
Abstract
To improve the dispersibility of graphene oxide (GO) in solvents, the grafting of polyacrylamide (PAM) from the GO surface was performed by redox polymerization system. The hydrophilic nature and high polarity of PAM have made effective dispersion of GO. Then, Pd nanoparticles (Pd NPs) were anchored on the surface of polyacrylamide grafted reduced graphene oxide nanosheets (PAM-g-rGO/Pd). The obtained nanocomposite was used for Suzuki–Miyaura cross-coupling reaction in an environmental friendly solvent under ambient conditions. The results showed that the prepared catalyst system exhibited high catalytic activity and stability which could be reused at least eight times without significant loss of its catalytic activity. This is mainly because of its small particle size, uniform dispersion of Pd NPs on the surface of PAM-g-GO/Pd, and lack of agglomeration of these nanoparticles during the preparation at room temperature.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Graphene and graphene oxide (GO) have been used extensively as supports for immobilizing active species and also metal-free catalysis due to their unique chemical, physical and surface properties such as high specific surface areas, chemical and electrochemical inertness, good biocompatibility and high adsorption capacity and easy surface modification [1–5].
Chemical functionalization of graphene sheets with polymer chains prevents significant π–π interaction between the sheets [6]. To date, many methods for chemical functionalization of graphene sheets have been reported, and these approaches could be divided into “graft onto” and “graft from” strategies [7, 8]. In “graft onto” strategy, polymer chains could be covalently linked to graphene by esterification or amidation reactions [9, 10]. In contrast, in the ‘‘grafting from’’ approach, the polymerization is initiated right on the surfaces. This technique produces substrate surfaces with high grafting ratio [11, 12]. So far, numerous graphene and grphene oxide-based polymer nanocomposites have been successfully prepared. The combination of them is suitable for real applications [6].
It is well known that the redox reaction systems, with Ce(IV) salts and reducing agents in aqueous solutions, are great initiators for vinyl polymerization. Homopolymers, block copolymers, and graft copolymers can be easily formed with this system. Suitable reducing agents include alcohols, aldehydes, ketones and amines [13, 14]. Polymer grafted GO sheets was reported using a redox system [15–18]. In related studies, Tu et al. obtained polymer grafted GO by using a Ce(IV)/HNO3 redox system in an aqueous solution [17]. Hu et al. Prepared poly-(acrylic acid) (PAA) and poly (N-isopropylacrylamide) (PNIPAM) functionalized GO by Ce(IV)-induced redox polymerization [12]. This reaction could be carried out in aqueous solution and mild temperature. Since the initiation sites are directly on the surface of GO sheets, free polymers can be neglected, and the materials are easily to be separated by filtration or centrifugation.
In the field of catalysis, Pd NPs plays an important role in heterogeneous catalysis for many applications including carbon–carbon coupling and alcohols oxidation reactions [19–21]. The three most important processes, the Suzuki–Miyaura coupling, the Heck reaction and the Sonogashira reaction typically catalyzed by palladium catalysts, have been widely studied in recent decades [22–27]. However, both their recovery and recycling are often difficult. To address these issues, immobilization of Pd catalysts on inorganic solid supports [28], organic polymers [29, 30], CNT [31–33], graphene and its derivatives have been studied [34–39]. Among these support systems, graphene and GO have been attracted significant attention due to the high specific surface area, thermal stability, and unique structural and electronic properties as potential support systems for palladium-catalyzed C–C coupling applications.
In the present work, for better immobilization of nanoparticles on the reduced graphene oxide (rGO), the surface of GO was functionalized by PAM. The chemical grafting of PAM onto the GO sheets was carried out using a redox system with ammonium cerium (IV) sulfate [(NH4)4Ce(SO4)4·2H2O] and GO sheets’ hydroxyl groups as redox initiating system. The hydrophilic nature and high polarity of PAM have made effective dispersion of GO. Then, the Pd nanoparticles were synthesized by reduction method on the surface of PAM-g-rGO. PAM-g-rGO/Pd nanocomposite was used for cross carbon–carbon coupling reactions (Miyaura–Suzuki). The nanocomposites were fully characterized by FT-IR, elemental analysis, transmission electron microscopy (TEM) and thermogravimetric analysis (TGA). The catalytic activity and recyclability of the PAM-g-rGO were also investigated for use in the Suzuki reactions of arylboronic acids and aryl halides.
2 Experimental
2.1 Materials
The reagents and solvents were obtained from Fluka, Merck or Aldrich chemical companies.
