
Abstract
Deuterium exchange in a deuterium oxide (D2O) atmosphere (95 % relative humidity), quantified by a dynamic vapor sorption (DVS) apparatus, was applied for assessing the accessibility of hydroxyl groups in birch kraft pulps. Achieving the maximum deuteration level exhibited slower kinetics than was earlier reported for experiments with ground wood and bacterial cellulose. The deuterium exchange process followed two parallel phenomena. Applying multiple drying and rewetting cycles gave kinetic information also on the hornification phenomenon occurring during these cycles. Dry birch pulps treated with sodium hydroxide solution of varying alkalinities at elevated temperatures were assessed for their accessible hydroxyl groups by DVS with deuterium exchange. This method was evaluated against deuteration combined with Fourier transform infra-red spectroscopy and water retention value (WRV). DVS measurements were in correlation with WRV and both the methods indicated that an alkaline treatment of dry birch pulp improves cellulose accessibility. The level of irreversible deuteration also decreased as the alkalinity was increased. DVS was shown to provide quantitative information on the accessibility but to be a time-consuming method for the pulp samples. A potential means to decrease the duration of the measurement is increased D2O exposure by excluding the drying phases.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cellulose accessibility, a term for the accessibility of the hydroxyl groups in cellulose, has been of increasing interest lately. Manufacture of novel cellulosic products, such as biofuels, nanocrystals and nanofibrils, require chemical modification of cellulose or even its dissolution in some cases. Cellulose accessibility can be considered a decisive factor during these heterogeneous chemical or biochemical treatments. In the past, cellulose accessibility was of interest mainly because of the importance of water retention properties of pulps in papermaking. Development of the reactivity of pulps during their processing has been studied widely (Pönni et al. 2012), e.g., during drying (Minor 1994; Nazhad and Paszner 1994; Weise 1998; Fernandes Diniz et al. 2004), chemical pulping (Hult et al. 2001; Fahlén and Salmén 2003, 2005; Pönni et al. 2014), and paper recycling (Howard 1990; Nazhad and Paszner 1994). The factors affecting the accessibility include pulp properties, such as hemicellulose (Oksanen et al. 1997) and acidic group content (Lindström and Carlsson 1982; Lindström 1992), and process parameters, such as temperature and pH (Maloney and Paulapuro 2000; Fahlén and Salmén 2003). Despite the vast research in the past, the question still remains: how to improve cellulose accessibility?
Alkaline treatments are known to improve cellulose accessibility (Klungness 1974; Lindström and Carlsson 1982; Lindström 1992; Weise et al. 1998). Especially strongly alkaline pretreatments for cellulosic raw materials have been studied since the mercerization process was developed (Mercer 1851) to improve the strength and appearance of cotton fibers. In mercerization (>8–9 % NaOH) the most severe changes occur in the crystallinity of cellulose (McKenzie and Higgins 1958; Philipp et al. 1959; Dinand et al. 2002; Kljun et al. 2011). The strong alkaline treatment swells the native cellulose I which after washing forms another allomorph, cellulose II (Klemm et al. 1998). In cellulose I the polymer chains are parallel, whereas cellulose II is considered to be formed of antiparallel chains (Nishiyama et al. 2000; Dinand et al. 2002).
Swelling of cellulose, in general, refers to the loosening of the intermolecular interactions relative to the competing interactions with the swelling agent, commonly water. Alkali improves the swelling of the aqueous cellulose suspensions due to ionization of the hydroxyl groups and cleavage of inter- and intra-molecular hydrogen bonds (Klemm et al. 1998). The maximum in swelling is obtained at approximately 10 % NaOH concentration, slightly depending on the cellulosic raw material (Saito 1939). In addition, the alkaline treatments are known to dissolve hemicelluloses (Sjöström and Enström 1967).
To date, there has not been a single method that could directly quantify the amount of accessible hydroxyl groups in cellulosic materials, i.e. cellulose accessibility. Jayme (1944) introduced the first widely accepted analytical method to approximate the accessibility of cellulose, namely the water retention value (WRV), lately standardized as the ISO 23714:2007 method. WRV describes the amount of water retained on pulp under standard centrifugal forces. Commonly, WRV is approximated to present the amount of water inside the cell wall (Jayme 1944, 1958). Fiber saturation point (FSP), measured as a volume that high molecular weight dextran cannot occupy, is another widely accepted method to quantify the cell wall bound water (Stone and Scallan 1967; Maloney et al. 1999). In addition, cellulose accessibility has been related with pore size distribution of fiber wall measured by a number of techniques including solute exclusion (Stone and Scallan 1967, 1968), thermoporosimetry conducted with differential scanning calorimetry (DSC) by the isothermal melting technique (Maloney et al. 1998; Wang et al. 2003), DSC combined with thermogravimetric analysis (TGA) (Park et al. 2006), nuclear magnetic resonance (NMR) cryoporosimetry (Gane et al. 2004; Östlund et al. 2010), and inverse size-exclusion chromatography (ISEC) (Berthold and Salmén 1997). In addition, many techniques that can visualize changes in the size and aggregation of microfibrils have been used to show reduction in cellulose accessibility. These methods include transmission electron microscopy (TEM) (Billosta et al. 2006), NMR spectroscopy, and atomic force microscopy (AFM) (Hult et al. 2001; Fahlén and Salmén 2003, 2005; Lee et al. 2007).
