
Abstract
2,2,6,6-Tetramethylpiperidine-1-oxyl radical (TEMPO)-mediated oxidation has been applied to bacterial cellulose (BC). The TEMPO-oxidized BC (TO-BC) gel particles were subjected to ultrasonication in water to prepare mechanically fibrillated TO-BC (TO-BC-U) samples. The carboxy contents of the TO-BC samples were 1.5‒1.6 mmol/g. X-ray diffraction patterns and solid-state 13C-nuclear magnetic resonance (NMR) spectra of the BC, TO-BC, and TO-BC-U samples showed that cellulose Iα was the dominant crystalline structure. The crystallinities of the samples calculated from the carbon signal areas in the NMR spectra were approximately the same between the BC and TO-BC samples, showing that TEMPO-mediated oxidation selectively occurred on the crystalline BC fibril surfaces. However, the crystallinities of the TO-BC-U samples were lower than those of the BC and TO-BC samples, indicating that ultrasonication of the TO-BC samples in water caused partial decreases in crystallinity. The TO-BC-U samples contained both single fibrils and fibril bundles; completely individualized TO-BC-U fibrils with homogeneous widths was not obtained. The average widths of the single TO-BC-U fibrils were ~ 3 nm, which are close to those of TO-cellulose nanofibrils prepared from wood-cellulose samples. Thus, the crystalline BC fibrils with widths of ~ 3 nm were the smallest crystalline elements. The lengths of the TO-BC samples were greater than 2‒3 µm, whereas the weight-average cellulose chain lengths of the cellulose/TEMPO-oxidized cellulose molecules in TO-BC-U samples were < 800 nm. Hence, each TO-BC-U fibril consisted of multiple cellulose and oxidized cellulose molecules, which were packed along the longitudinal direction.
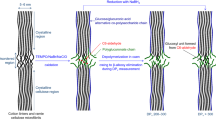
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cellulose is biologically synthesized by plants, tunicates, and aerobic bacteria, such as Gluconacetobacter xylinus. When bacterial cellulose (BC) is prepared by stationary cultivation, BC gel pellicles containing ~ 98% water are obtained at the air/culture medium interfaces (Iguchi et al. 2000; Klemm et al. 2005; Shoda and Sugano 2005; Lin et al. 2013; Campano et al. 2016). The BC gel pellicles consist of ribbon-like cellulose microfibrils of nanometer size, forming network structures (Iguchi 2000; Castro et al 2011; Pentilä et al. 2017). Thus, BC gel pellicles are composed of numerous, long, and almost pure cellulose fibrils that are formed by bacteria from low-molar-mass compounds through bottom-up processes. Various medical and material applications of BC have been reported based on the unique morphologies and properties of BC fibrils (Klemm et al. 2005; Czaja et al. 2007; Shah et al. 2013; Huang et al. 2014; Tajima et al. 2020; Illa et al. 2021).
One of the position-selective chemical reactions of crystalline cellulose microfibril surfaces is 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO)-mediated oxidation under aqueous conditions (Isogai et al. 2018). When wood-cellulose fibers are subjected to TEMPO-mediated oxidation under suitable conditions, the oxidized cellulose fibers can be converted to TEMPO-oxidized cellulose nanofibrils (TO-CNFs) with homogeneous ~ 3-nm widths by mechanical disintegration in water (Isogai et al. 2011; Isogai 2021). Sodium glucuronosyl units are densely and position-selectively formed from glucosyl units present on the crystalline wood-cellulose microfibril surfaces by oxidation. Counterion exchange from sodium to diverse metal and alkylammonium ions at the carboxylate groups abundantly present on the TO-CNF surfaces under aqueous conditions can add new functionalities to TO-CNF materials (Isogai et al. 2018; Isogai 2021).
TEMPO-mediated oxidation has been applied to various BC materials to introduce sodium carboxylate groups densely onto the crystalline BC-microfibril surfaces, and TEMPO-oxidized BC (TO-BC) materials have been applied to medical, antibacterial, and functional materials (Ifuku et al. 2009; Luo et al. 2013; Lai et al. 2014; Feng et al. 2014; Elayaraja et al. 2017; Wu et al. 2018; Jun et al. 2019). TO-BC-related compounds have also been used as oil-in-water emulsion stabilizers (Jia et al. 2016; Wu et al. 2021), supercapacitors (Zhang et al 2017), and metal-nanoparticle-supports for immobilized catalysts (Chen et al. 2017), some of which have been reported in review papers (Kim et al. 2015; Parte et al. 2020). Some fundamental studies of TEMPO-oxidized BC materials in terms of the carboxy contents, oxidation conditions, morphologies, and surface properties have also been reported (Lai et al. 2013; Wu and Cheng 2017; Jun et al. 2017; do Nascimento et al. 2019; Chitbanyong et al. 2020; Yang et al. 2021).
In this study, the chemical structures and molar-mass parameters of TO-BC samples with different carboxy contents and molar masses prepared under different conditions were comprehensively investigated. Size-exclusion chromatography with multi-angle laser-light scattering and refractive-index detection (SEC/MALLS/RI) was used to determine the molar mass and molar-mass distributions of the TO-BC samples. The chemical and crystalline structures of the TO-BC samples and those ultrasonicated in water (TO-BC-U) samples were analyzed by X-ray diffraction (XRD) and solid-state 13C nuclear magnetic resonance (13C-NMR) spectroscopy. The morphologies of the TO-BC-U samples were observed by transmission electron microscopy (TEM) and atomic force microscopy (AFM).
Experimental
Materials
BC pellicles were obtained by stationary cultivation of Gluconacetobacter xylinus (JCM10150) under reported conditions at 28 °C for 1 week (Kim et al. 2002; Sun et al. 2017). The BC pellicles were cut into small pieces and purified according to the conventional method (Kim et al. 2002; Sun et al. 2017). The pieces were converted to gel particles by mechanical disintegration in water using a domestic blender, and they were washed thoroughly with water by filtration. The excess water in the gel particles was removed by filtration to prepare wet BC gel particles with ~ 2.5% (w/w) solid content. TEMPO, NaBr, 1.33 M NaClO solution, ethylene diamine (EDA), LiCl, N,N-dimethylacetamide (DMAc), and the other chemicals and solvents were laboratory grade (FUJIFILM Wako Pure Chemical, Co., Tokyo, Japan) and used as received.