2.2 Synthesis of GO
Graphite oxide was synthesized from natural graphite using Hummer’s method [40]. Typically, graphite (1 g) and NaNO3 (0.5 g) were added into a 500-ml round-bottom flask followed by addition of H2SO4 (60 ml) with stirring in an ice water bath. Next, KMnO4 (3 g) was slowly added in 15 min. Cooling was completed in 2 h, and the mixture was allowed to stand for 5 days at room temperature with vigorous stirring. The obtained solution was added to aqueous solution of H2SO4 (100 ml, 5 wt%) in 1 h with stirring, and temperature was kept at 98 °C. The resultant mixture was further stirred for 2 h. Then, 80 ml of hot water (60 °C) and H2O2 (33 %, 15 ml) were added to reduce the residual KMnO4 until bubbling is disappeared. Finally, the solid product was separated by centrifuge. After that, the product was filtered and washed with HCl solution (5 %) twice to remove the metal ions. Next, the obtained product rinsed with deionized water to remove the acid in order to get graphite oxide powder after cake was completely dried in air. The product, graphite oxide, exfoliated in de-ionized water with ultrasonic to form GO nanosheets. Finally, Brown gel was obtained by freezing and drying of the suspension.
2.3 Synthesis of PAM-g-GO Nanocomposite
Graft polymerization was carried out for four different polymerization conditions. As the temperature and concentration of initiators are different in each condition, the products were named as PAM-g-GO1, PAM-g-GO2, PAM-g-GO3, and PAM-g-GO4. The overall procedure is as follows: GO (50 mg) was dispersed in distilled water (100 ml) in an ultrasonic bath for 30 min at room temperature to yield a homogeneous dispersion. The obtained suspension was loaded in a 250-ml three-neck round bottom flask and appropriate amount of Ce(IV) and K2S2O8 in 10 ml water solution were added to the suspension in dropping manner. Next, AM monomer (5 g) was added to the mixture. Then, the mixture was heated at 65 (or 30 °C) for 24 h. During the whole preparation process, the solution was under nitrogen atmosphere. After polymerization, the product were removed by centrifugation (12,000 rpm, 30 min), and the sediment was redispersed in THF or DMF. Repetition of this step was used to remove the unreacted monomer and free polymers until there is no precipitate formed when drops of colorless upper solution were added to methanol, which indicates no detectable free polymers remained in the solution. The solid product of PAM-g-GO nanocomposite was collected and dried by freeze drying.
2.4 Synthesis of PAM-g-rGO/Pd Nanocatalyst
PAM-g-GO2 (500 mg) was loaded in a 250-ml round bottom flask and water (100 ml) was added and stirred until a homogeneous dispersion was obtained. Then, PdCl2 (100 mg; 5.6 × 10−5 mol) was added to the PAM-g-GO2 solution. Next, NaBH4 (0.0075 g; 1.9 × 10−4 mol), as reduction agent for GO and Pd NPs, was slowly added in 15 min and the obtained solid product (PAM-g-rGO/Pd) was separated by centrifuge. Finally, the product was ultrasonicated in deionized water (100 ml) for 3 min to make a homogenous suspension and the black powder was obtained by freeze drying of the suspension. To determine the Pd content of the catalyst, PAM-g-rGO/Pd (20 mg) nanocatalysts was treated successively with mixture of concentrated H2SO4, HNO3 and HCl, and filtered. The filtrate was diluted to 50 ml with distilled water and subjected to ICP determination using calibration curve method. Pd content = 0.000154 mmol g−1. ICP analysis also showed that 6.78 wt% of Pd was loaded in the PAM-g-rGO catalyst.
2.5 Catalytic Activity of PAM-g-rGO/Pd Nanocatalyst
General protocol for Suzuki coupling is explained as follows: Aryl halide (1 mmol), phenylboronic acid (1.2 mmol), K2CO3 (1.5 mmol), PAM-g-rGO/Pd (20 mg; containing 0.000154 mmol Pd) were added to 4 ml of H2O as the solvent. The solution was stirred at room temperature for the time indicated. The reaction was monitored by TLC (or GC if necessary). After completion of the reaction, the reaction mixture had cooled to room temperature, the catalyst was easily recovered by filtration and the filtrate was extracted with ethyl acetate for three times. The organic fractions were washed with deionized water and then dried with magnesium sulfate. The mixture was then purified by column chromatography on silica gel using n-hexane or different mixtures of n-hexane–ethyl acetate as the eluents to afford a product with high purity. Characterization of the products was performed by comparison of their FT-IR, 1H NMR, 13C NMR, and physical data with those of the authentic samples.