A more direct method to analyze the changes in cellulose accessibility is deuteration combined with Fourier transform infra-red (FT-IR) spectroscopy, a technique that has been widely applied for native cellulose, cellulose derivatives, pulp, and wood (Jeffries 1963; Tsuchikawa and Siesler 2003a, b; Hofstetter et al. 2006). Lately, this method was successfully implemented to evaluate changes in cellulose accessibility during various treatments of wood and pulps including their drying and wet thermal treatments (Suchy et al. 2010a, b; Pönni et al. 2013, 2014). The method is based on deuteration of the accessible hydroxyl groups prior to the treatment to be studied (Frilette et al. 1948; Mann and Marrinan 1956). After the treatment in deuterium oxide (D2O) and subsequent washing with water, the extent of the irreversible deuteration can be considered as a measure of decrease in the accessibility of hydroxyl groups in the sample. However, this method has so far not been standardized to give a precise number for the change in the accessible hydroxyl groups.
Recently, dynamic vapor sorption (DVS) with deuterium exchange was applied for bacterial cellulose and wood to determine the amount of their accessible hydroxyl groups (Lee et al. 2011; Rautkari et al. 2013). When protons in the accessible hydroxyl groups are exchanged for deuterons, the mass of the sample increases and this increase can be quantified with a highly sensitive ultrabalance in the DVS apparatus. On contrast to other methods, DVS with deuterium exchange provides the actual number of accessible hydroxyl groups in the sample. The principle of the method was proposed already in the 1960s (Morrison 1960; Sepall and Mason 1961). Previously, DVS has been applied commonly to assess the water vapor sorption behavior of various materials including wood (Sharratt et al. 2011; Hill et al. 2012), natural fibers (Kohler et al. 2003; Xie et al. 2011; Hill et al. 2009; Okubayashi et al. 2004, 2005) and microcrystalline cellulose (Kachrimanis et al. 2006).
In this paper deuterium exchange in DVS was applied for quantifying accessible hydroxyl groups in birch kraft pulp. WRV and deuteration combined with FT-IR spectroscopy were used as reference methods. The potential of deuterium exchange coupled with DVS for studying drying induced changes in cellulosic materials is discussed. The advantages and disadvantages of the method are critically reviewed.
Experimental
Materials
Industrially dried fully bleached birch pulp (kappa number 0.7, viscosity 674 ml g−1) was obtained from a Finnish pulp mill. The dry pulp was soaked overnight in an excess of water and manually disintegrated. To set the alkalinity during alkaline pretreatments of the pulp, 8 M aqueous NaOH was prepared from solid NaOH pellets (VWR, Leuven, Belgium). For converting the pulp to the Na+-form for WRV analysis an aqueous solution of NaHCO3 was prepared from solid 99.5 % NaHCO3 (Merck, Darmstadt, Germany). In addition to NaHCO3, 0.1 M NaOH and HCl were applied (Merck, Darmstadt, Germany). Water was purified in a Milli-Q system (Millipore Corporation, resistivity 18.2 MΩ cm). D2O (99.9 atom% D, Sigma Aldrich, St. Louis, USA) was used for deuteration.
DVS with deuterium exchange
The amount of accessible hydroxyl groups in the pulp was quantified by measuring its mass increase during deuterium exchange in a DVS elevated temperature (ET) apparatus (Surface Measurement Systems, UK) with a measuring accuracy of 0.1 μg and chamber volume of 100 cm3. Approximately 10–30 mg of pulp was placed in the sample pan and preconditioned at 0 % relative humidity (RH) with dry nitrogen gas flow (200 cm3 min−1) at 25 °C until the change in the mass of the sample was <0.002 % min−1 over a 30 min period. Multiple (15–30) adsorption–desorption cycles with alternating RH between 0 and 95 % (dry N2 and D2O vapor) were then conducted. During the cycles, the DVS apparatus maintained a constant RH until the change in the sample mass was <0.002 % min−1 over a 10 min period to ensure a constant equilibrium condition after each RH step. The amount of accessible hydroxyl groups in the sample was calculated according the Eq. 1 with the approximation that atomic mass difference between deuterium (2H) and protium (1H) is 1 g mol−1:
where A is the amount of accessible hydroxyl groups per dry mass of the sample, mi is the dry weight of the sample prior to exposure to D2O vapor, and mf is the dry weight of the sample after the multiple DVS cycles. Scheme 1 illustrates the conversion of the accessible OH groups into OD groups as the RH of the mixture of dry N2 and D2O vapor is increased from 0 to 95 % in the DVS chamber.

