEJ repair mechanism, and by providing a repair template, it was possible to insert novel sequences via recombination events. Moreover, multiple genes could be edited simultaneously via matching spacer sequences and programming arrays (Brouns et al. 2008; Zhang et al. 2011; Sternberg and Doudna 2015). Thus, extending the application of CRISPR to mammalian cells opened up new avenues of translational research in cancer, neurodegenerative disorders, blood disorders, and AIDS. From food sciences to health sciences, almost every molecular biology laboratory around the world could apply CRISPR technology in their respective fields. One News Feature article in Nature stated “CRISPR, the disruptor, a powerful gene-editing technology is the biggest game changer to hit biology since PCR” (Ledford 2015). From the salt marshes of Spain, it is important to note that CRISPR is the consequence of a 20-year cumulative effort from scientists around the globe.
Principles and Mechanisms of the CRISPR-Cas9 System
In S. pyogenes, invading DNAs (e.g., phages) are cleaved into small chunks by Cas1 and Cas2 and are inserted as spacer sequences into the genome at the CRISPR loci (Doudna and Charpentier 2014). During transcription, the invading spacer portion and a portion of the repeat sequence are transcribed and processed into a single crRNA. The crRNA together with a tracrRNA form a duplex to direct a Cas9-induced DSB at sites complementary to the crRNA.
Current CRISPR-Cas9 systems based on S. pyogenes have engineered the tracrRNA:crRNA duplex to an gRNA (Doudna and Charpentier 2014) that serves two functions: the 20-nucleotide sequence at the 5′ end of gRNA can make Watson-Crick pairings to target genomic DNA sequences, while the 3′ side of the gRNA binds to Cas9 (Doudna and Charpentier 2014). This simplified single, two-component system allows modifications to the guide sequence (20 nucleotides in the native RNA) of the gRNA that program CRISPR-Cas9 to target any DNA sequence of interest, as long as it is adjacent to a PAM sequence. When the Cas9-gRNA complex forms base pairing with a complementary target DNA sequence, Cas9 introduces a site-specific DSB at three nucleotides upstream of the PAM site. While attempting to repair the DBS, NHEJ repair mechanisms often introduce indels, leading to gene disruption (Hsu et al. 2014). Spacer sequences within the CRISPR array are protected from cleavage where ever the direct repeat sequences lack PAM, which are otherwise extremely common throughout the genome.
The CRISPR-Cas9 System in Neurodegenerative Disease Research
Since the initial proof-of-concept studies in the uses of the CRISPR-Cas9 system for genome-editing in mammalian systems, experimental applications of CRISPR-Cas9 in neurodegenerative diseases, such as PD, AD, and Niemann-Pick disease type C1, have rapidly gained ground towards potential clinical applications. For example, in AD, mutations in amyloid precursor protein (APP) and presenilin-1 cause early-onset AD. Using CRISPR-Cas9, a group in the US generated AD-causing mutations in APP (APPswe) and presenilin-1 (PSEN1(M146 V)). Moreover, by establishing the relationship between a mutation’s incorporation rate after DSB repair and its distance to the DSB, they generated heterozygous and homozygous dominant mutations in APP and presenilin-1 in predictable frequencies (Paquet et al. 2016). More recently, Gyorgy et al. (2018) cultured fibroblasts isolated from patients exhibiting early-onset dominant familial AD and then transduced them with AAV-mediated CRISPR-Cas9 designed to disrupt the generation of the mutant APPswe, a key driver of neuroinflammation in AD patient brains (Heneka et al. 2015), and the resulting gene knockout reduced secreted Aβ by 60%.
Experimental Applications of CRISPR in PD
The premise of genome-editing in PD is that if cellular functions could be restored, and the balance between apoptotic and survival signals could be regained, the accumulated damages induced by chronic neuroinflammation and cell death could be stabilized or even restored. The current uses of CRISPR-Cas9 in experimental PD studies have been multifold: generating reporters, screening for susceptibility genes, building better models, and dissecting the pathways of neuronal inflammation and degeneration.
Generation of Reporters
Earlier studies of genome editing had already produced impressive results using ZFN to introduce reporter systems for gene expression. For example, the alpha-synuclein (SNCA) gene, under the endogenous control of SNCA gene expression, had high-throughput screening of compounds that could affect SNCA gene expression (Dansithong et al. 2015). More recently, the advantages conferred by CRISPR-Cas9 over other genome-editing tools have led to more rapid advancements in experimental applications for PD research. A proof-of-concept study using CRISPR-Cas9-mediated insertion of a nanoluciferase tag, as a reporter for endogenous SNCA transcriptional activity, has created an efficient drug screening tool for potential interventions affecting SNCA transcription (Basu et al. 2017).
Generation of Genomic Screens for Susceptibility Genes
The advantage of rapidly generated gRNA libraries was recently demonstrated in the use of a genome-wide CRISPR-Cas9 knockout screen to discover the regulators of Parkin, an important E3 ubiquitin ligase mutated in dominant familial PD. As Parkin participates in the normal mitophagy process, its loss of function leads to dysregulated mitophagy and associated neuroinflammation (Torres-Odio et al. 2017; Dionisio et al. 2018). Fifty-three positive and negative regulators of Parkin were found, among which THAP11 underwent further validation in a CRISPR-Cas9 knockout study using multiple cell types (Potting et al. 2018). This work highlighted the utility of unbiased, systematic knockout screens for discovering previously unknown regulatory networks. Other studies leveraged CRISPR-Cas9 systems to drive gene expression. A CRISPR-Cas9 transcription factor was used, for instance, to screen for protective genes during α-synuclein toxicity in a yeast model (Chen et al. 2017). The study demonstrated the construction of a gRNA-expressing plasmid library of 20-mer randomized nucleotides, which were co-expressed in a yeast system with CRISPR-Cas9-based transcription factors (crisprTFs) to activate expression of the gRNA-targeted gene. The gRNA-targeted genes that afforded protection against α-synuclein neurotoxicity, namely DJ-1, ALS2, GGA1, and DNAJB1, were validated in a human SH-SY5Y neuronal cell culture model of PD (Chen et al. 2017). However, the target of the gRNA that most dramatically reduced α-synuclein in yeast, named gRNA9–1, could not be readily identified with a human homolog.