2.6 Procedure for Recycling the PAM-g-rGO/Pd Nanocatalyst Using Iodobenzenes (Table 7)
Iodobenzene (1 mmol), phenylboronic acid (1.2 mmol), and PAM-g-rGO/Pd (20 mg; containing 0.000154 mmol Pd) were added to 4 ml of H2O as the solvent. The solution was stirred at room temperature and for the time indicated. When Suzuki coupling reactions were finished according to the procedures described in previous sections, the suspension was cooled down to room temperature and filtered off. After completion of the reaction, the procedure outlined above was followed. PAM-g-rGO/Pd nanocatalyst was washed with DMF, water and acetone. It was dried under vacuum and reused without any pretreatment for repeating cycles.
2.7 Characterization
The morphology of PAM-g-GO nanocomposite was examined by field emission scanning electron microscopy (FE-SEM; Hitachi S4160 Japan) and transmission electron microscopy (TEM, philips cm30 300kv) to explore the structural properties. X-ray diffraction analysis (XRD) was carried out with a X-pert philips, pw 3040/60 X-ray diffractometer. 1H NMR and 13C NMR spectra were recorded on a Bruker DRX400 (400 MHz). Progress of reactions was followed by thin layer chromatography (TLC) on silica-gel Polygram SIL/UV 254 plates or by Shimadzu GC 10-A instrument with hydrogen flame ionization detector. The spectroscopic analyses were carried out using a fourier transform infrared spectrometer (FT-IR-Nicolet IR 200) and a Micro Raman Spectrometer (SENTERRA 2009) using an Ar–ion laser at 640 nm as the excitation light source. Thermogravimetric/differential thermal analysis (TGA/DTA) were carried out using a thermobalance (InstSerial 0050–0307) from room temperature to 600 °C, at a rate of 10 °C/min in a continuous nitrogen flow. Atomic force microscopy (AFM) images were carried out by a Femtoscan SPM. AFM measurements were taken by aqueous solution of GO and PAM-g-GO/Pd. The Pd analysis and leaching test were carried out by inductively coupled plasma (ICP) analyzer (Varian, Vista-MPX). Elemental analysis was performed on a (ECS 4010) elemental analyzer. The X-ray photoelectron spectroscopy (XPS) experiments were carried out in an ultra-high vacuum system equipped by (Gammadte–scienta ESCA 200) XPS. XPS was done in an ESCA/AES system equipped with a Concentric Hemi-spherical analyzer (CHA, Specs model EA10 plus). XPS spectra were acquired using the Al Kα (hm = 1486.6 eV) radiation.
3 Results and Discussion
3.1 Grafting Mechanism of PAM from GO Nanosheets
In the present study, we report the preparation of PAM-g-rGO/Pd as a catalyst for different reactions (Scheme 1).

Preparation of PAM-g-GO nanocomposite and PAM-g-GO/Pd
PAM grafted GO sheets were prepared by redox polymerization of AM as monomer under mild experimental conditions. The mechanism of grafting is as follows: at first, free radicals are generated on GO sheets by the redox reaction of hydroxyl groups of GO sheets with Ce(IV), K2S2O8 and at 65 °C to initiate the polymerization. Then, polymer brushes formed on GO sheets by chain propagation. Finally, the termination steps include coupling of the growing chains with other radicals, disproportionation and chain transfer reactions. According to the previous studies [14, 17, 41–43] and obtained results from Table 1, it is suggested that the radical formation depends on three factors: temperature, concentration of Ce(IV) and K2S2O8 (Scheme 2). Since the initiation of polymerization is from the hydroxyl groups located mainly on the surface of GO sheets, the grafted polymers are expected to be on the surfaces of GO sheets. According to the obtained results, PAM-g-GO2 showed more grafting density compared to other similar samples and used for further studies.

Radical formation on the surface of GO sheets using Ce(IV), K2S2O8 as initiators of the polymerization
The percentage of polymer grafting was calculated by Eq. 1:
The effect of [Ce(IV)] concentration, K2S2O8 and temperature (30–65 °C) on graft polymerization was also studied. Elemental analysis was performed to evaluate the graft ratio of PAM on GO and the results are given in Table 2. The weight percentage of grafted PAM for PAM-g-GO2 sample was obtained 41.40 %.
3.2 Characterization of Synthesized Structures
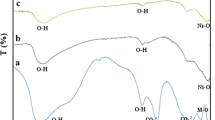
The FT-IR spectra of GO, PAM, PAM-g-GO2 and PAM-g-rGO/Pd are given in Fig. 1. As illustrated in Fig. 1a, the broad peak about 3200–3500 cm−1 is related to the –OH stretching vibration in GO structure. It also showed bands assigned to carboxyl (C=O, 1650 cm−1), epoxy (C–O, 1250 cm−1) and alkoxy (C–O, 1100 cm−1) groups situated at the edges of the GO nanosheets.