Principle of the deuteration of accessible hydroxyl groups during exposure to D2O vapor at 95 % RH in DVS chamber
Alkali treatment of pulp in D2O and water
The disintegrated pulp was treated in plastic bags for a period of 2 × 20 min with an excess of D2O, namely 20 ml of D2O per 1 g of dry pulp to convert all the accessible hydroxyl groups to OD groups. The sufficiency of time for the reversible deuteration and reprotonation was discussed in an earlier paper (Suchy et al. 2010b). The slurry was mixed twice during the 20 min by kneading. Between the deuteration treatments, D2O was squeezed out of the bag before fresh D2O was added. After the deuteration, the pulp was immediately subjected to an alkaline treatment.
The deuterium exchanged pulp was treated at 5 % consistency in D2O and varying alkalinity (0.25–2 M) in plastic bags in a temperature controlled oven at 90 °C for 4 h. The influence of the applied aqueous NaOH solution on the level of deuteration was taken into account when processing the data. After the treatment, the pulps treated were washed twice with 100 ml of water per 1 g of dry pulp, allowing the suspension to stand for 20 min at room temperature before filtration. The alkali treatments for WRV and DVS analysis were carried out similarly but using water instead of D2O. Scheme 2 illustrates the principle of deuteration of cellulose microfibrils, their aggregation in a subsequent treatment in the presence of D2O, and the final washing with H2O to remove deuterium in the non-aggregated areas (Suchy et al. 2010b; Pönni et al. 2013). The difference between the methods illustrated in Schemes 1 and 2 is the measured value, which in Scheme 1 is the amount of accessible hydroxyl groups in the sample, whereas Scheme 2 gives the amount of inaccessible OD formed during the studied treatment. Thus, FT-IR spectroscopy combined with deuteration has the limitation to detect only the changes within a certain treatment. In addition, the deuterium exchange is much slower in the vapor state compared to the liquid state (Mann and Marrinan 1956).

Deuteration of cellulose microfibrils, and their aggregation in a subsequent treatment in the presence of D2O followed by exchange of the accessible deuterium by water
FT-IR PAS spectroscopy
Prior to the FT-IR spectroscopy, the samples were dried at 40 °C in an oven. The spectra were collected using a Bio-Rad FTS 6000 spectrometer (Cambridge, MA, USA) with a Gasera PA301 photoacoustic cell (Turku, Finland) at a constant mirror velocity of 5 kHz, 1.2 kHz filter, and 8 cm−1 resolution. In the beginning of each set of measurements, a background spectrum was collected using a standard carbon black. The photoacoustic cell was purged with helium gas for 30 s prior to each measurement. 200 scans per spectrum were collected using the Win-IR Pro 3.4 software (Digilab, Holliston, MA, USA). Every sample was measured in duplicate and the averaged spectra were baseline corrected and normalized for cellulose band height at 1,200 cm−1 (Hofstetter et al. 2006), using Grams/AI 9.00 software (Thermo Fischer Scientific Inc., Waltham, MA, USA).
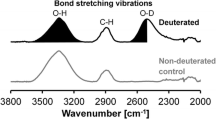
The area of the OD stretch band at around 2,500 cm−1 in the FT-IR spectrum was integrated using Grams/AI 9.00 software (Thermo Fischer Scientific Inc., Waltham, MA, USA). This band does not overlap with any other band in the FT-IR spectrum and, thus the normalized area of the OD stretch band measures the extent of irreversible deuteration (Suchy et al. 2010b).
WRV analysis
Prior to the analysis, the pulp samples were converted to their Na+-form. All of the subsequent treatments were done at 1 % consistency and each exchange of solution was done after filtration on a Büchner funnel. First, the samples were converted to their protonated form by a treatment in 0.01 M HCl for 1 h. Then, the samples were washed twice with water. Conversion to the Na+-form was done in 0.001 M NaHCO3 for 2 h with the pH adjusted to 9.5–10 with 0.1 M NaOH. Then, the samples were washed with water until the conductivity of the slurry was lower than 5 μS cm−1. The WRV analysis was done according to the standard SCAN-C 102 XE with a Jouan GR 4.22 centrifuge. The standard deviation was determined from four parallel measurements.