Oxidation of BC
The gel particles containing 0.8 g BC (on dry weight) were suspended in water (80 mL in total), and TEMPO-mediated oxidation was applied to the BC gel particles with NaBr and NaClO in water at pH 10 and room temperature (Shinoda et al. 2012; Hiraoki et al. 2015). The amounts of NaClO added were 5 and 10 mmol/g-BC. Oxidation was quenched by adding a small amount of ethanol to the reaction mixture for complete consumption of any NaClO that might have remained in the mixture. After oxidation, one-pot reduction with NaBH4 was performed in the same container at pH 10 and room temperature for 3 h (Takaichi et al. 2014). A small amount of acetic acid was added to the mixture to decompose NaBH4, and the pH of the mixture was adjusted to ~ 2.5 with dilute HCl solution. The TO-BC gel particles were filtered on a 20-mesh nylon filter and washed thoroughly with water by filtration. Some of the TO-BC gel particles were stored at 4 °C in the never-dried state. The rest of the TO-BC gel particles were freeze-dried to measure the solid content. The TO-BC samples prepared with NaClO of 5 and 10 mmol/g-BC are called TO-BC-5 and TO-BC-10, respectively.
Preparation of TO-BC-U samples
The never-dried TO-BC-5 and TO-BC-10 gel particles were suspended in water at a solid content of ~ 0.06% (w/w), and the mixtures were subjected to ultrasonication at 4 °C for a total of 8 min in an ice bath (Zhou et al. 2018) to prepare aqueous dispersions of TO-BC-U. In laboratory experiments, ultrasonication can efficiently convert the TO-BC/water mixtures (50‒100 mL) into individual BC nanofibril/water dispersions in high yields. The TO-BC-U samples prepared from TO-BC-5 and TO-BC-10 are called TO-BC-5U and TO-BC-10U, respectively. Part of the TO-BC-U/water dispersion was freeze-dried.
Carboxy-content and neutral-sugar-composition analyses
The carboxy contents of the freeze-dried TO-BC samples (~ 0.2 g each) were determined by the conductivity titration method (Zhou et al. 2018). The neutral-sugar compositions of the freeze-dried BC and TO-BC samples were determined by the conventional acid-hydrolysis method and subsequent liquid chromatography using myo-inositol as an internal standard (Ono et al. 2016b).
SEC/MALLS/RI analysis
The freeze-dried BC was soaked in EDA at room temperature for 4 d, and then solvent-exchanged to DMAc through methanol and dissolved in 8% (w/w) LiCl/DMAc according to a previously reported method (Ono et al. 2016b; Ono and Isogai 2020). It took ~ 2 weeks to completely dissolve the EDA-treated BC in LiCl/DMAc. The carboxy groups in the freeze-dried TO-BC sample were position-selectively methyl-esterified with trimethylsilyl diazomethane in DMAc in the presence of a small amount of methanol (Hiraoki et al. 2015; Ono and Isogai 2020). After centrifugation of the mixture to remove excess amounts of the reagent and solvents, the carboxy-group-methylated TO-BC gel was washed thoroughly with ethanol and then solvent-exchanged to t-butanol several times by centrifugation followed by freeze-drying. The methyl-esterified TO-BC sample was completely dissolved in 8% (w/w) LiCl/DMAc by stirring the mixture for 1 d. The BC or methyl-esterified TO-BC solution was diluted to 1% (w/v) LiCl/DMAc with fresh DMAc, and the solution (100 µL) with ~ 0.05% (w/v) sample concentration was subjected to SEC/MALLS/RI analysis according to a previously reported method (Ono et al. 2016b; Ono and Isogai 2020). The values of 0.131 and 0.121 mL/g were used as the specific refractive index increments (dn/dc) of BC and methyl-esterified TO-BC, respectively, to calculate their number- and weight-average molar masses (Mn and Mw, respectively) by SEC/MALLS/RI analysis (Hiraoki et al. 2015; Ono et al. 2016a).
Solid-state 13C-NMR and XRD analyses
The solid-state 13C-NMR spectra of the freeze-dried samples were obtained with a JNM-ECA II 500 spectrometer (JEOL, Tokyo, Japan) according to a previously reported method (Funahashi et al. 2017; Zhou et al. 2020; Ono et al. 2021). The XRD patterns of pellets (~ 0.1 g each) of the freeze-dried samples were recorded based on a previously reported method (Ono et al. 2021). PeakFit software (Hulinks Co., Japan) was used for deconvolution of each solid-state 13C-NMR spectrum or XRD pattern.
TEM and AFM analyses of the BC and TO-BC-U samples
The never-dried BC/water dispersion with ~ 0.01% solid content was ultrasonicated, and large particles were removed by centrifugation at 2000 or 12,600 g for 10 min. The TO-BC-U/water dispersion with ~ 0.06% solid content was diluted to ~ 0.01% solid content, and the mixture was centrifuged at 12,600 g for 10 min. The supernatant (10 µL) of the BC/water or TO-BC-U/water dispersion was deposited on a TEM grid and air-dried. The sample on the grid was stained with 2% uranyl acetate (5 µL), and the air-dried sample grid was observed by TEM (JEM-1010, JEOL, Japan) at 100 kV (Ono et al. 2021, 2021a). The TO-BC-10U/water dispersion was diluted to 0.001–0.0001% (w/v) solid content with water, and it was deposited on a freshly cleaved mica surface at 50 µL/cm2 followed by air drying and then vacuum drying (Ono et al. 2021, 2022a, 2022b). The AFM images of the sample were obtained in tapping mode according to a previously reported method (Ono et al. 2021, 2022a, 2022b).
Results
TEMPO-mediated oxidation of BC
To prepare TO-BC-5 and TO-BC-10, it took 3 h and 6 h of TEMPO-mediated oxidation of the BC sample, respectively, until no consumption of 0.4 M NaOH, which was continuously added to the reaction mixture to maintain the pH at 10, was observed. The TO-BC samples were obtained almost quantitatively, and some weight losses may have occurred during the isolation, purification, and/or handling processes of the TO-BC gel particles. The freeze-dried TO-BC samples contained protonated carboxy groups (i.e., TO-BC-COOH structures), because the wet TO-BC samples were suspended in acidic water at pH ~ 2.5 followed by thorough washing with water.
The carboxy contents of the TO-BC-5 and TO-BC-10 samples were determined to be 1.6 and 1.5 mmol/g, respectively, by the conductivity titration method (Fig. S1 in the Electronic Supplementary Material). Thus, almost the maximum amounts of carboxy groups were likely to form in the TO-BC samples by TEMPO-mediated oxidation under the conditions used in this study.