FT-IR spectrum of GO, PAM, PAM-g-GO2, PAM-g-rGO/Pd
In the spectrum of PAM-g-GO2 (Fig. 1c) and PAM-g-rGO/Pd (Fig. 1d), a new peak appeared at 2950 cm−1 associated to the asymmetric stretching vibration of CH2. The peak related to –NH stretching of the AM unit, carboxyl (C=O), and alkoxy (C–O) could also be found in the spectrum, but the adsorption band assigned to the –OH stretching vibration disappeared. Moreover, there is a significant decrease in the intensity of the adsorption bands of the oxygenated functional groups for the PAM/rGO-Pd sample. This can probably due to the existence of Pd NPs on the surface of GO and also slight reduction of GO by NaBH4 during the synthesis of the PAM/rGO-Pd nanocomposite.
Raman spectroscopy is a non-destructive optical technique. As can be seen in Fig. 2, two representative peaks around 1308 and 1584.11 cm−1 are observed for GO, which are generally assigned as D and G band, respectively. G band arises primarily from the presence of a sp2 carbon network, whereas D band is related to the vibrations of sp3 carbon atoms from defects and disorder inherent in the graphite and the edge effect of graphite crystallites [44, 45]. By comparing the relative intensities of D and G band, we can speculate the information about the disordered and ordered crystal structures of carbon. For our samples, the D/G ratios of GO, PAM-g-GO2 and PAM-g-rGO/Pd are 1.31, 1.61 and 1.90, respectively. Due to the decrease in the average size of the sp2 domains in PAM-g-GO2 and PAM-g-rGO/Pd [44, 45], the D/G intensity ratio of PAM-g-GO and PAM-g-rGO/Pd increased compared to GO. The 2D peak is also an important band in identifying graphene nanosheets. Defects on the graphene basal plane can induce the broadening of the 2D peak and the appearance of the combination mode. As shown in Fig. 2, an increase in the I (D)/I (G) ratio from GO (1.31) to PAM-g-rGO/Pd (1.90) as well as the appearance of 2D band (at 2637 cm−1) and the combination band (at 2880 cm−1) prove that the reduction of GO occurs simultaneously with the formation of Pd NPs. In addition, because of the significant difference observed between the Raman spectra of GO and PAM-g-GO2 (or between GO and PAM-g-GO/Pd), it can be concluded that PAM chains are covalently attached to GO nanosheets.

Raman spectra of graphene oxide, PAM-g-GO2 and PAM-g-GO/Pd
XPS analysis was performed for PAM-g-rGO/Pd sample, as shown in Fig. 3a–d. Typically, the high-resolution of PAM-g-rGO/Pd C1s spectrum showed six different curves (Fig. 3b). The binding energies at 284.7, 286.3, 287.2, 288.2, 289.4, and 291.4 eV were assigned to the C–C/C=C, C–O, C–H, C–N/C=O, O=C–O, and O=N–C, respectively [46, 47]. As shown in the inset of Fig. 3a, N1s band appeared at 402.2 eV which indicates the presence of PAM units in modified GO nanosheets.

XPS spectrum of a general, b curve fit of C1s spectra of PAM-g-rGO/Pd, c PAM-g-rGO/Pd(II) binding energies, and d PAM-g-rGO/Pd(0) binding energies
XPS measurements for the Pd 3d peak were also performed to explain the oxidation state of the Pd NPs (Fig. 3d). The as-prepared PAM-g-rGO/Pd2+ mainly consisted of +2 oxidation state, as evident from the measured binding energies of Pd 3d5/2 and Pd 3d3/2 electrons at 343.5 and 337.6 eV, respectively (Fig. 3c). After reduction of PAM-g-rGO/Pd2+, the peaks shifted to lower binding energy. The observed binding energy peaks of Pd 3d5/2 at 336.1 eV and Pd 3d3/2 at 341.3 eV clearly indicate the presence of Pd(0) in the PAM-g-rGO/Pd catalyst (Fig. 3d) [48].
3.3 Thermogravimetric Analysis
Thermogravimetric curves for GO, PAM, PAM-g-GO2 and PAM-g-rGO/Pd are illustrated in Fig. 4. Although GO is thermally unstable and starts to lose mass upon heating even below 100 °C, the major mass loss occurs at 200 °C, presumably due to pyrolysis of the labile oxygen-containing functional groups, yielding CO, CO2, and steam [49, 50]. The two mass loss peaks at about 200 and 420 °C of the PAM and PAM-g-GO2 are arising from the pyrolysis of the oxygen functional groups and PAM moieties, respectively [51].

