
Abstract
Carbonyl reductase, as biocatalyst, is very important to chiral alcohols production through asymmetric reduction of carbonyl compound. A novel thermal stable carbonyl reductase from Bacillus cereus (BcCR) dependant on NADPH was obtained through a new genome mining strategy proposed in this work. By analyzing its amino acid sequence and structure, the BcCR should be a thermal stability and wide pH tolerance carbonyl reductase. Its gene was cloned by PCR with B. cereus genomic DNA as template. Its heterologous expression system, E. coli BL21 (DE3) plysS/pET28a-bccr, was constructed, and BcCR was successfully expressed. Enzymatic properties show that at 57.5 °C and pH 7.0 it can reach maximum reaction rate. Its Km and Vmax to ethyl 4-chloroacetoacetate is 1.85 mmol/L and 0.22 µmol/(min·mgprotein), respectively. It can catalyze the asymmetric reduction of the β-carbonyl compound, such as ethyl 4-chloroacetoacetate to ethyl S-4-chloro-3-hydroxybutyrate. This paper proposes a practical method for discovery of new carbonyl reductases, and provides a novel enzyme as biocatalyst for asymmetric reduction of β-carbonyl compound.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Chiral alcohols serve as an important chiral blocks in the process of industrial synthetic drugs, agrochemicals, natural products and certain special materials [1,2,3]. Synthesis of chiral alcohols through asymmetric reduction of the corresponding prochiral ketones with carbonyl reductase is a promising route. Compared to chemical synthesis, it possesses inherent various advantages, such as high stereoselectivity, mild conditions, environmentally friendly and meeting green chemistry guidelines [3,4,5]. The carbonyl reductases were mainly classified in three families, i.e. short-chain dehydrogenases/reductase (SDRs), medium-chain dehydrogenases/reductase (MDRs) and Aldehyde ketone reductase (Aldo-keto reductase, AKRs). Some new carbonyl reductases were reported, which can catalyze the asymmetric reduction of various ketones to chiral alcohols with stereoselectivity [6,7,8], such as SDR (LCRIII) from Lactobacillus curieae S1L19 [9], MDR (CPCR from Candida parapsilosis ATCC 7330) [10] and AKRs (LEAKR 48, LEAKR 49, and LEAKR 50 from Lodderomyces elongisporus LH703) [11]. But obtaining a new high activity enzyme is still the most significant to a biocatalysis process. Therefore, discovery of the high efficient novel carbonyl reductases is always the hot spot in biocatalysis especial in biocatalytic asymmetric synthesis [7].
Traditionally, a new enzyme was discovered by screening from nature based on substrate specificity and stereoselectivity [12, 13]. This route is high time and labor cost. In recent years, with the rapid development of high-throughput gene sequencing technology, the number of gene data in bioinformatics database is explosively growing. Simultaneously, the protein databases provide abundant biological information for many enzymes, such as the relationship between structure and function. At the end of November 2018, the putative SDRs number is 233818 in UniProt database. But it is about just 1000, which has been confirmed by literatures or simulation analysis. To AKRs, these numbers are respectively 143550 and 324. And to MDRs, they are 82 and 30, respectively. These gene and protein databases are very valuable bioinformatics resource for the potential, novel and efficient enzyme discovery. With bioinformatics technology such as genome mining, it is possible to discover a potential high efficient novel enzyme [14,15,16]. Compared with the traditional route, genome mining can obtain new enzyme from a wider sources and various organisms. Also, it will be more cost saving in time and labor.
This paper proposes a genome mining strategy. This mining strategy was based on the reported thermal stability carbonyl reductase from an extreme thermophilic bacteria as probe to mine potential carbonyl reductase from protein database with laboring and time save. With this strategy, a novel carbonyl reductase from Bacillus cereus (BcCR) was discovered. Its gene was successfully cloned from Bacillus cereus genome DNA by PCR. An expression vector was constructed and was transformed into E. coli BL2l (DE3)(pLysS) for expression. Furthermore, its enzymatic properties were investigated. Its catalytic activity to the asymmetric reduction of β-carbonyl compound such as ethyl 4-chloroacetoacetate in the whole cell were also assessed.