The neutral-sugar compositions of the BC and TO-BC samples are given in Table 1. Neither galactose nor rhamnose was detected for BC. Mannose, arabinose, and xylose were detected in the chromatogram of the BC sample, although their amounts were significantly small. The 9.4% “others” fraction for the BC sample in Table 1 may have been caused by residual impurities, such as proteins, incomplete hydrolysis, and/or side reactions of, for example, formation of 5-hydroxymethyl furfural from glucose under acid-hydrolysis conditions.
The glucose contents of TO-BC-5 and TO-BC-10 were calculated to be 72% and 74%, respectively, based on their carboxy contents (1.6 and 1.5 mmol/g, respectively). However, complete hydrolysis of the glycoside bonds of the glucuronosyl units of TEMPO-oxidized native-cellulose samples is difficult (Fujisawa et al. 2010). Thus, not only glucuronosyl units, but also some glucosyl units linked to glucuronosyl units in the TO-BC samples could not be detected to be glucose by neutral sugar-composition-analysis. As a result, only glucose contents of ~ 60% were determined for the TO-BC samples, and the “others” contents were as high as ~ 40% (Ono et al. 2020, 2021a, 2021b).
SEC/MALLS/RI analysis of the BC and TO-BC samples
The SEC-elution patterns and corresponding molar-mass plots are shown in Fig. 1a. Since the molar-mass plots of the three samples linearly decreased with increasing SEC-elution volume, the BC and methyl-esterified TO-BC molecules were suitably separated in the SEC column depending on their molecular sizes. Figure 1a shows that each of the three samples showed a large main peak owing to the high-molar-mass (HMM) fraction and a small shoulder peak at 9‒10 mL SEC-elution volume. The peak position of the HMM fraction of BC shifted in the high-SEC-elution volume (or low molar mass) direction by TEMPO-mediated oxidation, which is reasonable because the presence of NaClO and/or NaBrO in the oxidation system causes partial depolymerization of cellulose molecules (Ono et al. 2021).

a SEC-elution patterns and corresponding molar-mass plots, and b double-logarithmic plots of the molar mass versus the root-mean-square radius (or conformation plots) for bacterial cellulose (BC), and TEMPO-oxidized and methyl-esterified BC (TO-BC-5 and TO-BC-10) samples
The double-logarithmic plots of the molar mass versus root-mean-square radius (or conformation plots) of the BC and methyl-esterified TO-BC samples are shown in Fig. 1b. All three samples exhibited almost straight lines with slope values of 0.57‒0.59, which indicates that the BC and methyl-esterified TO-BC molecules had linear random-coil conformations in 1% (w/w) LiCl/DMAc. The methyl-esterified TO-BC molecules had higher molar masses than the BC molecules at the same degree of polymerization. Thus, the conformation plots of the TO-BC samples shifted in the HMM direction compared with that of BC (Fig. 1b).
The Mw, Mn, and Mw/Mn values, and mass recovery ratios of the three samples calculated from the SEC/MALLS/RI data are summarized in Table 2. The molar mass decreased in the order of BC > TO-BC-5 > TO-BC-10. The molar mass of the TO-BC samples obtained by partial depolymerization was lower for higher added amount of NaClO and longer oxidation time. The ~ 0.05% (w/v) sample solution in 1% (w/v) LiCl/DMAc was prepared and filtered through a poly(tetrafluoroethylene) (PTFE) membrane with a 0.45-µm pore size. The filtered sample solution (100 µL) was then injected into the SEC/MALLS/RI system. Thus, the calculated mass or mass-recovery values of 43‒46 µg in Table 2 showed that the prepared BC and methyl-esterified TO-BC samples were almost quantitatively analyzed by the SEC/MALLS/RI system without significant mass loss by filtration with the PTFE membrane or SEC guard column.
XRD patterns of the BC, TO-BC, and TO-BC-U samples
The XRD patterns of the freeze-dried BC, TO-BC, and TO-BC-U samples are shown in Fig. S2 in the Electronic Supplementary Material. The crystal width (CW), which was calculated from the (2 0 0) diffraction peak by Scherrer’s equation (Alexander 1979), was approximately the same for the BC (5.4 nm) and TO-BC samples (5.5 nm), but it decreased to 5.0‒5.1 nm for the TO-BC-U samples. Thus, conversion from BC to TO-BC-U by TEMPO-mediated oxidation and successive ultrasonication in water resulted in a decrease in the CW when these values were calculated from the XRD patterns.
BC contains a mixture of crystal structures of cellulose Iα and Iβ (Wada et al. 1993, 2001; Castro et al. 2011). The d-spacings, which were calculated from the (1 − 1 0), (1 1 0), and (2 0 0) diffraction peak positions and the X-ray wavelength (0.1542 nm) of Cu Kα radiation using Bragg’s law (Wada et al. 2001), showed that all of the BC, TO-BC, and TO-BC-U samples had the cellulose-Iα-rich structures (Fig. S3 in the Supplementary Material).
Solid-state 13C-NMR analysis of the BC, TO-BC, and TO-BC-U samples
The solid-state 13C-NMR spectra of the five samples are shown in Fig. 2a, and the extended spectra for the C=O, C4, and C6 carbon atoms are shown in Fig. 2b. The crystalline cellulose Iα/Iβ ratios of various BC samples have been calculated from solid-state 13C-NMR spectra (Tokoh et al. 2002a, 2002b; Castro et al. 2011). All of the samples in Fig. 2a showed a single and sharp C1 signal, differing from the doublet C1 signals for cotton- and ramie-cellulose samples measured under the same conditions (Ono et al. 2021). The NMR and XRD results described in the above section showed that the cellulose Iα crystal structure was dominant in the BC, TO-BC, and TO-BC-U samples.

Solid-state 13C-NMR spectra of BC, TEMPO-oxidized BC (TO-BC) samples, and TEMPO-oxidized and then ultrasonicated BC (TO-BC-U) samples. a Wide and b extended chemical shift ranges
The peak of the C=O carbon atoms of the protonated carboxy groups of the TO-BC samples was located at ~ 172 ppm, while those of the sodium carboxylate groups of the TO-BC-U samples were located at ~ 175 ppm. The C=O carbon atoms of the TO-BC and TO-BC-U samples did not contain mixed C=O groups of protonated and sodium carboxylate groups from the single C=O peaks without a shoulder peak.