TGA curves of GO, PAM, PAM-g-GO2 and PAM-g-rGO/Pd
According to Fig. 4, PAM, PAM-g-GO2, and PAM-g-rGO/Pd showed a similar pattern of weight loss, and all had two major mass losses at about 325 and 420 °C which could be the result of decomposition and carbonization of PAM, respectively [52, 53]. The overall weight loss for GO is about 34.81 % at 600 °C, but it is 84.34, 40.98 and 39.92 % for PAM, PAM-g-GO2 and PAM-g-rGO/Pd, respectively. These results prove that the thermal stability of PAM-g-GO2 is greater than PAM which is due to grafting of PAM on the surface of GO. Moreover, final char value of PAM-g-rGO/Pd nanocatlysts is somewhat higher than PAM-g-GO2 nanocomposite which is mainly on account of the presence of Pd NPs in this sample.
3.4 Phase Structure Study of Synthesized Structures
XRD analysis was used to investigate the phase structure of synthesized samples compared to the pristine materials. Figure 5 shows powder XRD results of raw graphite, GO, PAM-g-GO2 and PAM-g-GO/Pd. For the pristine graphite (Fig. 5a), there is a feature diffraction peak at about 26° (002), corresponding to an interlayer d-spacing of 0.34 nm. However, the feature diffraction peak appears at 11.6° (002) for GO (Fig. 5a), corresponds to an interlayer d-spacing of 0.76 nm.

XRD patterns of a pristine graphite and GO nano sheets, b PAM-g-GO2 and PAM-g-GO/Pd
In Fig. 5b, the strong broad peak at 2θ = 23° can be attributed to the amorphous nature of the PAM-g-GO2. For PAM-g-rGO/Pd several peaks are observed in XRD pattern at around 39.8°, 43.6°, and 78.7°. These peaks correspond to the (111), (200) and (220) planes of face centered cubic (fcc) lattice, respectively. Therefore, XRD demonstrated the presence of Pd NPs on PAM-g-rGO2 surface. In addition, the XRD patterns of the obtained black powders demonstrate the existence of Pd NPs in PAM-g-rGO2. The major diffraction lines can be indexed to the Pd fcc phase. The broad diffraction peaks of Pd indicate relatively small crystal size. The average particle size of the deposited Pd NPs is calculated to be ca. 5 nm from the (111) peak in terms of the Scherrer’s equation. For PAM-g-rGO/Pd there is no peak at 11.6°, which confirms the reduction of GO nanosheets.
3.5 Morphology Studies
In the current case, we successfully obtained AFM images of samples by liquid method. Figure 6 shows the two and three-dimensional AFM images of GO and PAM-g-GO2 surfaces. As shown in Fig. 6, AFM images of both two samples displayed single layer GO nanosheets with lateral dimensions of several micrometers confirming that GO nanosheets remained isolated after grafting of polymers. The surface of the as prepared GO is clear and smooth and its thickness is about 0.91 nm, demonstrating that the GO is single layer [46]. The surfaces of PAM-g-GO2 is uniformly covered by a layer of PAM and much rougher than GO. The thickness of PAM-g-GO2 evidently increased to 11.42 nm. These results indicated that PAM was successfully grafted onto the surface of GO.

Tapping mode AFM images for GO and PAM-g-GO2
The control of the size and dispersion of Pd NPs on PAM-g-GO is very important for their application in C–C coupling reaction. In our study, Pd NPs with controllable size and dispersion supported on PAM-g-GO was achieved by controlling the chemical reduction pathway in aqueous solution.
PAM-g-GO nanocomposite was prepared by redox system. After grafting of PAM onto GO surface, the morphology of the synthesized PAM-g-GO nanocomposite was investigated by FE-SEM and TEM. The results are illustrated in Figs. 7 and 8, respectively. The morphology of nanocomposite before and after addition of Pd NPs was monitored by FE-SEM (Fig. 7a–d). By comparing the TEM images of GO (Fig. 8a) before and after polymerization reaction (Fig. 8), it can be concluded that many dark ball-like particles formed which are attached PAM segments on GO nanosheets. Also, FE-SEM (Fig. 7) and TEM (Fig. 8) images showed that the Pd NPs on GO had uniform size distribution with diameter of 5–10 nm (Fig. 8c, d).