2 Materials and Methods
2.1 Chemicals and Strains
Plasmid extraction kit and PCR product recovery kit were purchased from Tiangen Biotech Co., Ltd (Beijing, China). DNA gel recovery kit was purchased from Omega Bio-tek (Norcross GA, USA). T4 DNA ligase, PCR Mix, DNA marker and protein maker were purchased from Vazyme Biotech Co., Ltd (Nanjing, China). BamHI and XhoI restriction endonucleases were purchased from New England Biolabs Ltd (Beijing, China). Ethyl 4-chloroacetoacetate (COBE) and ethyl S-4-chloro-3-hydroxybutyate (CHBE) were purchased from Acros Organics (New Jersey, US). The other ordinary chemical regents were purchased from Aladdin regents (Shanghai, China).
Bacillus cereus was purchased from China Center for Type Culture Collection (CCTCC AB 2011085), which was used for genomic DNA extract as the PCR template. E. coli DH5α and E. coli BL2l (DE3)(pLysS) was preserved in our laboratory. They were respectively used for plasmid operation and BcCR expression.
2.2 Genome Mining for Carbonyl Reductase
In this work, the carbonyl reductase genome mining was based on the UniProt database. The main strategy is using an amino acid (AA) sequence as the probe to extract out as many proteins as possible from the UniProt database to form the potential enzyme pool. The select process is based on the conservative AA sequence for the carbonyl reductase, such as co-factor binding site and catalysis site. The genome mining procedure mainly include the flowing four steps [17]. The first step is to construct a carbonyl reductase probe bank and selecting a favorable probe. The probe enzymes in the bank were selected out the reported carbonyl reductases from the main bioinformation database such NCBI and UniProt. Their enzymatic properties and catalytic efficiency of these selected carbonyl reductases in the probe bank were compared and evaluated. The most suitable one was chosen as the probe based on its enzymatic properties and catalytic efficiency. The second step is to construct the carbonyl reductase candidate pool. The potential carbonyl reductase candidate was mined out from the protein database, such as UniProt, through homologous alignment (BLAST) with the probe AA sequence. These proteins, which AA sequence have a consistency more than 30% to the probe, were selected out as the potential enzyme candidates. The third step is to select out the target carbonyl reductase from the candidate pool. In this step, the AA sequence of candidate enzymes was analyzed, their secondary structure and tertiary structure were simulated, and their catalysis function was predicted based on the conserved sequence and structure comparing with the probe enzyme. After comparing the structure with the probe enzyme, the best candidate protein was chosen as the target enzyme. In the last step, the gene DNA of the chosen target enzyme will be cloned by PCR from its source microorganism genomic DNA.
2.3 Cloning of Carbonyl Reductase Gene from B. cereus
B. cereus genomic DNA was extracted by CTAB/NaCl method from its culture broth in Luria-Bertani medium (LB) [18]. The primers was designed by Primer Premier5 with the target gene sequence (GenBank: AAP10771.1, Gene ID: 1206194), which was obtained by the genome minging in the previous section. The foward primer is 5′-CGCG/GATCCATGTTAAAAGGGAAGGTAGC-3′ (with BamHI site, G/GATCC). And the reverse primer is 5′-CCGC/TCGAGCATTACCATACCGCCATCAAC-3′ (with XhoI site, C/TCGAG). The target gene was amplified by PCR using B. cereus genomic DNA as template. The reaction conditions were: 95 °C for 5 min initial denaturation, 35 amplification cycles (95 °C for 1 min denaturation, 71 °C for 15 s annealing, 72 °C for 1 min elongation), 72 °C for 10 min final elongation and stored at 4 °C.
2.4 Construction of BcCR Heterogeneous Expression System and Expression of BcCR
The vector, plasmid pET-28a, and BcCR gene (bccr) were digested with BamHI and XhoI. After purification of the digested product, the digested bccr was ligated into the digested pET-28a with T4 DNA ligase at 16 °C for overnight. The ligation product, pET-28a-bccr, was transformed into E. coli DH5α and screened on the LB/Kan50 (LB medium with 50 µg/mL of Kanamycin) resistant agar plate. The positive transformants were identified by PCR, and further confirmed by double enzymes digestion of the recombinant plasmid, which was extracted from the transformant. Also, it was sequenced to further certify the bccr. The recombinant plasmid pET-28a-bccr was further transformed into E. coli BL2l (DE3)(pLysS) for BcCR heterologous expression. Then, E. coli BL2l (DE3)(pLysS)/pET-28a-bccr was constructed, which was BcCR heterologous expression system. The recombinant E. coli was cultured in LB medium with Kan50 at 37 °C and induced with 0.5 mmol/L IPTG at 24 °C for 16 h. After expression, cell were collected by centrifugation from the culture broth, and washed twice with 0.01 mol/L phosphate buffer (PBS, pH 7.0). The cells were lysed by sonication and the supernatant was collected by centrifuged at 4 °C. The expression product was detected by SDS–PAGE.