By deconvolution using Gaussian‒Lorentzian functions (Fig. S3 in the Electronic Supplementary Material) (Wickholm et al. 1998; Larsson et al. 2005; Foston 2014; Brinkmann et al. 2016; Ono et al. 2021), the two C4 signals for each sample centered at ~ 89 and ~ 85 ppm are ascribed to crystalline and non-crystalline cellulose molecules, respectively. The broad signal centered at ~ 84 ppm (Fig. 2b, bottom) is ascribed to the C4 carbon atoms of non-crystalline cellulose molecules present on the crystalline BC fibril surfaces. The non-crystalline C4 signals of the TO-BC and TO-BC-U samples were sharper than that of BC, and they had different signal peak positions of ~ 83 and ~ 84 ppm, respectively. The different chemical structures of the protonated C6-carboxy and sodium C6-carboxylate groups for the TO-BC and TO-BC-U samples, respectively, can be explained in terms of the relatively high and low electron densities of the C4 carbon atoms. These different electron densities of the C4 carbon atoms for TO-BC-COOH and TO-BC-U-COONa were consistent with those of the C = O carbon atoms, which had chemical shifts of ~ 172 and ~ 174 ppm, respectively.
The C6 signals were roughly separated into two signals by deconvolution (Fig. 2b). The large peak located at ~ 65 ppm is ascribed to the C6‒OH groups with the trans‒gauche (tg) conformation of crystalline cellulose I (Horii et al. 1983; Viëtor et al. 2002; Newman and Davison 2004; Yang et al. 2018; Funahashi et al. 2017). A shoulder peak ascribed to C6‒OH groups with the gauche‒trans (gt) and gauche‒gauche (gg) conformations was present at 60‒64 ppm. The shoulder C6 signal of BC decreased for the TO-BC and TO-BC-U samples because most of the C6‒OH groups with the gg conformation, which were exposed on the crystalline BC fibril surfaces, were oxidized to C6-carboxy groups by TEMPO-mediated oxidation (Funahashi et al. 2017).
Although the solid-state 13C-NMR spectra were not measured in quantitative mode in this study, the relative signal areas of the carbon atoms provide some information about the structural changes of BC by TEMPO-mediated oxidation and successive ultrasonication in water. The carbon signal areas were separated by deconvolution using Gaussian‒Lorentzian functions and the least-square method (Fig. S4 in the Electronic Supplementary Material). The relative signal areas of (C2 + C3 + C5)/3/C1 and C4/C1 were approximately one (Fig. 3a), which is reasonable. However, the relative signal area of C6/C1 for BC and those of (C6 + C=O)/C1 for the TO-BC and TO-BC-U samples were 0.74 and 0.73‒0.82, respectively, showing that the signal areas of C6‒OH groups were smaller than the expected values. This is probably because of the different cross-polarization effects between the CH carbon atoms for C1‒C5 and CH2 carbon atoms for C6 (Sparrman et al. 2019; Zhou et al. 2020).

Relative signal areas of the solid-state 13C-NMR spectra of BC, TEMPO-oxidized BC (TO-BC), and TEMPO-oxidized and then ultrasonicated BC (TO-BC-U) samples. a The ratios of (C2 + C3 + C5)/3/C1, C4/C1, C6/C1, C=O/C1, and (C6 + C=O)/C1, and b those of C4cry/C1, C4cry/C4, Ctg/C1, Ctg/C6, and C6gt,gg/C6
The relative signal areas of C4cry/C1 and C4cry/C4 can be regarded as the C4 crystallinities of cellulose I, because the samples shown in Fig. 3 contained only glucosyl and glucuronosyl units without any hemicelluloses or other components (Wickholm et al. 1998; Larsson et al. 2005; Foston 2014; Brinkmann et al. 2016; Ono et al. 2021). The relative signal areas of C6tg/C1 also indicate relative crystallinities of cellulose I between the samples, because the C6‒OH groups with the tg conformation are ascribed to those in the crystalline cellulose I structure (Funahashi et al. 2017). The crystallinities were approximately the same between the BC and TO-BC samples, showing that the TEMPO-mediated oxidation occurred only the crystalline BC fibril surfaces. However, the crystallinities of the TO-BC-U samples were lower than those of the BC and TO-BC samples, indicating that the ultrasonication of TO-BC in water caused some decreases in crystallinity (Fig. 3b).
The C6gt,gg/C6 signal area ratio decreased from 26% for BC to 6‒7% for the TO-BC samples because most of the C6‒OH groups with the gg conformation exposed on the crystalline BC fibril surfaces were oxidized to C6-carboxy groups by TEMPO-mediated oxidation (Funahashi et al. 2017). These values increased to 12‒13% for the TO-BC-U samples, indicating that the C6‒OH groups with the crystalline tg conformation in the TO-BC samples partly turned to those with gt and/or gg conformations by the ultrasonication in water. Correspondingly, the C6tg /C6 signal ratios increased from 74% for BC to 93‒94% for the TO-BC samples. These C6tg/C6 signal ratios decreased to 87‒88% for the TO-BC-U samples by ultrasonication of the TO-BC samples in water (Fig. 3b).
TEM images of the BC and TO-BC-U samples
After the dispersion was centrifuged at 2000 g, the ultrasonicated BC sample contained some large unfibrillated bundles with widths of > 100 nm (Fig. 4, top). These large fibril bundles were removed by centrifugation at 12,600 g, and the smallest fibrils widths were ~ 10 nm on average. Some twisted ribbon-like fibrils were observed (Fig. 4) (Tokoh et al. 2002a; Yamamoto et al. 2006; Castro et al. 2011; Pentilä et al. 2017).

TEM images of bacterial cellulose (BC) fibrils prepared from the supernatant after ultrasonication of the BC/water dispersion and subsequent centrifugation at 2000 g and 12,600 g. The arrows indicate single fibrils
TEM images of TO-BC-5U and TO-BC-10U are shown in Figs. 5 and 6, respectively. The fibril morphologies of the two samples were similar. Some ribbon-like fibril bundles with cracks along the longitudinal direction and branches were observed as twisted structures. Apparent single fibrils (indicated by arrows in Figs. 5 and 6) were also observed, and their widths were 5‒11 nm from the TEM images. Thus, formation of sodium carboxylate groups on the BC fibril surfaces by TEMPO-mediated oxidation and successive ultrasonication in water enhanced further fibrillation to fibrils with smaller widths. However, complete fibrillation of BC to individual fibrils with homogeneous widths without unfibrillated bundles or branches was not achieved.