Field emission scanning electron microscopy (FE-SEM) images with different magnifications of a, b PAM-g-GO2 and c, d PAM-g-rGO/Pd

Transmission electron microscopy (TEM) images of a GO; b, c PAM-g-rGO/Pd and d PAM-g-rGO/Pd (180 day after in NRW)
Since most oxidized functional groups are removed during the reduction of GO, it is generally difficult to disperse the resulting graphene in solution. PAM is used as the stabilizer agent in the synthesis of the Pd-graphene hybrids. In this regard, the stability of the PAM-g-rGO/Pd composites in natural reservoir water (NRW) after exposure to natural light over a period of 180 days was investigated (Fig. 8d). The TEM images of PAM-g-rGO/Pd composites after 180 days exhibit the same phenomenon as uniform and monodispersed Pd NPs still anchored on a PAM-g-GO sheet without obvious changes. This implies that PAM-g-GO/Pd composites have excellent stability in NRW during a period of 180 days, even after exposure to natural light.
3.6 Catalytic Activity of PAM-g-rGO/Pd
The catalytic activity of PAM-g-GO/Pd was applied for C–C bond formation via a Suzuki–Miyaura coupling reaction. For this purpose, the reaction of iodobenzene (1.0 mmol) with phenylboronic acid (1.2 mmol) as a model reaction in the presence of K2CO3 (1.5 mmol) and 20 mg of PAM-g-rGO/Pd (this optimized amount of the catalyst was found through a study as shown in Table 5 and contains 0.000154 mmol of Pd as determined by ICP analysis) in different solvents at room temperature was studied (Table 3). The results of this experiment showed that H2O was the most suitable solvent for this reaction. The effect of different bases was also studied using the reaction of iodobenzene with phenylboronic acid. Among the studied bases, K2CO3 was found to be suitable (Table 4; entry 2). The results outlined in Table 5 indicate that the best result (entry 4) is obtained with the use of H2O as the solvent.
After optimizing the catalytic system for the Suzuki–Miyaura reaction, we performed the cross-coupling reactions using different aryl halides (Table 6). The coupling reaction of iodobenzene (entry 1) occurred in only 20 min. However, a long reaction time was needed to couple deactivated aryl bromides and chlorides. Table 6 also shows that both electron-rich and electron-poor aryl halides were efficiently coupled in the presence of PAM-g-rGO/Pd (entries 4–12), furnishing the cross-coupled products in excellent yields. Since PAM-g-rGO composites can stabilize the Pd NPs in H2O as a green solvent, it was our goal to determine their activity in this reaction medium. Hence, PAM-g-rGO/Pd was efficiently used for Suzuki coupling reactions under mild conditions and at a relatively low temperature in an aerobic environment and in H2O as the solvent.
The most attracting advantage of heterogeneous catalysts is their reusability. Thus, the recyclability and reusability of the supported catalyst have been investigated using iodobenzene with phenylboronic acid as model substrates. This catalyst was used successfully in eight subsequent reactions. The Pd catalyst exhibited only a slight loss in its activity and required a bit longer time to achieve full conversion even after eight cycles. The results are tabulated in Table 7. After each run, the amount of Pd leaching from 8th was estimated by performing ICP measurements on the supernatant solutions. The solutions were collected by filtering the catalyst from the reaction mixture after the reactions were complete. Based on the results from TEM (Fig. 8c) and ICP analyses, there was no significant agglomeration and leaching of Pd NPs after the recycling experiments. It is worth noting that bare Pd nanostructures or even supported Pd NPs in conventional Suzuki coupling reactions usually suffer from significant loss in the activity due to Pd leaching and agglomeration [20].
Generally, highly dispersed metal NPs on supports with smaller diameter are known to have higher catalytic activity due to the advantages of increased low coordination numbers in NPs and larger surface area, resulting in larger number of active sites [54]. Therefore, the high catalytic activity can be attributed to small particle size and high dispersion of Pd NPs. The size-dependent electronic structural change may also contribute to increased catalytic efficiency [55]. On the basis of obtained results, it could be suggested that the current PAM-g-rGO/Pd is very stable and efficient catalysts for Suzuki–Miyaura cross-coupling reactions.
The catalytic performance of different Pd-based catalysts in the coupling of bromobenzene and phenyl boronic acid are shown in Table 8. The PAM-g-rGO/Pd shows the highest performance in comparison to other catalysts. Most of the other published procedures (Table 8) need long times, high temperature and use toxic solvents with lower isolated yields.
The products of reaction were fully characterized through melt point, 1H NMR and 13C NMR experiments which were in accordance with the literatures. The results are as follows:
Biphenyl [62]: White solid, mp: 67–70 °C. 1H NMR (300 MHz, CDCl3, δ in ppm): 7.45 (m, 2H), 7.56 (m, 4H), 7.72 (m, 4H). 13C NMR (75.4 MHz, CDCl3, δ in ppm): 141.2, 128.7, 127.2, and 127.1.