2.5 Investigation of BcCR Enzymatic Properties
In this paper, the effects of temperature, pH and metal ions on the heterogeneous expressed BcCR activity were investigated. Its thermal stability and kinetic parameters were also assessed. The generally procedure was as following: after respectively incubating the BcCR solution and the reaction mixture solution at the test temperature, general 37 °C, for 30 s, then the 100 μL BcCR solution was added into 1.9 mL reaction mixture solution to catalyze the reduction reaction, and the corresponding enzyme activity was assayed. The 1.9 mL reaction mixture solution was composition of 1.865 mL PBS buffer, 10 µL of 400 mmol/L COBE and 25 µL of 8 mmol/L NADPH.
2.6 Asymmetric Reduction of COBE with Whole Cell
The asymmetric reduction of COBE is carried out with whole cell. The catalysis capability of the recombinant E. coli BL2l (DE3)(pLysS)/pET-28a-bccr, which expressed BcCR, was investigated. The recombinant cell was cultured and induced as the previous mentioned. The cell were collected by centrifugation and re-suspended after washing with PBS buffer. A certain amount of cell was added into the reaction mixture to a cell concentration of 0.1 g/mL. The reaction mixture contained 5% of glucose and 50 mmol/L COBE. Based on the BcCR enzymatic properties, the reaction temperature was 37 °C, and the pH was 7.0. The reaction samples was periodically withdrawn and extracted with ethyl acetate (1 mL of reaction sample was extracted with 1 mL ethyl acetate for two times). Finally, the concentrations of product and substrate were analyzed by GC, and the chemical yield and enantioselectivity were evaluated.
2.7 Analytical Techniques
Protein concentration, BcCR enzyme activity, substrate and product concentration were analyzed in this work.
The protein concentration was determined by the Bradford method with a spectrophotometer (GE Ultrospec 2100 pro, GE Healthcare Life Science, USA) at 595 nm using bovine serum albumin as the standard [19]. BcCR enzyme activity assay was based on the NAD(P)H consumption method [8]. One unit of the BcCR activity was defined as the amount of enzyme that catalyzed to consume 1 μmoL of NADPH per minute with the reduction of COBE, which was measured by the absorbance value at 340 nm with a spectrophotometer (Ultrospec 3300 pro, GE healthcare).
The reaction samples were extracted with ethyl acetate and were dried with anhydrous Na2SO4. The concentration of product and substrate were quantitatively analyzed by GC (SHIMADZU GC-2010 Plus, Japan) with an Rtx-Wax Capillary column (30 m × 0.32 mm) and FID. N2 was used as carrier gas with 1.5 mL/min flow rate. The accounts of S-form and R-form product were determined by high-performance liquid chromatography (HPLC, model 1100; Agilent Technologies Co., Ltd.) equipped with a Chiralcel OB column (4.6 mm × 250 mm) (Daicel Chemicals, Japan). The HPLC conditions were hexane/isopropyl alcohol (9/1, v/v) as mobile phase, flow rate of 1.5 mL/min, ambient column temperature, and ultraviolet (UV) detection set at 215 nm. The analytical approach was similar to our previous work [1].
3 Results and Discussion
3.1 Genome Mining for Carbonyl Reductase
By aligning and analyzing the reported carbonyl reductases, we found the conserved amino acids sequence for traditional SDRs family carbonyl reductases are usually composed of Tyr and Lys (Y-X3-K, X means variable amino acids), and the cofactor binding sites are usually composed of Thr and Gly (TG-X3-G-X-G) [20]. To the AKRs family, the conserved amino acids sequences are usually composed of Asp, Tyr, Lys and His (D-Y-K-H), and the coenzyme binding site are composed of Asp, Asn, Gln, Ser, Arg (D-N-Q-S-R) [6]. The corresponding results was present in Electronic supplementary material Figure S1 and S2. To genome mining, the probe is very key and important. It should directly dominate the mining efficiency and influence the target enzyme properties. By searching and analyzing for the reported carbonyl reductases, we constructed a probe enzyme bank for asymmetric reduction of β-carbonyl ester, which was present in Electronic supplementary material Table S1. Based on the requirement of the target enzyme and combining the reported carbonyl reductases, the carbonyl reductase ApCR (UniProt: Q9Y8Y1) from Aeropyrum pernix K1, an extreme thermophilic bacteria, was chosen as the probe. The ApCR belongs to the SDRs family and contains the typical conserved amino acids structure. It can catalyze the asymmetric reduction of β-carbonyl ester, for example COBE, with excellent enzymatic properties, such as high temperature tolerance, good stability and wide range of pH tolerance [21].