TEM images of the TO-BC-5U prepared from TO-BC-5 by ultrasonication in water

TEM images of TO-BC-10U prepared from TO-BC-10 by ultrasonication in water
The aqueous dispersions of TO-BC-5U and TO-BC-10U with ~ 0.06% solid content prepared by ultrasonication in water were transparent. These dispersions became hazy after the solid content was increased to ~ 0.2% using a rotary evaporator, probably because of formation of TO-BC fibril networks with sizes larger than the wavelengths of visible light. The long TO-BC-U fibrils with lengths of > 2‒3 µm may have caused formation of such entangled fibril network structures at solid content of ~ 0.2%. Birefringence behavior was, however, slightly observed between the cross polarizers for the hazy TO-BC-U/water dispersion (Fig. S5).
AFM images of TO-BC-10U
The AFM height and phase images of TO-BC-10U are shown in Fig. 7a‒d. Similar results to those of the TEM images (Fig. 6) were obtained. The height profiles of the apparent single fibrils were measured along each fibril length, and they are shown in Fig. 7e. Although the data were somewhat scattered, the average fibril heights were ~ 3 nm.

AFM a, b height and c, d phase images of TO-BC-10U prepared from TO-BC-10 by ultrasonication in water. e Height profiles of single TO-BC-U fibrils
Discussion
Fibril structures of BC from the carboxy contents
The carboxy contents of TEMPO-oxidized cellulose (TOC) samples prepared from air-dried and commercial cotton and ramie fibers prepared with NaClO of 10 mmol/g-cellulose are ~ 1.6 mmol/g (Ono et al. 2021). The carboxy contents of TOC samples prepared from never-dried softwood bleached kraft pulps and eucalyptus cellulose with NaClO of 10 mmol/g-cellulose are 1.7‒1.8 mmol/g (Shinoda et al. 2012; Hiraoki et al. 2015; Ono et al. 2022a). Thus, the carboxy contents of the TO-BC samples prepared in this study were close to or slightly lower than those of other TEMPO-oxidized plant-cellulose samples.
These results indicate that the widths of crystalline BC fibrils, the surfaces of which are mostly oxidizable by TEMPO-mediated oxidation, are close to or slightly larger than those of plant-cellulose microfibrils. When the single and surface-oxidizable crystalline BC fibrils are assumed to have homogeneous widths with a hexagonal cross section (Castro et al. 2011; Zhou et al. 2020), the maximum carboxy contents of 1.5‒1.6 mmol/g for the TO-BC-U samples indicate that each BC fibril with the smallest width consists of 30 or 36 cellulose chains in the cross section (Fig. S6 in the Electronic Supplementary Material).
Crystallinities of the BC, TO-BC, and TO-BC-U samples from their solid-state 13C-NMR spectra
The values of C4cry/C1, C4cry/C4, and C6tg/C1 in Fig. 3b were 0.56‒0.65 for the BC and TO-BC samples, showing that the crystallinity of BC was almost unchanged by TEMPO-mediated oxidation. However, the values slightly decreased to 0.46‒0.54 for the TO-BC-U samples, indicating that the ultrasonication of the TO-BC samples in water caused partial de-crystallization of cellulose I.
Compared with the TO-BC samples, the relative signal area of C6gt,gg/C1 for the TO-BC-U samples were higher. Thus, the crystalline C6‒OH groups with the tg conformation in the BC and TO-BC samples (Horii et al. 1983; Viëtor et al. 2002; Newman and Davison 2004; Yang et al. 2018) partly turned to those of gt and/or gg conformations by ultrasonication in water. Correspondingly the relative signal areas of C6tg/C6 for the TO-BC-U samples were lower than those for the TO-BC samples.
Lengths of the TO-BC-U samples
The lengths of TO-BC-5U and TO-BC-10U could not be measured from TEM images because they were longer than 2‒3 µm. The DPw values of TO-BC-5 and TO-BC-10 were 1530 and 1120, respectively (Table 2), which correspond to lengths of ~ 760 and ~ 560 nm, respectively, based on the length of one glucosyl unit (~ 0.5 nm) in cellulose I (Zhou et al. 2018). These lengths calculated from the DPw values were clearly smaller than the TO-BC-U fibril lengths measured from TEM images. Thus, each TO-BC-U fibril consisted of multiple BC and TO-BC molecules that were packed along the longitudinal direction. Similar results have been obtained for TO-CNFs prepared from wood-cellulose and microcrystalline-cellulose samples (Shinoda et al. 2012; Zhou et al. 2018).
Comparison of BC, ramie, and wood cellulose samples
The characteristics of TEMPO-oxidized cellulose samples or TEMPO-oxidized cellulose fibrils prepared from BC, air-dried commercial ramie fibers, and a never-dried wood-cellulose sample with NaClO of 10 mmol/g-cellulose are listed in Table 3. Although the carboxy contents of the samples were in the range 1.5‒1.8 mmol/g, the wood-cellulose sample gave the highest DPw values because of the shortest oxidation time. The DPw value is lower for longer oxidation time because the presence of NaClO and/or NaBrO in the oxidation system causes depolymerization without TEMPO (Ono et al. 2021). The never-dried samples were used for the BC and wood-cellulose samples to obtain the results in Table 3. However, for the wood-cellulose sample, the presence of sponge-like structures of disordered hemicellulose molecules between the crystalline wood-cellulose microfibrils probably improve the efficiency of covalent bond formation between cationic TEMPO+ molecules and the C6‒OH groups present on the crystalline microfibril surfaces during oxidation. This covalent-bond formation is required as the initial stage of TEMPO-mediated oxidation of native-cellulose samples for preparation of TEMPO-oxidized cellulose materials and fibrils (Isogai et al. 2018).