4-Nitrobiphenyl [63]: Brown solid, mp: 111–113 °C. FT-IR (cm−1): 3055, 2921, 1603, 1521, 1477, 1446, 1341, 1306, 1087, 1012, 845, 741. 1H NMR (400 MHz, CDCl3, δ in ppm): 7.44 (m, 3H), 7.63 (m, 2H), 7.77 (m, 2H), 8.46 (m, 2H). 13C NMR (75.4 MHz, CDCl3, δ in ppm): 147.9, 147.3, 139.0, 129.5, 129.2, 128.0, 127.7, 124.4.
4 Conclusion
In summary, PAM chains were successfully grafted to the GO surface via redox system. Then, the immobilization of Pd NPs was carried by reducing Pd(II) salts in the presence of PAM-g-GO. The formation of Pd NPs on the reduced graphene oxide surface was visually observed with the mixture change in color to black and it was also confirmed by FT-IR, TEM and X-ray analyses measurements. The PAM-g-rGO/Pd catalysts prepared showed high activity in Suzuki–Miyaura coupling reaction carried out in water under in aerobic condition. Recovery and reusability of the supported catalyst were achieved without significant loss of catalytic activity and low leaching of Pd. Generally, compared with literature examples of the Suzuki–Miyaura cross-coupling reaction, the notable features of our nanocatalyst are:
-
The reaction system is simple.
-
The Suzuki–Miyaura cross-coupling reaction is carried out in low temperature.
-
Organic solvents are not needed.
-
H2O was used as a green solvent.
-
Toxic and expensive ligands are not needed.
-
The yields of the products are very high (due to small particle size and high dispersion of Pd NPs).
-
The catalyst can be easily recovered.
-
PAM-g-rGO as a support for Pd NPs is biocompatible.
References
Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, Grigorieva IV, Firsov AA (2004) Science 306:666
Lee C, Wei X, Kysar JW, Hone J (2008) Science 321:385
Machado BF, Philippe S (2012) Catal Sci Technol 2:54
Gao L, Yue W, Tao S, Fan L (2013) Langmuir 29:957
Zhu Y, Murali S, Cai W, Li X, Suk JW, Potts JR, Ruoff RS (2010) Adv Mater 22:3906
Kuila T, Bose S, Mishra AK, Khanra P, Kim NH, Lee JH (2012) Prog Mater Sci 57:1061
Bielawski CV, Dreyer DR, Park S, Ruoff RS (2010) Chem Soc Rev 39:228
Jiang K, Ye CN, Zhang PP, Wang XS, Zhao YL (2012) Macromolecules 45
Lin Y, Jin J, Song M (2011) J Mater Chem 21:3455
Deng Y, Li YJ, Dai J, Lang MD, Huang XY (2011) J Polym Sci Polym Chem 49:1582
Kan L, Xu Z, Gao C (2011) Macromolecules 44:444
Yuan W, Wang J, Shen T, Ren J (2013) Mater Lett 107:243
Wang BD, Yang D, Zhang JZ, Xi CB, Hu JH (2011) J Phys Chem C 115:24636
Degirmenci M, Hicri S, Yilmaz H (2008) Eur Polym J 44:3776
Ramanathan T, Abdala AA, Stankovich S (2008) Nat Nanotechnol 3:327
Putz KW, Compton OC, Palmeri MJ, Nguyen ST, Brinson LC (2010) Adv Funct Mater 20:3322
Ma L, Yang X, Gao L, Lu M, Guo C, Li Y, Tu Y, Zhu X (2013) Carbon 53:269
Wang B, Yang D, Zhang JZ, Xi C, Hu J (2011) J Phys Chem C 115:24636
Balanta A, Godard C, Claver C (2011) Chem Soc Rev 40:4973
Molnar A (2011) Chem Rev 111:2251
Tamami B, Mohaghegh Nezhad M, Ghasemi S, Farjadian F (2013) J Org Chem 743:10
Guan ZH, Hu JL, Gu YL, Zhang HJ, Li GX, Li T, (2012) Green Chem 14:1964
Santra S, Hota PK, Bhattacharyya R, Bera P, Ghosh P, Mandal SK (2013) ACS Catal 3:2776
Park G, Lee S, Son SJ, Shin S (2013) Green Chem 15:3468