By genome mining from UniProt database with ApCR as the probe, we find out 8 candidate potential carbonyl reductases. Further comparing their AA sequence, structure and predicting function, an AA sequence from Bacillus cereus (UniProt Accession Number: Q819V6, GenBank: AAP10771.1, Gene ID: 1206194) was chosen as the potential target carbonyl reductase, which was named as BcCR. Its AA sequence was aligned with ApCR by homology alignment. The result was presented in Fig. 1. The amino acid sequence of the protein BcCR, was 38.4% identical to the probe ApCR. BcCR contains the active center conserved region Y-X3-K and the cofactor binding site conserved region TG-X3-G-X-G. These show that it is a typical SDR family reductase. Their structures was modelled by SWISS-MODEL (https://swissmodel.expasy.org/), the tertiary structures presented in Fig. 2 and the secondary structures given in Figure S3. Their secondary and tertiary structures are very similar. It can be speculated that the two enzymes possess similar enzymatic properties. This means that BcCR should have the potential catalytic activity of asymmetric reduction of β-carbonyl ester and have excellent enzymatic properties such as good thermal stability.

Amino acid sequence alignments of ApCR and BcCR

Tertiary structure of ApCR (left) and BcCR (right)
3.2 Cloning of Carbonyl Reductase Gene from B. cereus and Heterogeneous Expression of BcCR in E. coli
The target gene was cloned by PCR using the designed primers according to the DNA sequence of bccr (Gene ID: 1206194). The PCR product size was about 750 bps, the gel shown in Figure S4, which was consistent with the 741 bp of the gene shown in GenBank. The recombinant plasmid pET28a-bccr was constructed by ligating the double enzymes (BamHI and XhoI) digestion product of the PCR product and pET28a. The recombinant plasmid pET28a-bccr was verified by double enzymes digestion and DNA sequence. Figure 3 shows the result of the double enzymes digestion agarose gel. The target fragments of about 5300 bp and 750 bp size were obtained in the double enzymes digestion product, which was the size of pET28a and bccr fragments respectively. The cloned bccr was sequenced, and the result was presented in Figure S5 with the alignment of DNA sequence of Gene ID: 1206194.

Agarose gel of double enzyme digestion of recombinant plasmid pET 28a-bccr. Lane: M: DL5000 DNA Ladder, 1: Double enzyme digestion product
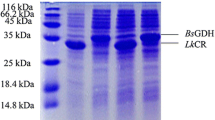
The recombinant plasmid pET28a-bccr was further transformed into E. coli BL2l (DE3)(pLysS), and the BcCR heterologous expression system, E. coli BL2l (DE3)(pLysS)/pET-28a-bccr, was obtained. After screening and identification, the positive transformant was selected for further culture, and induced by IPTG. After cell lysing and collecting the supernatant, the expression product was detected by SDS-PAGE, shown in Fig. 4. A target protein of about 32 kDa can be seen from the SDS-PAGE, which is consistent to the predicted size translated from the DNA sequence. This means that the BcCR was successfully expressed E. coli BL2l (DE3) (pLysS)/pET-28a-bccr in soluble status.

SDS–PAGE of induced expression products with IPTG. Lane: M: Protein Marker, 1–3: E. coli BL2l (DE3)(pLysS)/pET-28a, 4: E. coli BL2l (DE3)(pLysS)/pET-28a-bccr
3.3 BcCR Enzymatic Properties
Different enzymes from different sources will possess different enzymatic properties. Even though the structure of BcCR is similar to that of ApCR, their enzymatic properties are very likely different. The Enzymatic properties of the heterologous expressed BcCR were investigated. The effects of temperature, pH and metal ions on the BcCR activity were assessed. Its thermal stability and kinetic parameters were also evaluated. The corresponding results were presented in Fig. 5 and Table 1.