Results and discussion
Deuterium exchange
Deuterium exchange with multiple drying and rewetting cycles has been applied earlier for studying wood samples (Lee et al. 2011; Rautkari et al. 2013). It has also been shown that critical RH above which no further deuteration occurs is 60 % (Taniguchi et al. 1978). Figure 1 illustrates changes in the mass of birch pulp during the first and last three cycles of a 30 cycle DVS measurement. In order to quantify the amount of accessible hydroxyl groups, the increase in the dry mass of the sample after each cycle was recorded. Figure 2 illustrates this increase after the exposure to D2O vapor during these cycles. The increase in dry mass appeared to follow a multicomponent exponential function (parallel exponential kinetics). Thus, the following equation (Eq. 2) was fitted to the results:
where, Δm is the increase in mass at infinite time (x) of exposure to a constant RH, Δm0 is the increase in mass during the first cycle, Δm1 and Δm2 are the increases in mass at infinite time associated with fast and slow phenomena respectively, and 1/x1 and 1/x2 are the rate constants for these phenomena.

The first and last three cycles of a 30 cycle DVS experiment with birch kraft pulp. The lower and upper values express the sample mass in dry (RH 0 %) and humid (RH 95 %) conditions

Percentual increase in the dry mass (Δm) of birch kraft pulp as a function of the number of drying (N2, RH 0 %) and rewetting [D2O (square) and H2O (circle) vapor in N2, RH 95 %] cycles in DVS
The deuterium exchange was earlier suggested to consist of two parallel phenomena; a fast reaction corresponding to the exchange of protons in the amorphous regions and a slow reaction corresponding to the exchange on the surface of the crystalline regions (Mann and Marrinan 1956; Jeffries 1963). Hofstetter et al. (2006) proposed the two phenomena to be due to the exchange at the surfaces (fast) and at the interiors (slow) of the cellulose microfibrils. The deuteration of the less-ordered regions of cellulose, i.e. fast process, was considered to be complete and to occur within some minutes to 1 day depending on the conditions, e.g., temperature (Jeffries 1963). Jeffries (1963) suggested the main reason for the slow exchange in the crystalline regions to be the low concentration of D2O within these regions. Even exchange in the liquid state proceeds through fast and slow kinetics (Frilette et al. 1948). In this case, the fast exchange is considered to be diffusion controlled and to occur within a few minutes (Frilette et al. 1948; Tsuchikawa and Siesler 2003a, b). The slow exchange was shown to proceed even after 1 week of exposure to liquid D2O (Frilette et al. 1948). The slow reaction was not reported in previous studies on deuterium exchange in bacterial cellulose and wood powder (Lee et al. 2011; Rautkari et al. 2013). Thus, the more pronounced slow exchange in the birch kraft pulp is suggested to be due to the chemical processing and drying that the pulp has undergone.
In order to verify the deuterium exchange as the cause for the mass increase, a 30 cycle DVS trial for the birch pulp was conducted with water vapor. Prior to the experiment, the sample was dried over a period of time significantly longer to that applied between the cycles. Thus, the observed initial small mass increase could only indicate that the sample was not completely dried between the cycles (Fig. 2). As the water vapor cycles were continued, the dry mass of the sample slowly decreased. This might be due to hornification, which reduces the number of accessible hydroxyl groups and changes the fiber structure e.g., by pore closure (Higgins and McKenzie 1963; Scallan 1974). In any case, the mass increase during the first cycle with water vapor was only approximately 5 % of the mass increase obtained with D2O vapor (Fig. 2). Thus, the mass increase in D2O vapor must be mainly due to the deuterium exchange.
The necessity of the multiple drying and rewetting cycles was evaluated by applying a series of only two extended cycles having the overall exposure time with D2O comparable to that of the experiments with the shorter cycles. The amount of accessible hydroxyl groups determined after these extended cycles was within the standard deviation of the experiment conducted with 30 cycles but with similar overall exposure time for D2O (Table 1). Thus, the measurement is not dependent on the drying and rewetting, but on the extended exposure to D2O to allow also the slow exchange to occur. This was also found earlier by Jeffries (1963). More recently, the deuterium exchange of fully amorphous cellulosic films with D2O vapor was reported to occur within 10 h (Hishikawa et al. 1999).
Even though the extended cycles gave similar results, we chose to proceed with 15 cycles experiments, in order to have the measurement resemble the earlier work conducted on DVS with deuterium exchange (Lee et al. 2011; Rautkari et al. 2013). The duration for recording the 15 cycles was approximately 6 days. Three parallel measurements were completed for each sample. Thus, the overall duration for one sample was around 18 days. Moreover, the measurement was very sensitive to any unexpected disturbance and occasionally the measured value was out of range of the parallel measurements. These values were excluded from the data.
