
Abstract
The organic–inorganic hybrid catalyst L-Mn-PMoV was prepared simply by combining a schiff base Mn complex (L-Mn, L: N,N′-disalicylidene-1, 6-hexanediamine) with the Keggin-structured molybdovanadophosphoric heteropolyacid (PMoV). The proposed composition and structure of the catalyst were evidenced by TG, elemental analysis, FT-IR, and UV–Vis characterizations. Its catalytic performance was evaluated in the direct hydroxylation of benzene to phenol by molecular oxygen with ascorbic acid as the reducing agent. Various reaction parameters were changed to attain the optimal conditions. The hybrid catalyst has a formula [{Mn(C20H22N2O2)(Cl)}2(H4PMo11VO40)], with the two terminal oxygen atoms in the PMoV Keggin structure coordinately linked to the two Mn(III) ions in two L-Mn units, respectively. It exhibits a remarkably enhanced yield to phenol compared to the pure PMoV due to the synergy effect between the Schiff-base manganese complex and PMoV.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Phenol is one of the most important petrochemicals for the manufacture of fine chemicals, agrochemicals and plastics [1]. Currently, it is mainly produced by the three-stepped cumene process, which suffers from a vast amount of energy consumption and the creation of the equimolar by-product, acetone [2]. Therefore, the direct hydroxylation of benzene into phenol has been attracting great interest for tens of years [3, 4]. Using molecular oxygen as the oxidant in the hydroxylation of benzene is highly desirable because the resulting process would be green and economical [5]. For this process, Cu [6], Pt [7], Pd [8], and V [9] catalysts supported on porous carriers have been reported. Also, palladium itself was used as the catalyst with the heteropolyacid as the reoxidant for palladium acetate [10]. Liu et al. [5] used [(C4H9)4N]5[PW11CuO39(H2O)] as the catalyst for the liquid-phase hydroxylation of benzene to phenol by molecular oxygen with ascorbic acid as a reducing agent in an acetone/sulfolane/water-mixed solvent, showing 9.2% of benzene conversion and 91.8% of selectivity to phenol at 323 K and 12 h. More recently, the yield of phenol was remarkably enhanced by employing molybdovanadophosphoric acid as the catalyst and carbon monoxide as the reducing agent [11]. However, so far, studies on efficient catalysts for hydroxylation of benzene to phenol by molecular oxygen still remain as a huge challenge.
Organic–inorganic hybrid materials have recently drawn tremendous attention in the field of optics, electromagnetics, biology and catalysis [12, 13]. Polyoxometalates (POM) as the inorganic component in these hybrid materials is of great importance because of the structural diversity, as well as the activity in relation to catalysis, analytical chemistry, medicine and materials science [14]. In particular, Keggin-structured heteropolyacids have been widely used as catalysts in oxidation reactions due to their tailorable redox properties [15]. Various organic units such as aliphatic amines [16], metalloporphyrins [17], and bipyrimidinylplatinum [18] were doped into Keggin heteropolyacids to modify their catalytic performances in oxidation reactions. Recently, we observed the promotion effects of cyclodextrin [19] and pyridine [20, 21] in the hydroxylation of benzene to phenol by molecular oxygen and/or hydrogen peroxide when anchoring them to Keggin heteropolyacids.
On the other hand, Schiff base metal complexes are well-known catalysts for many oxidation reactions [22], thus, the combination of Schiff base metal compounds with heteropolyacids should lead to a kind of novel organic–inorganic hybrid materials, the catalytic property of which may be reinforced by the two moieties [17]. Nevertheless, they are largely overlooked in catalysis context [23]. Until recently, Mirkhani et al. [24] prepared a series of hybrid compounds (M-salen-POM, M = Fe, Co, Ni, Mn), revealing their good catalytic activities in the oxidation of alkane and alkene.
In this work, we prepare a new organic–inorganic hybrid catalyst by attaching a manganese Schiff base complex to Keggin-structured molybdovanadophosphoric heteropolyacid (PMoV), and use it as the catalyst in the hydroxylation of benzene to phenol with molecular oxygen as the oxidant, observing a remarkably enhanced yield of phenol due to the synergy effect between the Schiff base manganese complex and PMoV.
2 Experimental
2.1 Preparation of the Catalyst L-Mn-PMoV
All solvents and reagents (analytical grade) were purchased commercially and were used as it is. H4PMo11VO40 (PMoV) was prepared according to the procedure described in our previous report [20]. MoO3 (17.77 g, 120 mmol) and V2O5 (1.02 g, 5.61 mmol) were added to 250 mL of deionized water. The mixture was heated up to the reflux temperature, when the aqueous solution of H3PO4 (85 wt%) (1.29 g, 13.16 mmol) was added drop-wise to the reaction mixture. After a clear orange-red solution appeared, the solution was cooled to the room temperature. The orange-red powder PMoV product was obtained by evaporation of the solution to dryness, re-crystallizing for purification, and further drying in a vacuum oven at 373 K for 12 h.
