
Abstract
A graphene supported palladium (Pd) catalyst for Suzuki coupling reaction has been successfully prepared by immobilizing Pd(II) onto graphene oxide surface through the in situ coordination interaction with aminosilane ligand spacers. This catalyst showed high catalytic activities in the Suzuki coupling of various aryl halides and phenylboronic acid. Moreover, it could be readily recycled and reused for several times without discernible loss of its catalytic activity.
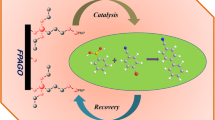
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Palladium (Pd)-catalyzed reactions play a key role in the synthesis of many important chemicals [1–4]. As an efficient catalyst in organic reactions, it can offer the most beneficial combination of activity and selectivity [5]. Among these reactions, Suzuki coupling is the most powerful tool for constructing the biaryl structures that exist in many biologically active compounds and natural products [6, 7]. Usually, the Suzuki coupling reactions were catalyzed by Pd complex under homogeneous condition. However, the high cost of the catalysts and their low efficiency in separation limit their applications on an industrial scale. Therefore, considerable efforts have been made to immobilize Pd complex on various separable organic, inorganic or hybrid supports for decades [8, 9]. Unfortunately, the supported Pd complex catalysts often suffer from reduced activity, inferior selectivity, and other problems such as metal leaching and high preparation complexity.
Recently, graphene [10] and graphene oxide (GO) [11, 12] have attracted extensive attention in heterocatalysis due to their unique two-dimensional structures, huge surface areas and other excellent properties [13, 14]. In fact, they have been demonstrated as promising supports for many metallic nanocatalysts, such as Pd [15–17], Au [18], Co3O4 [19], Fe3O4 [20], AucorePtshell nanoparticles [21] and PdAg nanorings [22].
In this study, we report a facile and efficient method to immobilize Pd complexes on GO. The supported Pd catalyst was prepared by the in situ coordination between Pd(II) and aminosilane ligand that was covalently immobilized on GO via the one-pot silylation (Scheme 1). We found that this catalyst afforded high catalytic activities for the Suzuki coupling of various halogenobenzene and phenylboronic acids.

Illustration for synthetic methodology of GO–2N–Pd(II) and its catalytic application for the Suzuki coupling reaction
2 Experimental
2.1 Reagents and Materials
N-[3-(Trimethoxysilyl)propyl] ethylenediamine (AAPTS, 95 %), Pd(II) chloride and graphite powder were purchased from Sigma-Aldrich Chemical Company (USA). Various aryl halide, phenylboronic acid and ethanol (99.8 %) were purchased from Aladdin Reagent Company (Shanghai, China). All other chemicals were of analytical grade and used as received without further purification.
2.2 Synthesis of GO and Ethylenediamine-Functionalized GO (GO–2N)
GO was prepared and purified by modified Hummers Method [23]. A suspension was obtained by dispersing GO (12.0 mg mL−1, 20.0 mL) in 120 mL ethanol with the aid of intensive sonication (100 W, 40 kHz, 0.5 h). Ethylenediamine-functionalized GO was processed by stirring the obtained GO suspension with excess AAPTS (373.6 mg, 1.68 mmol) and refluxed for 6 h. The suspension was filtrated and intensively washed with water, ethanol and methanol twice, respectively, followed by freeze-drying.
2.3 Synthesis of Ethylenediamine-Functionalized GO Immobilized Pd Complex [GO–2N–Pd(II)]
200 mg of the as-prepared GO–2N was added to a methanol solution of Pd(II) chloride (177 mg, 1 mmol). After the mixed system was stirred at room temperature for 24 h, the resulting precipitates were then thoroughly filtrated and washed with methanol, ethanol and water, twice, respectively. A black solid was obtained after a freeze-drying procedure.
2.4 Typical Procedures for the Suzuki Reaction
A mixture of aryl halide (1 mmol), phenylboronic acid (1.5 mmol), K2CO3 (2 mmol), ethanol (15 mL) and GO–2N–Pd(II) (10 mg, 0.5 mol%) were stirred at 80 °C in a 25 mL round bottom flask. Thereafter, the catalyst was simply separated from the mixture by simple centrifugation and could be reused after washed with ethanol three times. For the control experiments, samples containing the same amount of Pd species (0.005 mmol) were used as the catalyst. The resulting solution was analyzed by gas chromatography (GC) using a capillary column.
2.5 Instrumentations
The samples were characterized by Fourier transform infrared spectroscopy (FTIR, Thermo Nicolet Nexus FTIR), X-ray photoelectron spectroscopy (XPS, PerkinElmer, PHI 1600 spectrometer), Raman spectroscopy (NT-MDT NTEGRA Spectra), scanning electron microscopy (SEM, Hitachi S4800), energy dispersive X-ray spectroscopy (EDS, Hitachi S4800), transmission electron microscopy (TEM, Philips Tecnai G2 F20), thermogravimetric analysis (TGA, Shimadzu TGA-50), 1H and 13C NMR (Varian-INOVA, 500 MHz) and inductively coupled plasma optical emission spectroscopy (ICP-OES, Vista-MPX). The catalytic results were measured by Agilent 6890N GC using the standard curve method.
3 Results and Discussion
3.1 Characterization of GO–2N and GO–2N–Pd(II)
The typical FTIR absorption peaks of GO could be observed in Fig. 1. Comparison studies on the spectra of GO and GO–2N identified the incorporation of ethylenediamine group silanes onto the GO surface. Note that bands at around 3,400 cm−1 of both GO and GO–2N ascribed to the absorbed water. The doublet at 2,927 and 2,854 cm−1 were assigned to the stretching vibrations of CH2 that existed on the long silane chains which were obviously strengthened compared to GO. The obvious band at 1,400 cm−1 was assigned to C–H deformation vibrations of alkyl groups which could also verify the presence of long silane chains. The prominent bands at 3,240 and 1,589 cm−1 were ascribed to the stretching and bending vibrations of N–H which testified the successful introduction of amines group [24]. Stretching vibrations of C–N at around 1,631 cm−1 could also be observed. In addition, the strong absorption of Si–O (at 1,100 and 1,053 cm−1) and stretching vibrations of Si–O–C at 686 cm−1 provided direct evidences for the successful silylanization of GO [25]. Similar characteristic bands could be found in the FTIR of free AAPTS (Fig. S1).