BcCR enzymatic properties. a Influence of temperatures on BcCR activity, b BcCR thermal stability, c Influence of pH on BcCR activity, d BcCR Km and vmax by fitting curve of V-[S] with M-M model
Figure 5a indicated the effects of temperature on the enzyme activity. At 57.5 °C, BcCR could reach to its highest activity. This temperature is lower than that of the probe enzyme ApCR, which is about 70 °C. However, it is higher than most carbonyl reductases such as RpCR [7], CPE [22], ScCR [23], CgKR2 [24] about 10 °C. The higher temperature means better operating conditions and application performance. Thermal stability is another important property to an enzyme. The thermal stability of BcCR was investigated at 40 °C, 50 °C and 60 °C, the results were shown in Fig. 5b. Although incubated at 40 °C and 50 °C for 2 h, BcCR could still maintain its residual enzyme activity more than 70%. This means that BcCR possesses good thermal stability. The pH property of BcCR was determined, results shown in Fig. 5c. The results show that the optimum pH is 7.0. In pH range from 6.0 to 8.0, the relative activity of BcCR can keep more than 75%. The effect of metal ions on BcCR was determined, results were presented in Table 1. The results showed that no metal ions were found to promote the enzyme activity. In general, the carbonyl reductase of SDRs family is a kind of nonmetallic dependent dehydrogenase. This indicates that BcCR follow the SDRs family feature.
Its kinetic constant was determined through the initial rate at various concentration of COBE based on Michaelis–Menten equation. Km and Vmax were obtained by fitting the V-[S] data with least squares method. The experiment data and fitting curve is shown in Fig. 5d. By curve fitting, Km is 1.85 mmol/L and Vmax is 0.22 µmol/min/mg(protein) with R2 = 0.9973. The Km value indicates the affinity of enzyme to substrate, the smaller Km means higher affinity. Compared with the reported SDR carbonyl reductase, such as PsCRII(3.3 mmol/L) [25], CpCR(3.7 mmol/L) [7] and CHKRED20 (7.5 mmol/L) [26], the Km of BcCR is smaller than that of those enzymes. This means that BcCR possesses higher affinity to substrate COBE, which demonstrates BcCR can possess higher catalytic activity to COBE.
3.4 Asymmetric Reduction of COBE with Resting Cell
To assay BcCR catalytic ability for asymmetric reduction of β-carbonyl ester, the asymmetric reduction ethyl 4-chloroacetoacetate (COBE) by the recombinant E. coli BL2l (DE3) (pLysS)/pET-28a-bccr resting cell was preliminarily investigated. Reduction of COBE to chiral ethyl S-4-chloro-3-hydroxybutyrate catalyzed by the recombinant resting cell was chosen as the model reaction due to the product ethyl S-4-chloro-3-hydroxybutyrate is a key important chiral building block and which was widely researched. The product is main S-form configuration, and e.e. of S-form is more than 97%. The results were given in Fig. 6. With the increase of reaction time, the yield of whole cell reaction increased from 35.8% at 8 h to 61.7% at 32 h. Wei et al. reported a carbonyl reductase (AcCR) from Acetobacter sp. and co-expressed with GDH in E. coli BL21(DE3)pLysS (pETDuet-gaccr-gdh) [27]. The product also is S-form configuration, ethyl S-4-chloro-3-hydroxybutyate. But they reported an 80% yield. The higher yield to this work because they used a co-express system which can express GDH to enhance NADPH regeneration. Additionally, the cell (biocatalyst) amount was another factor to the higher yield. This shows that the recombinant cell has catalytic ability to β-carbonyl ester.

Asymmetric reduction of COBE with E. coli BL2l (DE3)(pLysS)/pET-28a-bccr resting cell
4 Concluding Remarks
Based on the protein database, a novel carbonyl reductase, BcCR from B. cereus, was discovered by the constructed gene mining strategy. The open reading frame of BcCR was 738 bp, and encodes 246 amino acids. The BcCR was successfully expressed in E. coli BL21 (DE3) (pLysS) with pET28a-bccr vector. Its optimum reaction temperature and pH were about 57.5 °C and 7.0, respectively. Also, the carbonyl reductase shows good thermal stability. Its kinetic parameters showed that the Km value was 1.85 mmol/L to COBE, and the corresponding maximum reaction rate Vmax was 0.22µmol/min/mg. These show that the BcCR is a good potential carbonyl reductase for asymmetric reduction of β-carbonyl ester to chiral alcohol. Also, the proposed gene mining strategy show good practical application prospects for discovery of new carbonyl reductases.