Generation of PD Animal Models
CRISPR-Cas9 has also revolutionized the production of knockout models for various diseases, including PD. Previously, selective breeding across multiple generations of offspring was needed to generate single or multiple gene knockouts in rodents or other higher animals, which was a time-consuming and sometimes fruitless process. By contrast, CRISPR-Cas9’s precise genome editing facilitated Parkin and PINK1 double-knockout pigs in combination with somatic cell nuclear transfer (SCNT) techniques (Zhou et al. 2015). Piglets with double knockouts born from this study were negative for immunofluorescence staining of anti-Parkin and anti-PINK1 antibodies. All piglets born were homozygous and no off-target mutagenesis was detected.
That work was closely followed by a study from Wang et al. (2016), who generated Parkin, DJ-1, and PINK1 triple-knockout Bama miniature pigs. In their study, CRISPR-Cas9 gRNAs against Parkin, DJ-1, and then PINK1, respectively, were co-injected together with Cas9 mRNA into pronuclear embryos and later transplanted. Immunostaining the fibroblasts of the gene-edited pigs revealed an absence of staining for DJ-1 protein expression, while Parkin and PINK1 gene expression were drastically decreased but not absent. More recently, Zhu et al. (2018) successfully introduced PD-causing missense mutations in the SNCA gene using CRISPR-Cas9 in Guangxi Bama mini-pigs. These proof-of-concept studies demonstrate both the time and cost savings achieved by the CRISPR-Cas9 system, but the models did not differ from wild-types in neuroinflammation or behavioral deficits. This is presumably because the piglets were still young. Nonetheless, the usefulness of double- or triple-knockout pigs in recapitulating human PD symptoms remains to be seen.
Identifying Novel Early Apoptotic Pathways Involved in PD
The scalability of CRISPR-Cas9 gRNA libraries can also help to identify previously unreported metabolic and biotransformation pathways, where uncovering such pathways that facilitate cell death will contribute to better understanding the progression of neurodegenerative disorders. For instance, the complex etiology of idiopathic PD likely results from interactions between genetic factors and environmental stressors, such as chronic exposure to certain agrochemicals. For example, paraquat is a widely used herbicide that has been shown to increase the risk of PD by 1.3- to 3.6-fold in humans. With a chemical structure similar to the classic Parkinsonian toxicant 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), paraquat is a potent inducer of neuroinflammation in neurotoxicant-based models of PD (Mitra et al. 2011). A recent study by Reczek et al. (2017) illustrates the usefulness of functional genomic screens to identify novel metabolic and biotransformation pathways mediating increased neurotoxicity. They transduced Jurkat cells, an immortalized cell line derived from human T cell acute lymphoblastic cells, with a library of 30,000 CRISPR-Cas9 gRNAs representing 3000 genes involved in metabolism, with 10 unique gRNA for each of the 3000 genes. The transduced Jurkat cells were treated with 110 μM paraquat to induce cell death. This positive-selection screen for metabolic genes, whose loss allowed cell survival in the presence of paraquat, yielded three genes required for paraquat-induced cell death: cytochrome P450 oxidoreductase (POR), copper transporter ATP7A, and sucrose transporter (SLC45A4). In addition to neurotoxicity models of PD demonstrating how environmental stressors contribute to protein aggregation (Kanthasamy et al. 2012) and mitochondrial dysfunction, transgenic PD models are yielding similar findings (Langley et al. 2017), and it is becoming increasingly likely that both proteinopathy and mitochondrial dysfunction are linked in PD pathogenesis (Ganguly et al. 2017). Indeed, using CRISPR-Cas9 gRNA to target telomere repeats, Kim et al. (2017) found that ablating SH-SY5Y neuronal cell telomeres not only reduced cell proliferation and viability, but importantly also increased protein aggregation while decreasing mitochondrial bioenergetics.
CRISPR in Dissecting Neuroinflammatory Mechanisms Involved in PD
Neuroinflammation has been implicated as a main driver of PD disease progression (Ghosh et al. 2016; De Virgilio et al. 2016; Sampson et al. 2016; Sarkar et al. 2017). In neurodegenerative disorders, such as PD, both microglia and astrocytes can be hyper-activated by various physical or chemical insults. Exposure to environmental neurotoxicants, such as Mn, perturbs complex pathophysiological signaling mechanisms between neurons and glial cells that cause neuroinflammation, particularly in the striatal pallidum and substantia nigra pars reticulata. Activated microglia adopt an amoeba-like morphology and dramatically increase the release of pro-inflammatory cytokines, while reactive astrocytes upregulate the surface expression of GFAP and secrete pro-inflammatory cytokines. Reactive gliosis is a neuroinflammatory response generally believed to be a supportive process to maintain homeostasis, a process that in PD becomes a chronically progressive self-sustaining cycle of neuroinflammation (Orr et al. 2002). It manifests itself as the proliferation of activated microglia and astrocytes, activation and nuclear translocation of NF-κB, and elevation of pro-inflammatory factors, including tumor necrosis factor-α (TNF-α), inducible nitric oxide synthase (iNOS), nitric oxide (NO), interleukin (IL)-1beta, IL-6, cyclooxygenase-2 (COX-2), and prostaglandins E2 (PGE2).