FTIR spectra of GO and GO–2N
These results were further confirmed by XPS analysis. Additional signals of Si and N were clearly observed in the XPS spectrum of GO–2N (Fig. S3b). C 1s XPS spectrum of GO–2N showed a significant decrease in the C–O–C and C–OH peaks at around 286.5 eV after the silylation reaction (Fig. S3d), which suggested a structure deprived of parts of hydroxy groups [26]. A pronounced peak at 101.7 eV corresponding to the binding energy of Si–O–C appeared at the same time (Fig. 2a), indicating that considerable amounts of C–OH were converted into Si–O–C through the silylation process [27]. Besides, compared with the N 1s XPS spectrum of GO–2N (Fig. 2b), the binding energy shifted from 399.36 to 399.52 eV after the coordination reaction, and this was attributed to the stronger coordination bond between the ethylenediamine group and the Pd centers [28]. The loading of the nitrogen atoms in GO–2N–Pd(II) was 3.94 wt% and the atom percentage of N–Pd is approximately 4.0, corresponding to the structure of the GO–2N–Pd(II) catalyst. Note that additional signals of Pd were clearly observed in the obtained GO–2N–Pd(II) (Fig. 2c) with the Pd weight percentage of 5.38 %, which was much higher than those on other supports [29, 30]. In addition, all the Pd species in the GO–2N–Pd(II) presented in +2 oxidation state, corresponding to the binding energy of 337.75 eV in Pd 3d5/2 level (Fig. 2d) [31].

a Si 2p XPS spectrum of GO–2N, N 1s XPS spectra of b GO–2N–Pd(II) and b, inset GO–2N, c XPS spectrum of GO–2N–Pd(II) and d Pd 3d XPS spectrum of GO–2N–Pd(II)
Raman spectra of GO, GO–2N and GO–2N–Pd(II) (Fig. 3) showed an obvious and step-by-step blue shift of the G band from 1,578 to 1,590 and 1,594 cm−1, probably due to the gradually increased compressive local stress caused by molecule intercalation. Besides, the intensity ratio of D and G bands (I D/I G) that related to the extent of disorder also increased [1.78, 1.96 and 2.35 for the GO, GO–2N and GO–2N–Pd(II), respectively], reflecting the formation of more edges and defects during the reaction processes [32].

Raman spectra of GO, GO–2N and GO–2N–Pd(II)
SEMs and corresponding quantitative EDS mapping were employed to determine the morphology of the obtained GO–2N–Pd(II). Figure 4a presented the typical planar structure of GO–2N–Pd(II) maintaining the two-dimensional structures with heavy crumpling features. Element distribution analysis displayed a homogeneous distribution of element Si, Cl and Pd (Fig. 4b–d) on the whole surface of GO, implying the consistent attachments of the silylation of GO and a desirable anchoring of the Pd complexes.

a SEM image of GO–2N–Pd(II), and corresponding quantitative EDS mapping of b Si, c Cl and d Pd
TGA analysis of the samples were also used to investigate the thermal stability of GO, GO–2N and GO–2N–Pd(II) (Fig. 5). For GO, the weight loss was about 38 % at the temperature under 240 °C. This major weight loss was due to pyrolysis of the labile oxidation functional groups on GO. By comparison, GO–2N and GO–2N–Pd(II) showed slighter decrease in this region because some of the hydroxyl groups have been treated with silanization. In addition, GO–2N and GO–2N–Pd(II) showed weight loss from 240 to 550 °C which was attributed to the oxidation of ethylenediamine groups. At the same time, there was no obvious weight loss above 600 °C. The results indicated that the ethylenediamine groups were chemically bonded to the surface of GO and the coordination between the Pd and ethylenediamine groups exhibited a relatively high stability.