Hornification
In addition to the increase in dry mass due to the exchange of protons with deuterons, the DVS data showed a decrease in the wet mass of the pulp, i.e. its equilibrium moisture content (EMC), with time during the repeated humidity cycles as well as a decrease in the duration to reach the EMC at the high humidity. The decrease in the wet mass is most likely due to the drying induced hornification. Even though the raw material was industrially dried birch kraft pulp, the cellulose microfibril aggregation, i.e. hornification, proceeds as the drying cycles are continued. Previously, hornification was reported to be most severe during the first cycle but to proceed to a smaller extent with multiple drying and rewetting cycles (Laivins and Scallan 1993; Wistara and Young 1999). Changes in WRV of regenerated cellulosic fibers were reported to be most pronounced during the first five drying and rewetting cycles and to stabilize after around 15 cycles (Siroká et al. 2012). The decrease in the obtained moisture content at 95 % RH appears to follow an exponential function (Fig. 3). Thus, a corresponding equation (Eq. 3) was fitted with the results:
where EMC is the moisture content at infinite time of exposure to a constant RH, EMC0 is the initial moisture content, EMC1 is the moisture contents at infinite time associated with hornification, and 1/x1 is the rate constant related to the rate of the hornification. As expected, the parameters were hardly affected by the sorbate (water or D2O) and the exponential function (Eq. 3) approached EMC = 14 % in both cases.

Reduction in EMC of birch kraft pulp at 95 % RH after repeated DVS cycles in D2O vapor
The DVS cycles have earlier been modeled with parallel exponential kinetics (Kohler et al. 2003; Kachrimanis et al. 2006; Hill et al. 2010; Xie et al. 2011). The hornification phenomenon can thus be observed by applying the model (Eq. 4) for individual DVS cycles:
where EMC is the moisture content at infinite time of exposure to a constant RH, EMC0 is the initial moisture content, EMC1 and EMC2 are the moisture contents at infinite time associated with the fast and slow phenomena respectively, and 1/t1 and 1/t2 are rate constants for the fast and the slow phenomena, respectively. Information on the dynamics of hornification can be obtained by observing changes in parameters EMC1, EMC2, 1/t1 and 1/t2 as the function of repeated DVS cycles (Fig. 4). Both rates increase with repeated DVS cycles. Interestingly, the extent of the slower phenomenon (EMC2) is unaffected by the continuing repetitions on contrary to the faster hydration the extent of which (EMC1) decreases linearly with time. Thus, the slow phenomenon, unlike the faster one, appears not to be related with hornification.

Effect of repeated DVS cycles on dynamics of hydration and EMC of birch kraft pulp. The constants of the PEK model; the infinite moisture contents a for the fast reaction (EMC1) (square) and for the slow reaction (EMC2) (circle) and the reaction rates b for the fast reaction (1/t1) (square) and for the slow reaction (1/t2) (circle); applied to 30 DVS cycles for a dry birch pulp sample
Comparison between pulps
The idea in developing the DVS analysis coupled with deuterium exchange was to have a method to assess the reactivity of different pulps. An alkaline treatment series was performed for the dry bleached birch pulp because such treatments are often used to activate pulps for increased reactivity. The series had varying alkalinities between 0.25 and 2 M NaOH concentrations. The temperature during the treatments was 90 °C, duration 4 h, and consistency 5 % in all of the experiments. Figure 5 illustrates the extent of irreversible deuteration (alkali treatments in D2O) as observed by the integrated area of the FT-IR band at 2,500 cm−1 and WRV (alkali treatments in water) for the treated pulps. The effect of the aqueous NaOH solution applied in the D2O experiments was taken into account according to the applied molar ratios. WRV increased as a function of alkalinity while the level of irreversible deuteration was decreased. From previous research, it is known that WRV increases during an alkali treatment of dry pulps, through increased swelling (Weise et al. 1998). Irreversible deuteration, measured after washing the alkali treated pulp with water, was previously shown to be mostly reversed in a second alkali treatment in water (Pönni et al. 2012). Thus, the alkali treatment was suggested to induce an equilibrium state between the microfibrils and microfibril aggregates. It was also shown, that during kraft pulping, where cellulose microfibrils aggregate due to the removal of lignin and hemicelluloses, the extent of irreversible deuteration decreases as the alkali concentration approaches the mercerization level (Pönni et al. 2014). Thus, the irreversible deuteration under the conditions applied in this study should present an equilibrium state between the microfibrils and microfibril aggregates. Obviously, the equilibrium is shifted towards decreased aggregation as the alkalinity is increased.