MPTP is another potent inducer of neuroinflammation and neurodegeneration, and brain inflammatory factors can remain elevated even years after the initial neurotoxicant exposure (Orr et al. 2002; Tansey et al. 2007; Ghosh et al. 2016), suggesting that a cascade of events involving neuronal cell death can sustain glia activation in a cycle of self-propelling neurotoxicity. Postmortem examinations of brains of PD patients have revealed the presence of extensive neuroinflammation with distinct elevation of pro-inflammatory cytokines such as TNF-α, IL-12, IL-6, and IL-1β, and sustained neuroinflammation in the SN and elsewhere has been shown to promote disease progression. Mn also induces excessive expression of TNF-α, IL-12, IL-6, IL-10, and IL-1β and stimulates iNOS protein levels through activation of the NF-κB (p50/p65) transcription factor signaling pathway (Filipov et al. 2005; Verina et al. 2011; Sarkar et al. 2019). A major intracellular signaling mechanism driving Mn-induced apoptotic cell death involves activation of the protein kinase C delta (PKCδ), a non-canonical member of the protein kinase C family of kinases. PKCδ can be proteolytically activated by caspase-3 in the apoptotic signaling pathway (Kanthasamy et al. 2006; Kitazawa et al. 2005; Latchoumycandane et al. 2005), as well as in neuroinflammatory processes in PD models (Wilkins and Swerdlow 2016; Singh et al. 2018). To better understand the PKCδ downstream pathway leading to apoptosis, we recently used the CRISPR-Cas9 system to knockdown PKCδ expression in dopaminergic neuronal cells, revealing that downregulation of PKCδ significantly blocked Mn-induced DNA fragmentation (Song et al. 2019).
Neuroinflammatory processes and mitochondrial dysfunction are intricately linked during the process of neurodegeneration, and both are thought of as common denominators in a variety of neurodegenerative diseases (Witte et al. 2010; Wilkins and Swerdlow 2016; Gong et al. 2018). Indeed, some studies including our own suggest that mitochondrial dysfunction contributes to neuroinflammation, hastening cell death (Wilkins and Swerdlow 2016; Sarkar et al. 2017).
Several groups have shown approaches for nuclear and mitochondrial genome editing using CRISPR-Cas9. For example, Cas9 protein expressed with gRNAs against Cox1 and Cox3 can localize to mitochondria and initiate cleavage at the mitochondrial mtDNA loci (Jo et al. 2015). CRISPR-Cas9 systems have since been used to dissect cellular regulation of mitochondrial biogenesis, especially during neurodegeneration. Jiang et al. (2016) found that adult conditional knockout of peroxisome proliferator-activated receptor γ (PPAR γ) coactivator protein-1α (PGC-1α), a master regulator of mitochondrial biogenesis, leads to dopaminergic neuronal cell death. Another regulator of mitochondrial biogenesis is mitochondrial transcription factor A (TFAM), a key activator of mitochondrial transcription. TFAM aids transcription by regulating mitochondrial copy number, via binding to mitochondrial promoter DNA, as well as calcium and reactive oxygen species (Kunkel et al. 2018). Similar to a PGC-1α knockout, since a complete TFAM knockout depletes mitochondrial DNA, it is embryonically lethal in mice (Stiles et al. 2016). Mice with a conditional knockout of mitochondrial TFAM using the Cre/LoxP system in dopaminergic neurons present with neuroinflammation in the SN, characterized by upregulation of the microglial activation marker IBA1, resulting in the progressive loss of these neurons (Ekstrand and Galter 2009; Good et al. 2011). The ensuing motor symptoms recapitulate human PD symptoms. Given this mitochondrial-based induction of PD-like symptoms, these mice are called MitoPark. Our mechanistic studies of the TFAM knockout using CRISPR-Cas9 gRNA targeted against TFAM in rat dopaminergic neuronal cultures (Fig. 2) revealed that TFAM-knockout neurons were more susceptible to Mn neurotoxicity, exhibiting lower mitochondrial basal and ATP-linked respiration (Langley et al. 2018).

Experimental application of CRISPR-Cas9 in elucidating the role of PKCδ in neuronal apoptosis and neuroinflammation. Oxidative stress-induced reactive oxygen species (ROS) can lead to activation of caspase-3, which induces the phosphorylation of inhibitor of κB (IκB), causing nuclear translation of the p50/p65 complex. In the nucleus, it participates in the transcription of PKCδ, which can be proteolytically cleaved, thereby inducing neuroinflammation and neuronal cell death. However, CRISPR-Cas9 knockout of PKCδ made cells more resistant to oxidative stress-induced cell death
Our recent studies have demonstrated a major role of Prokineticin 2 (PK2) in mitochondrial biogenesis (Gordon et al. 2016). PK2 belongs to the prokineticin family, which consists of Prokineticin 1 and Prokineticin 2, a pair of chemokines that signals through the Prokineticin receptors PKR1 and PKR2. They were named for their ability to contract gastrointestinal smooth muscle from isolated ileal segments (Wade et al. 2010). PK2 has since been attributed with wide-ranging functional roles, including hematopoiesis, pain reception, neurogenesis, and circadian rhythm (Ng et al. 2005). We reported that PK2 signaling was increased in PD post-mortem brains, and acted as a protective compensatory response against neurodegeneration in PD cell culture and animal models (Gordon et al. 2016). During neurotoxic stress, overexpression of PK2 preserved mitochondrial bioenergetics by rescuing neurotoxicant-induced loss in ATP production and increasing levels of PGC-1α and BCL2 (Gordon et al. 2016). Importantly, during MPTP-induced gliosis, AAV-mediated delivery of PK2 reduced the processes of reactive astrocytes (Neal et al. 2018) and increased the expression of genes associated with the alternative, neuroprotective A2 astrocyte phenotype (Neal et al. 2018), which can counter neuroinflammation induced by reactive A1 astrocytes (Liddelow et al. 2017). Our mechanistic cell culture studies (Fig. 3) have shown that a PK2 knockout using CRISPR-Cas9, in contrast, increased neuronal susceptibility to neurotoxicant-induced cell death (Gordon et al. 2016). Furthermore, CRISPR-Cas9-mediated knockout of PKR1, a receptor for the PK2 chemokine, abolished the effects of PK2 on alternative A2 astrocyte activation (Neal et al. 2018). Collectively, these studies further link mitochondrial dysfunction with neuroinflammation and demonstrate the utility of CRISPR-Cas9 in elucidating these mechanisms.