TGA graphs of GO, GO–2N and GO–2N–Pd(II)
3.2 GO–2N–Pd(II) Catalyzing Suzuki Reaction
The catalytic performance of the prepared catalyst was systematically examined through Suzuki coupling reaction. We used ethanol as a medium because it was not only safe, cost-effective but also efficiently promoted the Suzuki reaction. The Suzuki reaction of iodobenzene and phenylboronic acid was chosen as the first model reaction, and the results were summarized in Table 1. We found that GO–2N–Pd(II) at a loading of 0.5 mol% Pd (with respect to iodobenzene) afforded biphenyl in 100 % within only 0.5 h at 80 °C (Table 1, Entry 3). The yield went down when the catalyst loading was reduced to 0.1 mol%. Otherwise, the yield remained unchanged when the catalyst loading rose to 1 mol%. As for the influence of temperature, 80 °C was found to be the optimal choice. The yield went down with the drop of temperature while the reaction effectively occurred at 80 °C as the same as 100 °C. Therefore, the optimized reaction conditions were also employed in later Suzuki coupling reactions with variation of different substituents.

To investigate the effective component of the catalyst, a series of control experiments were conducted. The results were summarized in Table 2. When no catalyst was used or GO–2N was used as a control, hardly any reaction occurred (Entries 1 and 2). The results indicated that the Suzuki reaction could not proceed with the absence of Pd. When a physical mix of GO, AAPTS and PdCl2 or a mix of charcoal, AAPTS and PdCl2 was used to catalyze the reaction, poor yield was obtained and this might be due to the absence of coordinating ligands (Entries 3 and 4). When the supported catalyst GO–2N–Pd(II) was used, the yield was fond to be 100 % (Entry 5). It seemed that the linkage ligand between GO and the Pd complex promoted the reaction and made the catalyst achieve excellent catalytic performance.
As shown in Table 3, GO–2N–Pd(II) was also found to be highly effective for different substituents. Various electron-donating and electron-withdrawing groups, such as –OCH3 and –CH3 on the aryl iodide were well tolerated (Entries 1–3), and a satisfactory yield for bromides containing –CH3, –NH2, and –OH (Entries 4–7) was achieved. Biphenyl-4-carbaldehyde gave 53 % in yield when 4-bromo-benzaldehyde was coupled with phenylboronic acid (Entry 8). Note that aryl bromide containing electron-withdrawing substituents reacted faster than those with electron-donating substituents which is due to the electronic effects of the substituent (Entry 5 vs. 6). Further functional group tolerance of the catalyst was tested with the heteroaryl halides delivering good yield of 78 and 75 %, respectively (Entries 9 and 10). Additionally, the catalyst showed high activity towards aryl chlorides but with much longer time compared with aryl iodides and aryl bromides (Entries 11 and 12). This result could be attributed to the different strengths of the C–I, C–Br and C–Cl bonds, as well as the different electron-withdrawing abilities of the halogen substituents [33].

The reusability of GO–2N–Pd(II) was evaluated between iodobenzene and phenylboronic acid by repeating the same procedure. The solid catalyst was readily recovered from the reaction mixture by direct filtration and reused in sequential runs. As shown in Fig. 6, the catalytic activity almost did not reduce after reuse of six times. Comparison TEM study of GO–2N–Pd(II) before and after repeated experiments were shown in Fig. 7. Compared to the fresh one, Pd nanoparticles dispersed on the GO sheets could be observed after several runs. This was in agreement with the previously reported results that the formation of Pd nanoparticles as an active spices were evolved during the catalytic reaction. To further investigate the stability of the used catalyst, ICP-OES analysis was also employed. We found that only about 0.5 % of the Pd species leached off after six cycles, suggesting that the loss of the active Pd(II) sites could be neglected. The high stability and excellent reusability of the catalyst should be attributed to the strong coordination bonds.