The level of deuteration observed as the OD band area at 2,500 cm−1 (square) and WRV (circle) for dry birch pulps treated at various NaOH concentrations for 4 h at 90 °C
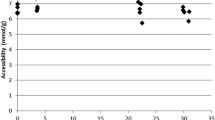
The number of accessible hydroxyl groups was determined for the dry birch pulp and the pulps treated with 0.25 and 2 M NaOH and correlated with WRV (Fig. 6). Only these few samples were tested due to the long time needed for the determination of the accessible hydroxyl groups by DVS. Regardless, the correlation provides supporting evidence for WRV as an approximate measure of cellulose accessibility for dry and alkaline treated birch pulps. In addition, Fig. 6 suggests an increase in the amount of accessible hydroxyl groups during the alkaline treatment. This increase is visible even though strong alkali treatments are known to remove the highly accessible hemicelluloses (Sjöström and Enström 1967).

The amount of accessible hydroxyl groups as a function of WRV for dry birch kraft pulp and the same pulp treated with 0.25 and 2 M NaOH at 90 °C for 4 h
Conclusions
The use of a DVS apparatus with deuterium exchange is a powerful analytical tool to quantify the accessible hydroxyl groups in chemical pulps. The weakness of the measurement, at the moment, is its long duration. The measurement does not depend on the number of drying and rewetting cycles, but on the overall time of exposure to D2O vapor. The deuteration follows parallel exponential kinetics, which is in agreement with the earlier finding on fast and slow exchange of protons in the amorphous regions and crystallite surfaces, respectively. In addition to the number of accessible hydroxyl groups, DVS gives also information on the dynamics and extent of hornification during repeated humidity ramps. The number of accessible hydroxyl groups in dry pulp correlates with the traditional measure of cell wall bound water, the WRV. In future, DVS in D2O vapor could be used to calibrate OD group measurement by FT-IR spectroscopy. This would enable a quantitative assessment of deuterium intake in liquid D2O, too.
References
Berthold J, Salmén L (1997) Inverse size exclusion chromatography (ISEC) for determining the relative pore size distribution of wood pulps. Holzforschung 51:361–368
Billosta V, Brändström J, Cochaux A, Joseleau J-P, Ruel K (2006) Ultrastructural organization of the wood cell wall can explain modifications caused in fibers during the pulping process. Cell Chem Technol 40:223–229
Dinand E, Vignon M, Chanzy H, Heux L (2002) Mercerization of primary wall cellulose and its implication for the conversion of cellulose I → cellulose II. Cellulose 9:7–18
Fahlén J, Salmén L (2003) Cross-sectional structure of the secondary wall of wood fibers as affected by processing. J Mater Sci 38:119–126
Fahlén J, Salmén L (2005) Pore and matrix distribution in the fiber wall revealed by atomic force microscopy and image analysis. Biomacromolecules 6:433–438
Fernandes Diniz JMB, Gil MH, Castro JAAM (2004) Hornification—its origin and interpretation in wood pulps. Wood Sci Technol 37:489–494
Frilette VJ, Hanle J, Mark H (1948) Rate of exchange of cellulose with heavy water. J Am Chem Soc 70:1107–1113
Gane PAC, Ridgway CJ, Lehtinen E, Valiullin R, Furó I, Schoelkopf J, Paulapuro H, Daicic J (2004) Comparison of NMR cryoporometry, mercury intrusion porosimetry, and DSC thermoporosimetry in characterizing pore size distributions of compressed finely ground calcium carbonate structures. Ind Eng Chem Res 43:7920–7927
Higgins HG, McKenzie AW (1963) The structure and properties of paper XIV. Effects of drying on cellulose fibres and the problem of maintaining pulp strength. Appita 16:145–164
Hill CAS, Norton A, Newman G (2009) The water vapor sorption behavior of natural fibers. J Appl Polym Sci 112:1524–1537
Hill CAS, Norton A, Newman G (2010) The water vapour sorption properties of Sitka spruce determined using a dynamic vapour sorption apparatus. Wood Sci Technol 44:497–514
Hill CAS, Ramsay J, Keating B, Laine K, Rautkari L, Hughes M, Constant B (2012) The water vapour sorption properties of thermally modified and densified wood. J Mater Sci 47:3191–3197
Hishikawa Y, Togawa E, Kataoka Y, Kondo T (1999) Characterization of amorphous domains in cellulosic materials using a FTIR deuteration monitoring analysis. Polymer 40:7117–7124