Experimental application of CRISPR-Cas9 in elucidating the role of PK2 in protective, anti-apoptotic responses against neurotoxic stress in neurons. Neurotoxic stress can induce a variety of transcription factors, including HIF1α, which can bind to putative binding sites on the promoter of the PK2 gene. The upregulation of PK2 can induce cell survival via increases in mitochondrial biogenesis and upregulation of PGC1α and BCL-2. Moreover, astrocytes react to secreted PK2 by upregulating genes associated with the anti-inflammatory alternative activation (A2) phenotype. CRISPR-Cas9 knockout of the PK2 gene produces undesirable effects, increasing cell susceptibility to a neurotoxicant
CRISPR-Cas9 for Genetic Mutation Repair
SNCA has long been known to be a strong risk locus for sporadic PD. Combined with induced pluripotent stem cell (iPSC) techniques, the genome of human (h) patient-specific hiPSCs was isolated and corrected using ZFN that targeted a disease-causing mutation (A53T) in the SNCA gene (Soldner et al. 2011). Importantly, the repair of the A53T mutation in the patient-derived hiPSCs using CRISPR did not compromise the differentiation of TH-positive dopaminergic neurons (Soldner et al. 2011). Leucine-rich repeat kinase 2 (LRRK2) is another PD-susceptibility gene, with G2019S and R1441C mutations in the gene resulting in mitochondrial impairments. Attempts at using ZFN to “repair” PD patient-specific iPSCs carrying G2019S and R1441C mutations in the LRRK2 gene successfully achieved healthy differentiated neuroprogenitor and neural cells with no detectable mtDNA damage (Sanders et al. 2014). GWAS had previously been used to identify several microsatellite repeat regions upstream of the SNCA gene that were suspected to enhance SNCA expression (Nalls et al. 2014). A shorter repeat region Rep1–257 or Rep1–259 conferred significantly lower risk of developing PD, compared to individuals carrying Rep1–261 or Rep1–263 (Farrer et al. 2001). Based on these data, Soldner et al. (2016) used CRISPR-Cas9 to delete the entire repeat region, and re-inserted representative alleles for each of the 4 reported repeat-length alleles in hiPSCs. However, after terminal differentiation of hiPSC into neurons, Soldner’s team found no cis-regulatory effects on SNCA expression due to repeat length. On the other hand, Chen et al. (2019) used CRISPR-Cas9 to delete the endogenous SNCA gene in human embryonic stem cells (hESCs). When treated with recombinant α-synuclein preformed fibrils (PFFs), the hESCs that differentiated into dopaminergic neurons exhibited significant resistance to pS129-αSyn-positive protein aggregation, a pathological hallmark of Lewy body synucleinopathies.
Challenges and Drawbacks
Despite many advantages, several areas associated with CRISPR-Cas9 technology’s practical use need to be improved to maximize its utility. A major challenge is the highly frequent off-target modifications of the genome by CRISPR-Cas9. Single- and double-base mismatches could be tolerated to varying degrees at the gRNA-DNA interface, leading to unwanted mutagenesis (Fu et al. 2013). Furthermore, Cas9- and gRNA-encoding plasmids constitutively express functional Cas9-gRNA complexes that could lead to the accumulation of off-target mutagenesis. Cloud-based tools are now available for designing unique target sequences to avoid potential off-target sites in the genome. Another option is the use of recombinant Cas9 proteins, which quickly induce mutations at intended on-target sites after delivery into cells and are eventually degraded by cellular protease machinery to reduce off-target effects (Koo et al. 2015). Due to the relative novelty of this technology, long-term consequences from undesirable mutagenesis induced by CRISPR-Cas9 is hard to predict in humans, as illustrated by the discussion among experts on the potential long-term health benefits and negative side effects surrounding the first-ever CRISPR-Cas9-edited baby born in China in 2018 (Cyranoski 2018). Such ethical and safety concerns need to be addressed before moving this technology to clinical applications. A second major challenge of CRISPR-Cas9-based therapies lies in its variable efficiency of delivery to target cells. Common delivery methods (reviewed by Lino et al. 2018) for the CRISPR-Cas9 system include microinjection, electroporation, AAV, lentivirus, and nanoparticle or liposomal delivery methods, only a few of which in their current form could be utilized for central nervous system delivery of CRISPR-Cas9-based therapy (Maguire et al. 2014; Banks 2016; Lino et al. 2018). Other groups have had some success in utilizing a neuron-preferring chimeric AAV-based CRISPR-Cas9 system administered into the cerebrospinal fluid to mediate brain-specific gene deletion, with no detectable events in other non-target organs, including the liver (Murlidharan et al. 2016).
Conclusion
CRISPR-Cas9-mediated therapies have been embraced for their role in identifying essential genes for cancer immunotherapy and in treating a variety of genetic diseases (Patel et al. 2017; Demirci et al. 2018). Several recently approved clinical trials are underway for applications of CRISPR to treat sickle cell anemia and retinitis pigmentosa (Demirci et al. 2018; Cai et al. 2018). CRISPR-Cas9 is also poised to transform biomedical research on complex neurodegenerative diseases by dissecting key pathways of neuroinflammation and neuronal cell death. As the research and engineering of CRISPR-Cas9 systems expand, it is likely that yet unknown applications might help find cures for neurodegenerative diseases such as PD.