Conclusions
TEMPO-mediated oxidation has been applied to never-dried BC gel particles with NaClO of 5 and 10 mmol/g-BC. The carboxyl contents of the TO-BC samples were 1.5‒1.6 mmol/g. These carboxy contents are likely to be the maximum values for oxidation of the C6‒OH groups on the crystalline BC fibril surfaces that are accessible to the TEMPO+ molecules in oxidation. The lengths of TO-BC-5U and TO-BC-10U measured from TEM images were greater than 2‒3 µm, and thus each TO-BC-U fibril consisted of multiple cellulose and oxidized-cellulose molecules that were packed along the longitudinal direction. The XRD patterns and solid-state 13C-NMR spectra of the BC, TO-BC, and TO-BC-U samples showed that cellulose Iα was the dominant crystalline structure, and no clear conversion from cellulose Iα to cellulose Iβ occurred during TEMPO-mediated oxidation of BC and subsequent ultrasonication in water. The solid-state 13C-NMR spectra showed that the crystallinities calculated from the C4cry/C1, C4cry/C4, and C6tg/C1 signal ratios were approximately the same between the BC and TO-BC samples, indicating that TEMPO-mediated oxidation occurred position-selectively at the C6‒OH groups on the crystalline BC fibril surfaces. However, the crystallinities slightly decreased for the TO-BC-U samples, indicating that ultrasonication of the TO-BC samples in water caused partial de-crystallization of cellulose I. Correspondingly, the C6‒OH groups with the crystalline tg conformation in the TO-BC samples partly turned to those with the gt and/or gg conformations by ultrasonication in water. From TEM and AFM images, the TO-BC-U samples contained not only single fibrils but also ribbon-like fibril bundles with twisted structures. Thus, complete individualization of BC to TO-BC-U fibrils with homogeneous widths was not achieved by TEMPO-mediated oxidation and successive ultrasonication in water, even though significant amounts of sodium carboxylate groups (1.5‒1.6 mmol/g) were present in the TO-BC samples. The single TO-BC-U fibril widths measured from AFM height images were ~ 3 nm, which are close to those of TO-CNFs prepared from wood cellulose. Thus, the fibrils with ~ 3-nm widths are probably the smallest crystalline elements in BC, the surfaces of which are selectively convertible to oxidized-cellulose molecules by TEMPO-mediated oxidation.
References
Alexander LE (1979) X-ray diffraction methods in polymer science. Robert E (Ed), Kreiger Publishing, Humington, pp 423–424
Brinkmann A, Chen M, Couillard M, Jakubek ZJ, Leng T, Johnston LJ (2016) Correlating cellulose nanocrystal particle size and surface area. Langmuir 32:6105–6114. https://doi.org/10.1021/acs.langmuir.6b01376
Campano C, Balea A, Blanco A, Negro C (2016) Enhancement of the fermentation process and properties of bacterial cellulose: a review. Cellulose 23:57–91. https://doi.org/10.1007/s10570-015-0802-0
Castro C, Zuluaga R, Putaux JL, Caro G, Mondragon I, Ganan P (2011) Structural characterization of bacterial cellulose produced by Gluconacetobacter swingsii sp. from Colombian agroindustrial wastes. Carbohydr Polym 84:96–102. https://doi.org/10.1016/j.carbpol.2010.10.072
Chen Y, Chen S, Wang B, Yao J, Wang H (2017) TEMPO-oxidized bacterial cellulose nanofibers-supported gold nanoparticles with superior catalytic properties. Carbohydr Polym 160:34–42. https://doi.org/10.1016/j.carbpol.2016.12.020
Chitbanyong K, Pisutpiched S, Khantayanuwong S, Theeragool G, Puangsin B (2020) TEMPO-oxidized cellulose nanofibril film from nano-structured bacterial cellulose derived from the recently developed thermotolerant Komagataeibacter xylinus C30 and Komagataeibacter oboediens R37–9 strains. Int J Biol Macromol 163:1908–1914. https://doi.org/10.1016/j.ijbiomac.2020.09.124
Czaja WK, Young D, Kawecki M, Bron RM Jr (2007) The future prospects of microbial cellulose in biomedical applications. Biomacromol 8:1–12. https://doi.org/10.1021/bm060620d
do Nascimento ES, Pereira ALS, de Oliveira BM et al (2019) TEMPO oxidation and high-speed blending as a combined approach to disassemble bacterial cellulose. Cellulose 26:2291–2302. https://doi.org/10.1007/s10570-018-2208-2
Elayaraja S, Zagorsek K, Li F, Xiang J (2017) In situ synthesis of silver nanoparticles into TEMPO-mediated oxidized bacterial cellulose and their antimicrobial activity against shrimp pathogens. Carbohydr Polym 166:329–337. https://doi.org/10.1016/j.carbpol.2017.02.093
Feng J, She Q, Li W, Shu X, Chen A, Xie X, Huang X (2014) Antimicrobial activity of silver nanoparticles in situ growth on TEMPO-mediated oxidized bacterial cellulose. Cellulose 21:4557–4567. https://doi.org/10.1007/s10570-014-0449-2
Foston M (2014) Advances in solid-sate NMR of cellulose. Curr Opin Biotechnol 27:176–184. https://doi.org/10.1016/j.copbio.2014.02.002
Fujisawa S, Isogai T, Isogai A (2010) Temperature and pH stability of cellouronic acid. Cellulose 17:607–615. https://doi.org/10.1007/s10570-010-9407-9
Funahashi R, Okita Y, Hondo H, Zhao M, Saito T, Isogai A (2017) Different conformations of surface cellulose molecules in native cellulose microfibrils revealed by layer-by-layer peeling. Biomacromol 18:3687–3694. https://doi.org/10.1021/acs.biomac.7b01173
Hiraoki R, Ono Y, Saito T, Isogai A (2015) Molecular mass and molecular-mass distribution of TEMPO-oxidized celluloses and TEMPO-oxidized cellulose nanofibrils. Biomacromol 16:675–681. https://doi.org/10.1021/bm501857c