Chen W, Zhong, L-X, Peng X-W, Wang K, Chena Z-F, Sun R-C (2014) Catal Sci Technol 4:1426
Jawale DV, Gravel E, Boudet C, Shah N, Geertsen V Li H, Namboothiri INN, Doris E (2015) Catal Sci Technol 5:2388
Deraedt C, Salmon L, Astruc D (2014) Adv Synth Catal 356:2525
Kim H, Abdala AA, Macosko CW (2010) Macromolecules 43:6515
Tamami B, Ghasemi S (2010) J Mol Catal A Chem 322:98
Golubeva ND, Dyusenalin BK Selenovab BS, Pomogailo SI, Zharmagambetova AK, Dzhardimalieva GI, Pomogailo AD (2011) Kinet Catal 52:242
Pan HB, Yen CH, Yoon B, Sato M, Wai CM (2006) Commun 36:3473
Sullivan JA, Flanagan AF, Hain H (2009) Catal Today 145:108
Siamaki AR, Lin Y, Woodberry K, Connell JW, Gupton BF (2013) J Mater Chem A 1:12909
Gao S, Shang N, Feng C, Wang C, Wang Z (2014) RSC Adv 4:39242
Yamamoto S-I, Kinoshita H, Hashimotob H, Nishin Y (2014) Nanoscale 6:6501
Hoseini SJ, Dehghani M, Nasrabadi H (2014) Catal Sci Technol 4:1078
Putta C, Sharavath V, Sarkara S, Ghosh S (2015) RSC Adv 5:6652
He D, Kou Z, Xiong Y, Cheng K, Chen X, Pan M, Mu S (2014) Carbon 66:312
Lee KH, Han S-W, Kwon K-Y, Park JB (2013) J Colloid Interf Sci 403:127
Hummers WS, Offeman RE (1958) J Am Chem Soc 80:1339
Hebeish A, Bayazeed A, El-Alfy E, Khalil M (1988) Starch/Starke 40:223
Huang Y-L, Tien H-W, Ma C-CM., Yang S-Y Wu S-Y, Liu H-Y, Mai Y-W (2011) J Mater Chem 21:18236
Shanmugharaj AM, Yoon JH, Yang WJ, Ryu SH (2013) J Colloid Interf Sci 401:148
Stankovich S, Dikin DA, Piner RD, Kohlhaas KA, Kleinhammes A, Jia Y, Wu Y, Nguyen ST, Ruoff RS (2007) Carbon 45:1558
Ferrari AC (2007) Solid State Commun 143:47
Zhang FB, Fan XB, Peng WC, Li Y, Li XY, Wang SL, Zhang GL (2008) Adv Mater 20:4490
Tasis D, Papagelis K, Prato M, Kallitsis I (2007) Macromol Rapid Commun 28:1553
Parambhath VB, Nagar R, Ramaprabhu S (2012) Langmuir 28:7826
Wang G, Yang Z, Li X, Li C (2005) Carbon 43:2564
Paredes JI, Villar-Rodil S, Solis-Fernandez P, Martinez-Alonso A, Tascon JD (2009) Langmuir 25:5957
Sun S, Wu P (2011) J Mater Chem 21:4095
Yang MH (2002) J Appl Polym Sci 86:1540
Ke Y, Wang YJ, Ren L, Wu G, Xue W (2010) J Appl Polym Sci 118:390
Remediakis IN, Lopez N, Norskov JK (2005) Angew Chem Int Ed 44:1824
Santra AK, Yang F, Goodman DW (2004) Surf Sci 548:324
Ren L, Yang F, Li Y, Liu T, Zhang L, Ning G, Liu Z, Gao J, Xu C, (2014) RSC Adv 4:26804
Putta C, Sharavath V, Sarkar S, Ghosh S (2015) RSC Adv 5:6652
Moussa S, Siamaki AR, Gupton BF, El-Shall MS (2012) ACS Catal 2:145
Niu J, Liu M, Wang P, Long Y, Xie M, Li R, Ma J (2014) New J Chem 38:1471
Shendage SS, Patil BU, Nagarkar MJ (2013) Tetrahedron Lett 54:3457
Ba C, Zhao Q, Li Y, Zhang G, Zhang F, Fan X (2014) Catal Lett 144:1617
Nandurkar NS, Bhange BM (2008) Tetrahedron 64:3655
Riggleman S, DeShong P (2003) J Org Chem 68:8106
Acknowledgments
We are thankful to the Research Council of the University of Tehran.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mahdavi, H., Rahmani, O. Polyacrylamide-g-Reduced Graphene Oxide Supported Pd Nanoparticles as a Highly Efficient Catalyst for Suzuki–Miyaura Reactions in Water. Catal Lett 146, 2292–2305 (2016). https://doi.org/10.1007/s10562-016-1851-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-016-1851-1