References
Banks WA (2016) From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov 15:275–292. https://doi.org/10.1038/nrd.2015.21
Barrangou R (2012) RNA-mediated programmable DNA cleavage. Nat Biotechnol 30:836–838
Barrangou R, Marraffini LA (2014) CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol Cell 54:234–244. https://doi.org/10.1016/j.molcel.2014.03.011
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. https://doi.org/10.1126/science.1138140
Barzilai A, Melamed E (2003) Molecular mechanisms of selective dopaminergic neuronal death in Parkinson’s disease. Trends Mol Med 9:126–132
Basu S, Adams L, Guhathakurta S, Kim Y-S (2017) A novel tool for monitoring endogenous alpha-synuclein transcription by NanoLuciferase tag insertion at the 3’end using CRISPR-Cas9 genome editing technique. Sci Rep 8:45883. https://doi.org/10.1038/srep45883
Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, van der Oost J (2008) Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964. https://doi.org/10.1126/science.1159689
Cai B, Sun S, Li Z, Zhang X, Ke Y, Yang J, Li X (2018) Application of CRISPR/Cas9 technologies combined with iPSCs in the study and treatment of retinal degenerative diseases. Hum Genet 137:679–688. https://doi.org/10.1007/s00439-018-1933-9
Chen Y-C, Farzadfard F, Gharaei N et al (2017) Randomized CRISPR-Cas transcriptional perturbation screening reveals protective genes against alpha-Synuclein toxicity. Mol Cell 68:247–257.e5. https://doi.org/10.1016/j.molcel.2017.09.014
Chen Y, Dolt KS, Kriek M, Baker T, Downey P, Drummond NJ, Canham MA, Natalwala A, Rosser S, Kunath T (2019) Engineering synucleinopathy-resistant human dopaminergic neurons by CRISPR-mediated deletion of the SNCA gene. Eur J Neurosci 49:510–524
Cyranoski D (2018) CRISPR-baby scientist fails to satisfy critics. Nature 564:13–14
Dansithong W, Paul S, Scoles DR, Pulst SM, Huynh DP (2015) Generation of SNCA cell models using zinc finger nuclease (ZFN) technology for efficient high-throughput drug screening. PLoS One 10:e0136930. https://doi.org/10.1371/journal.pone.0136930
De Virgilio A, Greco A, Fabbrini G et al (2016) Parkinson’s disease: autoimmunity and neuroinflammation. Autoimmun Rev 15:1005–1011. https://doi.org/10.1016/j.autrev.2016.07.022
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607. https://doi.org/10.1038/nature09886
Demirci S, Uchida N, Tisdale JF (2018) Gene therapy for sickle cell disease: an update. Cytotherapy 20:899–910. https://doi.org/10.1016/j.jcyt.2018.04.003
Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S (2008) Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190:1390–1400. https://doi.org/10.1128/JB.01412-07
Dionisio PEA, Oliveira SR, Amaral JSJD, Rodrigues CMP (2018) Loss of microglial Parkin inhibits necroptosis and contributes to Neuroinflammation. Mol Neurobiol. https://doi.org/10.1007/s12035-018-1264-9
Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM (2007) Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68:384–386
Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096. https://doi.org/10.1126/science.1258096
Duffy MF, Collier TJ, Patterson JR, Kemp CJ, Luk KC, Tansey MG, Paumier KL, Kanaan NM, Fischer DL, Polinski NK, Barth OL, Howe JW, Vaikath NN, Majbour NK, el-Agnaf OMA, Sortwell CE (2018) Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J Neuroinflammation 15:129. https://doi.org/10.1186/s12974-018-1171-z
Ekstrand MI, Galter D (2009) The MitoPark mouse - an animal model of Parkinson’s disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat Disord 15(Suppl 3):S185–S188. https://doi.org/10.1016/S1353-8020(09)70811-9
Farrer M, Maraganore DM, Lockhart P, Singleton A, Lesnick TG, de Andrade M, West A, de Silva R, Hardy J, Hernandez D (2001) Alpha-Synuclein gene haplotypes are associated with Parkinson’s disease. Hum Mol Genet 10:1847–1851
Filipov NM, Seegal RF, Lawrence DA (2005) Manganese potentiates in vitro production of proinflammatory cytokines and nitric oxide by microglia through a nuclear factor kappa B-dependent mechanism. Toxicol Sci 84:139–148. https://doi.org/10.1093/toxsci/kfi055
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD (2013) High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31:822–826. https://doi.org/10.1038/nbt.2623
Ganguly G, Chakrabarti S, Chatterjee U, Saso L (2017) Proteinopathy, oxidative stress and mitochondrial dysfunction: cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des Devel Ther 11:797–810. https://doi.org/10.2147/DDDT.S130514
Garneau JE, Dupuis M-E, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468:67–71. https://doi.org/10.1038/nature09523
Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 109:E2579–E2586. https://doi.org/10.1073/pnas.1208507109
Ghosh A, Langley MR, Harischandra DS, Neal ML, Jin H, Anantharam V, Joseph J, Brenza T, Narasimhan B, Kanthasamy A, Kalyanaraman B, Kanthasamy AG (2016) Mitoapocynin treatment protects against neuroinflammation and dopaminergic neurodegeneration in a preclinical animal model of Parkinson’s disease. J NeuroImmune Pharmacol 11:259–278. https://doi.org/10.1007/s11481-016-9650-4
Gong Z, Pan J, Shen Q, Li M, Peng Y (2018) Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J Neuroinflammation 15:242. https://doi.org/10.1186/s12974-018-1282-6
Good CH, Hoffman AF, Hoffer BJ, Chefer VI, Shippenberg TS, Bäckman CM, Larsson NG, Olson L, Gellhaar S, Galter D, Lupica CR (2011) Impaired nigrostriatal function precedes behavioral deficits in a genetic mitochondrial model of Parkinson’s disease. FASEB J Off Publ Fed Am Soc Exp Biol 25:1333–1344. https://doi.org/10.1096/fj.10-173625