Horii F, Hirai A, Kitamaru R (1983) Solid-state 13C-NMR study of conformations of oligosaccharides and cellulose - conformation of CH2OH group about the exo-cyclic C–C bond. Polym Bull 10:357–361. https://doi.org/10.1007/BF00281948
Huang Y, Zhu C, Yang J, Nie Y, Chen C, Sun D (2014) Recent advances in bacterial cellulose. Cellulose 21:1–30. https://doi.org/10.1007/s10570-013-0088-z
Ifuku S, Tsuji M, Morimoto M, Saimoto H, Yano H (2009) Synthesis of silver nanoparticles templated by TEMPO-mediated oxidized bacterial cellulose nanofibers. Biomacromol 10:2714–2717. https://doi.org/10.1021/bm9006979
Iguchi M, Yamanaka S, Budhiono A (2000) Review: bacterial cellulose—a masterpiece of nature’s arts. J Mater Sci 35:261–271. https://doi.org/10.1023/A:1004775229149
Illa MP, Peddapapannagari K, Raghavan SC, Khandelwal M, Sharma CS (2021) In situ tunability of bacteria derived hierarchical nanocellulose: current status and opportunities. Cellulose 28:10077–10097. https://doi.org/10.1007/s10570-021-04180-3
Isogai A (2021) Emerging nanocellulose technologies: recent developments. Adv Mater 33:2000630. https://doi.org/10.1002/adma.202000630
Isogai A, Saito T, Fukuzumi H (2011) TEMPO-oxidized cellulose nanofibers. Nanoscale 3:71–85. https://doi.org/10.1039/C0NR00583E
Isogai A, Hänninen T, Fujisawa S, Saito T (2018) Catalytic oxidation of cellulose with nitroxyl radicals under aqueous conditions. Prog Polym Sci 86:122–148. https://doi.org/10.1016/j.progpolymsci.2018.07.007
Jia Y, Zhai X, Fu W, Liu Y, Li F, Zhong C (2016) Surfactant-free emulsions stabilized by tempo-oxidized bacterial cellulose. Carbohydr Polym 151:907–915. https://doi.org/10.1016/j.carbpol.2016.05.099
Jun SH, Lee SH, Kim S, Park SG, Lee CK, Kang NK (2017) Physical properties of TEMPO-oxidized bacterial cellulose nanofibers on the skin surface. Cellulose 24:5267–5274. https://doi.org/10.1007/s10570-017-1508-2
Jun SH, Park SG, Kang NG (2019) One-pot method of synthesizing TEMPO-oxidized bacterial cellulose nanofibers using immobilized TEMPO for skincare applications. Polymers 11:1044. https://doi.org/10.3390/polym11061044
Kim DY, Nishiyama Y, Kuga S (2002) Surface acetylation of bacterial cellulose. Cellulose 9:361–367. https://doi.org/10.1023/A:1021140726936
Kim SS, Jeon JH, Kim HI, Kee CD, Oh IK (2015) High-fidelity bioelectronic muscular actuator based on graphene-mediated and TEMPO-oxidized bacterial cellulose. Adv Funct Mater 25:3560–3570. https://doi.org/10.1002/adfm.201500673
Klemm D, Heublein B, Fink HP, Bohn A (2005) Cellulose: fascinating biopolymer and sustainable raw material. Angew Chem Int Ed 44:3358–3393. https://doi.org/10.1002/anie.200460587
Lai C, Zhang S, Sheng L, Liao S, Xi T, Zhang Z (2013) TEMPO-mediated oxidation of bacterial cellulose in a bromide-free system. Colloid Polym Sci 291:2985–2992. https://doi.org/10.1007/s00396-013-3033-7
Lai C, Zhang S, Chen X, Sheng L (2014) Nanocomposite films based on TEMPO-mediated oxidized bacterial cellulose and chitosan. Cellulose 21:2757–2772. https://doi.org/10.1007/s10570-014-0330-3
Larsson PT, Westlund PO (2005) Line shapes in CP/MAS 13C NMR spectra of cellulose I. Spectrochim Acta Part A 62:539–546. https://doi.org/10.1016/j.saa.2005.01.021
Lin SP, Calvar IL, Catchmark JM, Liu JR, Demirci A, Cheng KC (2013) Biosynthesis, production and applications of bacterial cellulose. Cellulose 20:2191–2219. https://doi.org/10.1007/s10570-013-9994-3
Luo H, Xiong G, Hu D, Ren K, Yao F, Zhu Y, Gao C, Wan Y (2013) Characterization of TEMPO-oxidized bacterial cellulose scaffolds for tissue engineering applications. Mater Chem Phys 143:373–379. https://doi.org/10.1016/j.matchemphys.2013.09.012
Newman RH, Davidson TC (2004) Molecular conformations at the cellulose-water interface. Cellulose 11:23–32. https://doi.org/10.1023/B:CELL.0000014778.49291.c6
Ono Y, Isogai A (2020) Analysis of celluloses, plant holocelluloses, and wood pulps by size-exclusion chromatography/multi-angle laser-light scattering. Carbohydr Polym 251:117045. https://doi.org/10.1016/j.carbpol.2020.117045
Ono Y, Ishida T, Soeta H, Saito T, Isogai A (2016a) Reliable dn/dc values of cellulose, chitin, and cellulose triacetate dissolved in LiCl/N, N-dimethylacetamide for molecular mass analysis. Biomacromol 17:192–199. https://doi.org/10.1021/acs.biomac.5b01302
Ono Y, Tanaka R, Funahashi R, Takeuchi M, Saito T, Isogai A (2016b) SEC–MALLS analysis of ethylenediamine-pretreated native celluloses in LiCl/N, N-dimethylacetamide: softwood kraft pulp and highly crystalline bacterial, tunicate, and algal celluloses. Cellulose 23:1639–1647. https://doi.org/10.1007/s10570-016-0948-4
Ono Y, Takeuchi M, Zhou Y, Isogai A (2021) TEMPO/NaBr/NaClO and NaBr/NaClO oxidations of cotton linters and ramie celluloses. Cellulose 28:6035–3049. https://doi.org/10.1007/s10570-021-03944-1
Ono Y, Takeuchi M, Zhou Y, Isogai A (2022a) Characterization of cellulose and TEMPO-oxidized celluloses prepared from Eucalyptus globulus. Holzforschung 76:169–178. https://doi.org/10.1515/hf-2021-0159
Ono Y, Takeuchi M, Isogai A (2022b) Changes in neutral sugar composition, molar mass and molar mass distribution, and solid-state structures of birch and Douglas fir by repeated sodium chlorite delignification. Cellulose. https://doi.org/10.1007/s10570-022-04448-2
Parte FGB, Santoso SP, Chou CC, Verma V, Wang HT, Ismadji S, Cheng KC (2020) Current progress on the production, modification, and applications of bacterial cellulose. Crit Rev Biotechnol 40:397–414. https://doi.org/10.1080/07388551.2020.1713721
Pentilä PA, Imai T, Sugiyama J (2017) Fibrillar assembly of bacterial cellulose in the presence of wood-based hemicelluloses. Int J Biol Macromol 102:111–118. https://doi.org/10.1016/j.ijbiomac.2017.04.010