References
Yang Z-H, Luo L, Chang X, Zhou W, Chen G-H, Zhao Y, Wang Y-J (2012) J Ind Microbiol Biotechnol 39:835–841
Xu GC, Yu HL, Shang YP, Xu JH (2015) RSC Adv 46:22703–22711
Luo W, Deng XX, Huo J, Ruan T, Gong ZW, Yan JB, Yang Z-H, Quan C, Cui ZF (2018) Catal Lett 148:1714–1722
Yang Z-H, Zeng R, Yang G, Wang Y, Li LZ, Lv ZS, Yao M, Lai B (2008) J Ind Microbiol Biotechnol 35:1047–1051
Wang YJ, Chen XP, Shen W, Liu ZQ, Zheng YG (2017) Biochem Eng J 128:54–62
Penning TM (2015) Chem-Biol Interact 234:236–246
Guo CX, Tang MH, Ni Y (2016) J Mol Catal B: Enzym 123:67–72
Wang YJ, Ying BB, Shen W, Zheng RC, Zheng YG (2017) Enzyme Microb Technol 107:32–40
Zhang YP, Wang HL, Chen LF, Wu K, Xie JL, Wei DZ (2016) J Mol Catal B: Enzym 134:51–60
Aggarwal N, Ananthathamula R, Kumar KV, Doble M, Chadha A (2018) Mol Catal 460:40–45
Ning CX, Su EZ, Wei DZ (2014) Arch Biochem Biophys 564:219–228
He YC, Zhang DP, Tao ZC, Zhang X, Yang ZX (2014) Bioresour Technol 172:342–348
Chen R, Liu X, Lin J, Wei D (2014) J Agric Chem Soc Japan 78:1350–1356
Guo R, Nie Y, Mu XQ, Xu Y, Xiao R (2014) J Mol Catal B: Enzym 105:66–73
Zaparucha A, de Berardinis V, Vaxelaire-Vergne C (2018) In: Williams G, Hall M (eds) Modern biocatalysis: advances towards synthetic biological systems. Royal Society of Chemistry, Cambridge
Yu SZ, Li HX, Lu Y, Zheng GJ (2017) Appl Biochem Biotechnol 184:1319–1331
Yang Z-H, Ruan T, Luo W, Wu Y, Zuo ZY, Hou YL, Li LL (2018) Chinese patent 201810218384.2
Wilson K (2001) Curr Protoc Mol Biol 56:241–245
Cheng Y, Wei H, Sun R, Tian Z, Zheng X (2016) Anal Biochem 494:37–39
Kavanagh KL, Jornvall H, Persson B, Oppermann U (2008) Cell Mol Life Sci 65:3895–3906
Fukuda Y, Sakuraba H, Araki T, Ohshima T, Yoneda K (2016) Enzyme Microb Technol 91:17–25
Wang Q, Shen L, Ye T, Cao D, Chen R, Pei X, Xie T, Li Y, Gong W, Yin X (2012) Bioresour Technol 123:690–694
Wang LJ, Li CX, Ni Y, Zhang J, Liu X, Xu JH (2011) Bioresour Technol 102:7023–7028
Huang L, Xu JH, Yu HL (2015) J Biotechnol 203:54–61
Cao H, Mi L, Ye Q, Zang G, Yan M, Wang Y, Zhang Y, Li X, Xu L, Xiong J, Ouyang P, Ying H (2011) Bioresour Technol 102:1733–1739
Zhao FJ, Pei XQ, Ren ZQ, Wu Z-L (2016) Appl Microbiol Biotechnol 100:3567–3575
Wei P, Gao JX, Zheng GW, Wu H, Zong MH, Lou WY (2016) J Biotechnol 230:54–62
Acknowledgements
The authors acknowledge all the financial supports for this research by the National Natural Science Foundation of China (Grant No. 21376184), the Scientific Research Foundation for the Returned Overseas Chinese Scholars (State Education Ministry), Foundation from the Educational Commission of Hubei Province of China (Grant No. D20121108), the Ministry of Science and Technology of China (2017YFF0205803), and the National Institute of Metrology of China (21-AKY1615).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest Statement
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Qin, YL., Ruan, T., Hou, HS. et al. A Novel Thermal Stable Carbonyl Reductase from Bacillus cereus by Gene Mining as Biocatalyst for β-Carbonyl Ester Asymmetric Reduction Reaction. Catal Lett 149, 610–618 (2019). https://doi.org/10.1007/s10562-018-2645-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-018-2645-4