Gordon R, Neal ML, Luo J, Langley MR, Harischandra DS, Panicker N, Charli A, Jin H, Anantharam V, Woodruff TM, Zhou QY, Kanthasamy AG, Kanthasamy A (2016) Prokineticin-2 upregulation during neuronal injury mediates a compensatory protective response against dopaminergic neuronal degeneration. Nat Commun 7. https://doi.org/10.1038/ncomms12932
Gyorgy B, Loov C, Zaborowski MP, Takeda S, Kleinstiver BP, Commins C, Kastanenka K, Mu D, Volak A, Giedraitis V, Lannfelt L, Maguire CA, Joung JK, Hyman BT, Breakefield XO, Ingelsson M (2018) CRISPR/Cas9 Mediated Disruption of the Swedish APP Allele as a Therapeutic Approach for Early-Onset Alzheimer's Disease. Molecular therapy Nucleic acids 11:429–440
Heneka MT, Carson MJ, El Khoury J et al (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14:388–405
Horvath P, Romero DA, Coute-Monvoisin A-C, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, Barrangou R (2008) Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol 190:1401–1412. https://doi.org/10.1128/JB.01415-07
Hsu PD, Lander ES, Zhang F (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278. https://doi.org/10.1016/j.cell.2014.05.010
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169:5429–5433
Jansen R, van Embden JDA, Gaastra W, Schouls LM (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43:1565–1575
Jarraya B, Boulet S, Ralph GS et al (2009) Dopamine gene therapy for Parkinson’s disease in a nonhuman primate without associated dyskinesia Sci Transl Med 1. https://doi.org/10.1126/scitranslmed.3000130
Jiang H, Kang SU, Zhang S, Karuppagounder S, Xu J, Lee YK, Kang BG, Lee Y, Zhang J, Pletnikova O, Troncoso JC, Pirooznia S, Andrabi SA, Dawson VL, Dawson TM (2016) Adult Conditional Knockout of PGC-1alpha Leads to Loss of Dopamine Neurons. eNeuro 3
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. https://doi.org/10.1126/science.1225829
Jo A, Ham S, Lee GH, Lee YI, Kim SS, Lee YS, Shin JH, Lee Y (2015) Efficient mitochondrial genome editing by CRISPR/Cas9. Biomed Res Int 2015:305716–305710. https://doi.org/10.1155/2015/305716
Kanthasamy AG, Anantharam V, Zhang D, Latchoumycandane C, Jin H, Kaul S, Kanthasamy A (2006) A novel peptide inhibitor targeted to caspase-3 cleavage site of a proapoptotic kinase protein kinase C delta (PKCdelta) protects against dopaminergic neuronal degeneration in Parkinson’s disease models. Free Radic Biol Med 41:1578–1589. https://doi.org/10.1016/j.freeradbiomed.2006.08.016
Kanthasamy AG, Choi C, Jin H, Harischandra DS, Anantharam V, Kanthasamy A (2012) Effect of divalent metals on the neuronal proteasomal system, prion protein ubiquitination and aggregation. Toxicol Lett 214:288–295. https://doi.org/10.1016/j.toxlet.2012.09.008
Kitazawa M, Anantharam V, Yang Y, Hirata Y, Kanthasamy A, Kanthasamy AG (2005) Activation of protein kinase Cδ by proteolytic cleavage contributes to manganese-induced apoptosis in dopaminergic cells: protective role of Bcl-2. Biochem Pharmacol 69:133–146. https://doi.org/10.1016/j.bcp.2004.08.035
Kim H, Ham S, Jo M, Lee GH, Lee Y-S, Shin J-H, Lee Y (2017) CRISPR-Cas9 Mediated Telomere Removal Leads to Mitochondrial Stress and Protein Aggregation. Int J Mol Sci 18:2093
Koo T, Lee J, Kim J-S (2015) Measuring and reducing off-target activities of programmable nucleases including CRISPR-Cas9. Mol Cells 38:475–481. https://doi.org/10.14348/molcells.2015.0103
Kowal SL, Dall TM, Chakrabarti R, Storm MV, Jain A (2013) The current and projected economic burden of Parkinson’s disease in the United States. Mov Disord 28:311–318. https://doi.org/10.1002/mds.25292
Kunkel GH, Chaturvedi P, Thelian N, Nair R, Tyagi SC (2018) Mechanisms of TFAM-mediated cardiomyocyte protection. Can J Physiol Pharmacol 96:173–181. https://doi.org/10.1139/cjpp-2016-0718
Lander ES (2016) The heroes of CRISPR. Cell 164:18–28. https://doi.org/10.1016/j.cell.2015.12.041
Langley M, Ghosh A, Charli A, Sarkar S, Ay M, Luo J, Zielonka J, Brenza T, Bennett B, Jin H, Ghaisas S, Schlichtmann B, Kim D, Anantharam V, Kanthasamy A, Narasimhan B, Kalyanaraman B, Kanthasamy AG (2017) Mito-Apocynin prevents mitochondrial dysfunction, microglial activation, oxidative damage, and progressive neurodegeneration in MitoPark transgenic mice. Antioxid Redox Signal 27:1048–1066. https://doi.org/10.1089/ars.2016.6905
Langley MR, Ghaisas S, Ay M, Luo J, Palanisamy BN, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG (2018) Manganese exposure exacerbates progressive motor deficits and neurodegeneration in the MitoPark mouse model of Parkinson’s disease: relevance to gene and environment interactions in metal neurotoxicity. Neurotoxicology 64:240–255. https://doi.org/10.1016/j.neuro.2017.06.002
Latchoumycandane C, Anantharam V, Kitazawa M, Yang Y, Kanthasamy A, Kanthasamy AG (2005) Protein kinase Cdelta is a key downstream mediator of manganese-induced apoptosis in dopaminergic neuronal cells. J Pharmacol Exp Ther 313:46–55. https://doi.org/10.1124/jpet.104.078469
Ledford H (2015) CRISPR, the disruptor. Nature 522:20–24
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. https://doi.org/10.1038/nature21029
Lino CA, Harper JC, Carney JP, Timlin JA (2018) Delivering CRISPR: a review of the challenges and approaches. Drug Deliv 25:1234–1257. https://doi.org/10.1080/10717544.2018.1474964
Maguire CA, Ramirez SH, Merkel SF, Sena-Esteves M, Breakefield XO (2014) Gene therapy for the nervous system: challenges and new strategies. Neurotherapeutics 11:817–839. https://doi.org/10.1007/s13311-014-0299-5