Shah N, Ul-Islam M, Khattak WA, Park JK (2013) Overview of bacterial cellulose composites: a multipurpose advanced material. Carbohydr Polym 98:1585–1598. https://doi.org/10.1016/j.carbpol.2013.08.018
Shinoda R, Saito T, Okita Y, Isogai A (2012) Relationship between length and degree of polymerization of TEMPO-oxidized cellulose nanofibrils. Biomacromol 13:842–849. https://doi.org/10.1021/bm2017542
Shoda M, Sugano Y (2005) Recent advances in bacterial cellulose production. Biotechnol Bioproc E 10:1–8. https://doi.org/10.1007/BF02931175
Sparrman T, Svenningsson L, Sahlin-Sjovold K, Nordstierna L, Westman G, Bernin DA (2019) Revised solid-state NMR method to assess the crystallinity of cellulose. Cellulose 26:8993–9003. https://doi.org/10.1007/s10570-019-02718-0
Sun SJ, Imai T, Sugiyama J, Kimura S (2017) CesA protein is included in the terminal complex of Acetobacter. Cellulose 24:2017–2027. https://doi.org/10.1007/s10570-017-1237-6
Tajima K, Tahara K, Ohba J, Kusumoto R, Kose R, Kono H, Matsushima T, Fushimi K, Isono T, Yamamoto T, Satoh T (2020) Detailed structural analyses of nanofibrillated bacterial cellulose and its application as binder material for a display device. Biomacromol 21:581–588. https://doi.org/10.1021/acs.biomac.9b0132
Takaichi S, Saito T, Tanaka R, Isogai A (2014) Improvement of nanodispersibility of oven-dried TEMPO-oxidized celluloses in water. Cellulose 21:4093–4103. https://doi.org/10.1007/s10570-014-0444-7
Tokoh C, Takabe K, Sugiyama J, Fujita M (2002a) Cellulose synthesized by Acetobacter xylinum in the presence of plant cell wall polysaccharides. Cellulose 9:65–74. https://doi.org/10.1023/A:1015827121927
Tokoh C, Takabe K, Sugiyama J, Fujita M (2002b) CP/MAS 13C NMR and electron diffraction study of bacterial cellulose structure affected by cell wall polysaccharides. Cellulose 9:351–360. https://doi.org/10.1023/A:1021150520953
Viëtor RJ, Newman RH, Ha MA, Apperley DC, Jarvis MC (2002) Conformational features of crystal-surface cellulose from higher plants. Plant J 30:721–731. https://doi.org/10.1046/j.1365-313X.2002.01327.x
Wada M, Sugiyama J, Okano T (1993) Native celluloses on the basis of two crystalline phase (Iα/Iβ) system. J Appl Polym Sci 49:1491–1496. https://doi.org/10.1002/app.1993.070490817
Wada M, Okano T, Sugiyama J (2001) Allomorphs of native crystalline cellulose I evaluated by two equatorial d-spacings. J Wood Sci 47:124128. https://doi.org/10.1007/BF00780560
Wickholm K, Larsson PT, Iversen T (1998) Assignment of noncrystalline forms in cellulose I by CP/MAS 13C NMR spectroscopy. Carbohydr Res 312:123–129. https://doi.org/10.1016/S0008-6215(98)00236-5
Wu CN, Cheng KC (2017) Strong, thermal-stable, flexible, and transparent films by self-assembled TEMPO-oxidized bacterial cellulose nanofibers. Cellulose 24:269–283. https://doi.org/10.1007/s10570-016-1114-8
Wu CN, Fuh SC, Lin SP, Lin YY, Chen HY, Liu JM, Cheng KC (2018) TEMPO-oxidized bacterial cellulose pellicle with silver nanoparticles for wound dressing. Biomacromol 19:544–554. https://doi.org/10.1021/acs.biomac.7b01660
Wu Y, Zhang X, Qiu D, Pei Y, Li Y, Li B, Liu S (2021) Effect of surface charge density of bacterial cellulose nanofibrils on the rheology property of O/W Pickering emulsion. Food Hydrocolloid 120:106944. https://doi.org/10.1016/j.foodhyd.2021.106944
Yamamoto H, Horii F, Hirai A (2006) Structural studies of bacterial cellulose through the solid-phase nitration and acetylation by CP/MAS 13C NMR spectroscopy. Cellulose 13:327–342. https://doi.org/10.1007/s10570-005-9034-z
Yang H, Wang T, Oehme D, Petridis L, Hong M, Kubicki JD (2018) Structural factors affecting 13C NMR chemical shifts of cellulose: a computational study. Cellulose 25:23–36. https://doi.org/10.1007/s10570-017-1549-6
Yang KY, Wloch D, Lee KY (2021) TEMPO-oxidized nanocellulose hydrogels and self-standing films derived from bacterial cellulose nanopaper. RSC Adv 11:28352-28360. https://doi.org/10.1039/D1RA04190H
Zhang T, Lang J, Liu L, Liu L, Li H, Gu Y, Yan X, Ding X (2017) Effect of carboxylic acid groups on the supercapacitive performance of functional carbon frameworks derived from bacterial cellulose. Chin Chem Lett 28:22122–22218. https://doi.org/10.1016/j.cclet.2017.08.013
Zhou Y, Saito T, Bergström L, Isogai A (2018) Acid-free preparation of cellulose nanocrystals by TEMPO oxidation and subsequent cavitation. Biomacromol 19:633–639. https://doi.org/10.1021/acs.biomac.7b01730
Zhou Y, Fujisawa S, Saito T, Isogai A (2019) Characterization of concentration-dependent gelation behavior of aqueous 2,2,6,6-tetramethylpiperidine-1-oxyl-cellulose nanocrystal dispersions using dynamic light scattering. Biomacromol 20:750–757. https://doi.org/10.1021/acs.biomac.8b01689
Zhou Y, Ono Y, Takeuchi M, Isogai A (2020) Changes to the contour length, molecular chain length, and solid-state structures of nanocellulose resulting from sonication in water. Biomacromol 21:2346–2355. https://doi.org/10.1021/acs.biomac.0c00281
Acknowledgments
The authors thank Edanz for editing a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. The manuscript was approved by all authors for publication.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ono, Y., Takeuchi, M., Kimura, S. et al. Structures, molar mass distributions, and morphologies of TEMPO-oxidized bacterial cellulose fibrils. Cellulose 29, 4977–4992 (2022). https://doi.org/10.1007/s10570-022-04617-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-022-04617-3