Hofstetter K, Hinterstoisser B, Salmén L (2006) Moisture uptake in native cellulose—the roles of different hydrogen bonds: a dynamic FT-IR study using Deuterium exchange. Cellulose 13:131–145
Howard RC (1990) The effects of recycling on paper quality. J Pulp Pap Sci 16:J143–J149
Hult E-L, Larsson PT, Iversen T (2001) Cellulose fibril aggregation—an inherent property of kraft pulps. Polymer 42:3309–3314
Jayme G (1944) Micromeasurements of the swelling of pulps. Papierfabr Wochenbl Papierfabr 6:187–194
Jayme G (1958) Properties of wood celluloses. II. Determination and significance of water-retention value. Tappi 41:180A–183A
Jeffries R (1963) An infra-red study of the deuteration of cellulose and cellulose derivatives. Polymer 4:375–389
Kachrimanis K, Noisternig MF, Griesser UJ, Malamataris S (2006) Dynamic moisture sorption and desorption of standard and silicified microcrystalline cellulose. Eur J Pharm Biopharm 64:307–315
Klemm D, Philipp B, Heinze U, Heinze W (1998) Comprehensive cellulose chemistry. Vol. 1. Fundamentals and analytical methods, vol 1. Wiley-VCH, Weinheim
Kljun A, Benians TAS, Goubet F, Meulewaeter F, Knox JP, Blackburn RS (2011) Comparative analysis of crystallinity changes in cellulose I polymers using ATR-FTIR, X-ray diffraction, and carbohydrate-binding module probes. Biomacromolecules 12:4121–4126
Klungness JH (1974) Recycled fiber properties as affected by contaminants and removal processes. Tappi 57:71–75
Kohler R, Dück R, Ausberger B, Alex R (2003) A numeric model for the kinetics of water vapor sorption on cellulosic fibers. Compos Interfaces 10:255–276
Laivins GV, Scallan AM (1993) The mechanism of hornification of wood pulps. Products of papermaking, transactions of the Xth fundamental research symposium, Oxford, 1235–1260
Lee JM, Heitmann JA, Pawlak JJ (2007) Technique for the measurement of dimensional changes of natural microfibril materials under variable humidity environments. Mater Sci Eng A 445–446:632–640
Lee K-Y, Quero F, Blaker JJ, Hill CAS, Eichhorn SJ, Bismarck A (2011) Surface only modification of bacterial cellulose nanofibres with organic acids. Cellulose 18:595–605
Lindström T (1992) Chemical factors affecting the behaviour of fibres during papermaking. Nord Pulp Pap Res J 4:181–192
Lindström T, Carlsson G (1982) The effect of carboxyl groups and their ionic form during drying on the hornification of cellulose fibers. Sven Papperstidn 85:R146–R151
Maloney TC, Paulapuro H (2000) The effect of drying conditions on the swelling and bonding properties of bleached kraft hardwood pulp. Appita Annu Conf Proc 1:41–46
Maloney TC, Paulapuro H, Stenius P (1998) Hydration and swelling of pulp fibers measured with differential scanning calorimetry. Nord Pulp Pap Res J 13:31–36
Maloney TC, Laine JE, Paulapuro H (1999) Comments on the measurement of cell wall water. Tappi J 82:125–127
Mann J, Marrinan HJ (1956) The reaction between cellulose and heavy water. Part 1. Qualitative study by infra-red spectroscopy. Trans Faraday Soc 52:481–487
McKenzie AW, Higgins HG (1958) The structure and properties of paper. Part II. The influence of alkali on the infra-red spectra, bonding capacity and beating response of wood and cotton fibres. Sven Papperstidn 61:893–901
Mercer J (1851) Improvement in chemical processes for fulling vegetable and other textures. US Patent 8303
Minor JL (1994) Hornification—its origin and meaning. Progr Pap Recycl 3:93–95
Morrison JL (1960) Deuterium–hydrogen exchange between water and macromolecules: accessibility of cellulose. Nature 185:160–161
Nazhad M, Paszner L (1994) Fundamentals of strength loss in recycled paper. Tappi J 77:171–179
Nishiyama Y, Kuga S, Okamo T (2000) Mechanism of mercerization revealed by X-ray diffraction. J Wood Sci 46:452–457
Oksanen T, Buchert J, Viikari L (1997) The role of hemicelluloses in the hornification of bleached kraft pulps. Holzforschung 51:355–360
Okubayashi S, Griesser UJ, Bechtold T (2004) A kinetic study of moisture sorption and desorption on lyocell fibers. Carbohydr Polym 58:293–299
Okubayashi S, Griesser UJ, Bechtold T (2005) Moisture sorption/desorption behavior of various manmade cellulosic fibers. J Appl Polym Sci 97:1621–1625
Östlund Å, Köhnke T, Nordstierna L, Nydén M (2010) NMR cryoporometry to study the fiber wall structure and the effect of drying. Cellulose 17:321–328