Mangold M, Siller M, Roppenser B, Vlaminckx BJM, Penfound TA, Klein R, Novak R, Novick RP, Charpentier E (2004) Synthesis of group a streptococcal virulence factors is controlled by a regulatory RNA molecule. Mol Microbiol 53:1515–1527. https://doi.org/10.1111/j.1365-2958.2004.04222.x
Marraffini LA, Sontheimer EJ (2008) CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322:1843–1845. https://doi.org/10.1126/science.1165771
Menken M, Munsat TL, Toole JF (2000) The global burden of disease study: implications for neurology. Arch Neurol 57:418–420
Mitra S, Chakrabarti N, Bhattacharyya A (2011) Differential regional expression patterns of alpha-synuclein, TNF-alpha, and IL-1beta; and variable status of dopaminergic neurotoxicity in mouse brain after Paraquat treatment. J Neuroinflammation 8:163. https://doi.org/10.1186/1742-2094-8-163
Mojica FJ, Juez G, Rodriguez-Valera F (1993) Transcription at different salinities of Haloferax mediterranei sequences adjacent to partially modified PstI sites. Mol Microbiol 9:613–621
Mojica FJ, Ferrer C, Juez G, Rodriguez-Valera F (1995) Long stretches of short tandem repeats are present in the largest replicons of the archaea Haloferax mediterranei and Haloferax volcanii and could be involved in replicon partitioning. Mol Microbiol 17:85–93
Mojica FJ, Diez-Villasenor C, Soria E, Juez G (2000) Biological significance of a family of regularly spaced repeats in the genomes of archaea, Bacteria and mitochondria. Mol Microbiol 36:244–246
Mojica FJM, Diez-Villasenor C, Garcia-Martinez J, Soria E (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60:174–182. https://doi.org/10.1007/s00239-004-0046-3
Murlidharan G, Sakamoto K, Rao L, Corriher T, Wang D, Gao G, Sullivan P, Asokan A (2016) CNS-restricted transduction and CRISPR/Cas9-mediated gene deletion with an engineered AAV vector. Mol Ther Nucleic Acids 5:e338. https://doi.org/10.1038/mtna.2016.49
Nakai W, Westmoreland J, Yeh E, Bloom K, Resnick MA (2011) Chromosome integrity at a double-strand break requires exonuclease 1 and MRX. DNA Repair (Amst) 10:102–110. https://doi.org/10.1016/j.dnarep.2010.10.004
Nalls MA, Pankratz N, Lill CM et al (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46:989–993. https://doi.org/10.1038/ng.3043
Nassif DV, Pereira JS (2018) Fatigue in Parkinson’s disease: concepts and clinical approach. Psychogeriatrics 18:143–150. https://doi.org/10.1111/psyg.12302
Neal M, Luo J, Harischandra DS, Gordon R, Sarkar S, Jin H, Anantharam V, Désaubry L, Kanthasamy A, Kanthasamy A (2018) Prokineticin-2 promotes chemotaxis and alternative A2 reactivity of astrocytes. Glia 66:2137–2157. https://doi.org/10.1002/glia.23467
Ng KL, Da Li J, Cheng MY et al (2005) Neuroscience: dependence of olfactory bulb neurogenesis on prokineticin 2 signaling. Science (80- ) 308:1923–1927. https://doi.org/10.1126/science.1112103
Orr CF, Rowe DB, Halliday GM (2002) An inflammatory review of Parkinson’s disease. Prog Neurobiol 68:325–340
Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, Olsen KM, Gregg A, Noggle S, Tessier-Lavigne M (2016) Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533:125–129. https://doi.org/10.1038/nature17664
Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM, Yamamoto TN, Merchant AS, Mehta GU, Chichura A, Shalem O, Tran E, Eil R, Sukumar M, Guijarro EP, Day CP, Robbins P, Feldman S, Merlino G, Zhang F, Restifo NP (2017) Identification of essential genes for cancer immunotherapy. Nature 548:537–542. https://doi.org/10.1038/nature23477
Potting C, Crochemore C, Moretti F, Nigsch F, Schmidt I, Manneville C, Carbone W, Knehr J, DeJesus R, Lindeman A, Maher R, Russ C, McAllister G, Reece-Hoyes JS, Hoffman GR, Roma G, Müller M, Sailer AW, Helliwell SB (2018) Genome-wide CRISPR screen for PARKIN regulators reveals transcriptional repression as a determinant of mitophagy. Proc Natl Acad Sci U S A 115:E180–E189. https://doi.org/10.1073/pnas.1711023115
Pourcel C, Salvignol G, Vergnaud G (2005) CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151:653–663. https://doi.org/10.1099/mic.0.27437-0
Reczek CR, Birsoy K, Kong H, Martinez-Reyes I, Wang T, Gao P, Sabatini DM, Chandel NS (2017) A CRISPR screen identifies a pathway required for paraquat-induced cell death. Nature chemical biology 13:1274-1279Reczek CR, Birsoy K, Kong H, Martinez-Reyes I, Wang T, Gao P, Sabatini DM, Chandel NS (2017) A CRISPR screen identifies a pathway required for paraquat-induced cell death. Nature chemical biology 13:1274–279
Sampson TR, Debelius JW, Thron T et al (2016) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167:1469–1480.e12. https://doi.org/10.1016/j.cell.2016.11.018
Sanders LH, Laganière J, Cooper O, Mak SK, Vu BJ, Huang YA, Paschon DE, Vangipuram M, Sundararajan R, Urnov FD, Langston JW, Gregory PD, Zhang HS, Greenamyre JT, Isacson O, Schüle B (2014) LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiol Dis 62:381–386. https://doi.org/10.1016/j.nbd.2013.10.013
Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V (2011) The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res 39:9275–9282. https://doi.org/10.1093/nar/gkr606
Sarkar S, Malovic E, Harishchandra DS, Ghaisas S, Panicker N, Charli A, Palanisamy BN, Rokad D, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG (2017) Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Park Dis 3:30. https://doi.org/10.1038/s41531-017-0032-2