Recycle efficiency of GO–2N–Pd(II) in Suzuki coupling reaction between iodobenzene and phenylboronic acid

TEM images of GO–2N–Pd(II) a, b before and c, d after repeated experiments
4 Conclusions
In summary, we successfully developed a novel, practical and economic GO supported Pd complex catalyst. The GO–2N–Pd(II) catalyst showed extraordinary performances in the Suzuki coupling reaction and could be readily recycled and reused without discernible loss of its catalytic activity. This strategy could be used to prepare other GO supported metal complexes which may offer more opportunities for developing powerful and reusable catalysts for green organic synthesis.
References
Chen Q-A, Ye Z-S, Duan Y, Zhou Y-G (2013) Chem Soc Rev 42:497
Santra S, Ranjan P, Bera P, Ghosh P, Mandal SK (2012) RSC Adv 2:7523
Sore HF, Galloway WRJD, Spring DR (2012) Chem Soc Rev 41:1845
Szőri K, Puskás R, Szőllősi G, Bertóti I, Szépvölgyi J, Bartók M (2013) Catal Lett 143:539
Jie X, Shang Y, Hu P, Su W (2013) Angew Chem Int Ed 52:3630
Han F-S (2013) Chem Soc Rev 42:5270
Shang N, Gao S, Feng C, Zhang H, Wang C, Wang Z (2013) RSC Adv 3:21863
Borah BJ, Dutta DK (2013) J Mol Catal A 366:202
Fang M, Fan G, Li F (2014) Catal Lett 144:142
Liu K, Chen T, Hou Z, Wang Y, Dai L (2014) Catal Lett 144:314
Chua CK, Pumera M (2014) Chem Soc Rev 43:291
Meyer JC, Geim AK, Katsnelson MI, Novoselov KS, Booth TJ, Roth S (2007) Nature 446:60
Dreyer DR, Park S, Bielawski CW, Ruoff RS (2010) Chem Soc Rev 39:228
Tan R, Li C, Luo J, Kong Y, Zheng W, Yin D (2013) J Catal 298:138
Morimoto N, Yamamoto S-i, Takeuchi Y, Nishina Y (2013) RSC Adv 3:15608
Santra S, Hota PK, Bhattacharyya R, Bera P, Ghosh P, Mandal SK (2013) ACS Catal 3:2776
Shang N, Feng C, Zhang H, Gao S, Tang R, Wang C, Wang Z (2013) Catal Commun 40:111
Qin Y, Li J, Kong Y, Li X, Tao Y, Li S, Wang Y (2014) Nanoscale 6:1281
Yao Y, Yang Z, Sun H, Wang S (2012) Ind Eng Chem Res 51:14958
Feng C, Zhang H-Y, Shang N-Z, Gao S-T, Wang C (2013) Chin Chem Lett 24:539
Qi J, Lv W, Zhang G, Li Y, Zhang G, Zhang F, Fan X (2013) Nanoscale 5:6275
Liu M, Lu Y, Chen W (2013) Adv Funct Mater 23:1289
Hummers WS, Offeman RE (1958) J Am Chem Soc 80:1339
Ou X, Jiang L, Chen P, Zhu M, Hu W, Liu M, Zhu J, Ju H (2013) Adv Funct Mater 23:2422
Zhao Q, Li Y, Liu R, Chen A, Zhang G, Zhang F, Fan X (2013) J Mater Chem A 1:15039
Kim S, Zhou S, Hu Y, Acik M, Chabal YJ, Berger C, de Heer W, Bongiorno A, Riedo E (2012) Nat Mater 11:544
Ma PC, Kim J-K, Tang BZ (2006) Carbon 44:3232
Kannari N, Ozaki J-i (2012) Carbon 50:2941
Zhang Q, Su H, Luo J, Wei Y (2013) Tetrahedron 69:447
Wei SY, Ma ZY, Wang P, Dong ZP, Ma JT (2013) J Mol Catal A 370:175
Bera P, Patil KC, Jayaram V, Subbanna GN, Hegde MS (2000) J Catal 196:293
Fang M, Wang K, Lu H, Yang Y, Nutt S (2009) J Mater Chem 19:7098
Alonso F, Beletskaya IP, Yus M (2008) Tetrahedron 64:3047
Acknowledgments
This study was supported by the National Natural Science Funds for Excellent Young Scholars (No. 21222608), Program for New Century Excellent Talents in University (No. NCET-12-0392), Research Fund of the National Natural Science Foundation of China (No. 21106099), Foundation for the Author of National Excellent Doctoral Dissertation of China (No. 201251), the Tianjin Natural Science Foundation (No. 11JCYBJC01700) and the Programme of Introducing Talents of Discipline to Universities (No. B06006).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bai, C., Zhao, Q., Li, Y. et al. Palladium Complex Immobilized on Graphene Oxide as an Efficient and Recyclable Catalyst for Suzuki Coupling Reaction. Catal Lett 144, 1617–1623 (2014). https://doi.org/10.1007/s10562-014-1299-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-014-1299-0