Park S, Venditti RA, Jameel H, Pawlak JJ (2006) Changes in pore size distribution during the drying of cellulose fibers as measured by differential scanning calorimetry. Carbohydr Polym 66:97–103
Philipp B, Lehmann R, Ruscher C (1959) Zum Einfluß des Cellulosematerials auf den Verlauf der Alkalicellulosebildung. Faserforschung und Textiltechnik 10:22–35
Pönni R, Vuorinen T, Kontturi E (2012) Proposed nano-scale coalescence of cellulose in chemical pulp fibers during technical treatments. BioResources 7:6077–6108
Pönni R, Kontturi E, Vuorinen T (2013) Accessibility of cellulose: structural changes and their reversibility in aqueous media. Carbohydr Polym 93:424–429
Pönni R, Galvis L, Vuorinen T (2014) Changes in accessibility of cellulose during kraft pulping of wood in deuterium oxide. Carbohydr Polym 101:792–797
Rautkari L, Hill CAS, Curling S, Jalaludin Z, Ormondroyd G (2013) What is the role of the accessibility of wood hydroxyl groups in controlling moisture content? J Mater Sci 48:6352–6356
Saito G (1939) Das Verhalten der Zellulose in Alkalilösungen. Kolloid-Beih 49:365–454
Scallan AM (1974) The structure of the cell wall of wood—a consequence of anisotropic inter-microfibrillar bonding? Wood Sci 6:266–271
Sepall O, Mason SG (1961) Hydrogen exchange between cellulose and water II. Interconversion of accessible and inaccessible regions. Can J Chem 39:1944–1955
Sharratt V, Hill CAS, Jalaludin Z, Kint DPR (2011) The influence of photodegradation and weathering on the water vapour sorption kinetic behavior of scots pine earlywood and latewood. Polym Degrad Stab 96:1210–1218
Siroká B, Manian AP, Noisternig MF, Henniges U, Kostic M, Potthast A, Griesser UJ, Bechtold T (2012) Wash-dry cycle induced changes in low-ordered parts of regenerated cellulosic fibers. J Appl Polym Sci 126:E396–E407
Sjöström E, Enström B (1967) Characterization of acidic polysaccharides isolated from different pulps. Tappi 50:32–36
Stone JE, Scallan AM (1967) The effect of component removal upon the porous structure of the cell wall of wood. II. Swelling in water and the fiber saturation point. Tappi 50:496–501
Stone JE, Scallan AM (1968) A structural model for the cell wall of water-swollen wood pulp fibres based on their accessibility to macromolecules. Cell Chem Technol 2:343–358
Suchy M, Kontturi E, Vuorinen T (2010a) Impact of drying on wood ultrastructure: similarities in cell wall alteration between native wood and isolated wood-based fibers. Biomacromolecules 11:2161–2168
Suchy M, Virtanen J, Kontturi E, Vuorinen T (2010b) Impact of drying on wood ultrastructure observed by deuterium exchange and photoacoustic FT-IR spectroscopy. Biomacromolecules 11:515–520
Taniguchi T, Harada H, Nakato K (1978) Determination of water adsorption sites in wood by a hydrogen–deuterium exchange. Nature 272:230–231
Tsuchikawa S, Siesler HW (2003a) Near-infrared spectroscopic monitoring of the diffusion process of deuterium-labeled molecules in wood. Part I: softwood. Appl Spectrosc 57:667–674
Tsuchikawa S, Siesler HW (2003b) Near-infrared spectroscopic monitoring of the diffusion process of deuterium-labeled molecules in wood. Part II: Hardwood. Appl Spectrosc 57:675–6681
Wang X, Maloney TC, Paulapuro H (2003) Internal fibrillation in never-dried and once-dried chemical pulps. Appita J 56:455–459
Weise U (1998) Hornification—mechanisms and terminology. Pap Puu 80:110–115
Weise U, Hiltunen E, Paulapuro H (1998) Verhornung von Zellstoff und Maßnahmen zu ihrer Reversion. Das Papier 52:V14–V19
Wistara N, Young RA (1999) Properties and treatments of pulps from recycled paper. Part I. Physical and chemical properties of pulps. Cellulose 6:291–324
Xie Y, Hill CAS, Jalaludin Z, Curling SF, Anandjiwala RD, Norton AJ, Newman G (2011) The dynamic water vapour sorption behavior of natural fibres and kinetic analysis using the parallel exponential kinetics model. J Mater Sci 46:479–489
Acknowledgments
Financial support from Academy of Finland and UPM are acknowledged. Mrs. Mirja Reinikainen is gratefully acknowledged for her help in experimental work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pönni, R., Rautkari, L., Hill, C.A.S. et al. Accessibility of hydroxyl groups in birch kraft pulps quantified by deuterium exchange in D2O vapor. Cellulose 21, 1217–1226 (2014). https://doi.org/10.1007/s10570-014-0166-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-014-0166-x