Sarkar S, Rokad D, Malovic E, Luo J, Harischandra DS, Jin H, Anantharam V, Huang X, Lewis M, Kanthasamy A, Kanthasamy AG (2019) Manganese activates NLRP3 inflammasome signaling and propagates exosomal release of ASC in microglial cells. Sci Signal 12:eaat9900. https://doi.org/10.1126/scisignal.aat9900
Siderowf A, Lang AE (2012) Premotor Parkinson’s disease: concepts and definitions. Mov Disord 27:608–616. https://doi.org/10.1002/mds.24954
Singh N, Lawana V, Luo J, Phong P, Abdalla A, Palanisamy B, Rokad D, Sarkar S, Jin H, Anantharam V, Kanthasamy AG, Kanthasamy A (2018) Organophosphate pesticide chlorpyrifos impairs STAT1 signaling to induce dopaminergic neurotoxicity: implications for mitochondria mediated oxidative stress signaling events. Neurobiol Dis 117:82–113. https://doi.org/10.1016/j.nbd.2018.05.019
Soldner F, Laganiere J, Cheng AW et al (2011) Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146:318–331. https://doi.org/10.1016/j.cell.2011.06.019
Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, Jaenisch R (2016) Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 533:95–99
Song C, Charli A, Luo J et al (2019) Mechanistic interplay between autophagy and apoptotic signaling in Endosulfan-induced dopaminergic neurotoxicity: relevance to the adverse outcome pathway in pesticide neurotoxicity. Toxicol Sci
Sternberg SH, Doudna JA (2015) Expanding the biologist’s toolkit with CRISPR-Cas9. Mol Cell 58:568–574. https://doi.org/10.1016/j.molcel.2015.02.032
Stiles AR, Simon MT, Stover A, Eftekharian S, Khanlou N, Wang HL, Magaki S, Lee H, Partynski K, Dorrani N, Chang R, Martinez-Agosto JA, Abdenur JE (2016) Mutations in TFAM, encoding mitochondrial transcription factor A, cause neonatal liver failure associated with mtDNA depletion. Mol Genet Metab 119:91–99. https://doi.org/10.1016/j.ymgme.2016.07.001
Tansey MG, McCoy MK, Frank-Cannon TC (2007) Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol 208:1–25
Torres-Odio S, Key J, Hoepken H-H, Canet-Pons J, Valek L, Roller B, Walter M, Morales-Gordo B, Meierhofer D, Harter PN, Mittelbronn M, Tegeder I, Gispert S, Auburger G (2017) Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J Neuroinflammation 14:154. https://doi.org/10.1186/s12974-017-0928-0
van der Oost J, Westra ER, Jackson RN, Wiedenheft B (2014) Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat Rev Microbiol 12:479–492. https://doi.org/10.1038/nrmicro3279
Verina T, Kiihl SF, Schneider JS, Guilarte TR (2011) Manganese exposure induces microglia activation and dystrophy in the substantia nigra of non-human primates. Neurotoxicology 32:215–226. https://doi.org/10.1016/j.neuro.2010.11.003\rS0161-813X(10)00221-4 [pii]
Wade PR, Palmer JM, Mabus J et al (2010) Prokineticin-1 evokes secretory and contractile activity in rat small intestine. Neurogastroenterol Motil 22:e152–e161. https://doi.org/10.1111/j.1365-2982.2009.01426.x
Wang X, Cao C, Huang J, Yao J, Hai T, Zheng Q, Wang X, Zhang H, Qin G, Cheng J, Wang Y, Yuan Z, Zhou Q, Wang H, Zhao J (2016) One-step generation of triple gene-targeted pigs using CRISPR/Cas9 system. Sci Rep 6:20620
Wilkins HM, Swerdlow RH (2016) Relationships between mitochondria and neuroinflammation: implications for Alzheimer’s disease. Curr Top Med Chem 16:849–857
Witte ME, Geurts JJG, de Vries HE, van der Valk P, van Horssen J (2010) Mitochondrial dysfunction: a potential link between neuroinflammation and neurodegeneration? Mitochondrion 10:411–418. https://doi.org/10.1016/j.mito.2010.05.014
Zhang F, Cong L, Lodato S, Kosuri S, Church GM, Arlotta P (2011) Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat Biotechnol 29:149–153. https://doi.org/10.1038/nbt.1775
Zhou X, Xin J, Fan N, Zou Q, Huang J, Ouyang Z, Zhao Y, Zhao B, Liu Z, Lai S, Yi X, Guo L, Esteban MA, Zeng Y, Yang H, Lai L (2015) Generation of CRISPR/Cas9-mediated gene-targeted pigs via somatic cell nuclear transfer. Cell Mol Life Sci 72:1175–1184. https://doi.org/10.1007/s00018-014-1744-7
Zhu XX, Zhong YZ, Ge YW, Lu KH, Lu SS (2018) CRISPR/Cas9-Mediated Generation of Guangxi Bama Minipigs Harboring Three Mutations in alpha-Synuclein Causing Parkinson's Disease. Sci Rep 8:12420
Acknowledgments
The writing of this review was primarily supported by the National Institutes of Health R01 grants ES027245, ES026892, NS100090 and NS088206. AAW and QW were supported by the College of Human Sciences at Iowa State University, National Institutes of Health R00 grant AG047282, and Alzheimer’s Association Research Grant to Promote Diversity (AARG-D) AARGD-17-529552. The W. Eugene and Linda Lloyd Endowed Chair and Eminent scholar and Armbrust Endowment to A.G.K. and the Salisbury chair to A.K. are also acknowledged. The support from the Presidential Interdisciplinary Research Initiative for the Big Data Brain Research from Iowa State University is also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
A.G.K and V.A have an equity interest in PK Biosciences Corporation located in Ames, IA. The terms of this arrangement have been reviewed and approved by Iowa State University in accordance with its conflict of interest policies. Other authors declare no actual or potential competing financial interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Luo, J., Padhi, P., Jin, H. et al. Utilization of the CRISPR-Cas9 Gene Editing System to Dissect Neuroinflammatory and Neuropharmacological Mechanisms in Parkinson’s Disease. J Neuroimmune Pharmacol 14, 595–607 (2019). https://doi.org/10.1007/s11481-019-09844-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-019-09844-3