The manganese Schiff base complex that is designated as L-Mn (L: N,N′-disalicylidene-1, 6-hexanediamine) was prepared following the previous literature [25]. The ethanol (40 mL) solution of salicylaldehyde (21 g, 86.20 mmol) was added to the ethanol (20 mL) solution of 1, 6-hexanediamine (10 g, 86.20 mmol) under stirring, and the mixture was refluxed at 353 K for 6 h. After cooled to room temperature, the resulting precipitate was filtered off, re-crystallized in ethanol, and dried in a vacuum oven at 353 K for 12 h to obtain the yellow crystals, the L ligand. Afterwards, the solution of MnCl2·4H2O (1.90 g, 9.57 mmol) in ethanol (40 mL) was added to the solution of L (3.10 g, 9.57 mmol) in hot ethanol (50 mL). The obtained mixture was stirred further at room temperature for 3 h; the dark green precipitate, L-Mn, was collected by filtration, washing with ethanol, and drying in a vacuum oven at 353 K for 12 h.
For preparing L-Mn-PMoV, a molar ratio of L-Mn to PMoV of 2:1 was used. In detail, the solution of L-Mn (0.25 g, 0.56 mmol) in methanol (40 mL) was added to the solution of H4PMo11VO40 (0.05 g, 0.28 mmol) in methanol (10 mL) under vigorous stirring, and the mixture was stirred at room temperature for 12 h. The resulting yellow precipitate was collected by filtration, washing with methanol, and drying in a vacuum oven at 333 K for 12 h to give the solid catalyst, L-Mn-PMoV. Another sample L-Mn-PMoV# was obtained by the same way as described above, except that the molar ratio of L-Mn to PMoV was 1:1 rather than 2:1.
2.2 Characterizations of Catalysts
C and N elemental analyses were obtained on a Vario EL III elemental analyzer. Thermal gravimetric analysis (TGA) was conducted on a TA Instrument (Netzsch, TG/209/F3) operated under the air atmosphere. The temperature program was a simple linear ramp from 303 to 873 K with the ramp rate of 10 K min−1. Infrared spectra (IR) were collected using KBr pellets on a Nexus 870 Fourier transform infrared spectrophotometer. To make the KBr pellet, 1 mg of sample was mixed with approximately 80 mg of KBr. The spectrum of air was subtracted from the spectrum for the tested sample. UV–Vis absorption spectra were recorded on a PE Lambda 35 UV–Vis spectrometer using dimethyl sulphoxide (DMSO) as the solvent.
2.3 Catalytic Tests
The hydroxylation of benzene was carried out in a customer-designed temperature controllable pressured titanic reactor (100 mL) equipped with a mechanical stirrer. In a typical experiment, 0.1 g (0.038 mmol) catalyst, 0.60 g ascorbic acid, and 2 mL benzene were added into 25 mL of the aqueous solution of acetonitrile (50 vol%) successively. After the system was charged with 2.0 MPa O2 at room temperature, the hydroxylation reaction was conducted at 373 K for 10 h with vigorous stirring. After the reaction, 1,4-dioxane was added into the product mixture as an internal standard for product analysis. The mixture was analyzed by a gas chromatograph (GC) with a FID and a capillary column (SE-54; 30 m × 0.32 mm × 0.25 μm). Under the reaction conditions, phenol was the only product detected by GC, and the commonly seen by-products (catechol, hydroquinone and benzoquinone) were not found.
3 Results and Discussion
3.1 Characterization of the Catalyst
Regarding the mode of interaction in organometallic-POM hybrid materials, both the electrostatic interaction [18, 26, 27] and the coordinate one [28, 29] have been reported, which are very helpful to understand the newly synthesized composite here. Summarizing the interpretations described in the previous works and used in the present study, we suppose that the L-Mn-PMoV sample should have a formula of [{Mn(C20H22N2O2)(Cl)}2(H4PMo11VO40)]. In this case, it is a dinuclear complex formed by the coordinate bonding of the two terminal oxygen atoms in the PMoV Keggin structure with two Mn(III) ions in two L-Mn units, respectively; the Mn(III) ion locates at the center of an octahedral geometry.
In order to verify the above-proposed composition and structure for L-Mn-PMoV, it was subjected to characterizations of TG, organic elemental analysis, FT-IR, and UV–Vis. In TG analysis (Fig. 1), the slight weight loss at early heating stage around 423 K is due to the release of moisture and constitutional water. Whereafter, the large weight loss up to 823 K is attributed to the decomposition of both organic and inorganic moieties of the hybrid catalyst into their individual oxides (P2O5, MoO3, V2O5, and Mn2O3), which accounts for 26.90% of the weight loss. This value is very near to the calculated one: 28.12%, following the proposed formula, which indicates the reasonableness of that composition. Data of the organic elemental analysis for the L-Mn-PMoV sample in Table 1 shows 18.62% for the weight percentage of C and 2.18% for the weight percentage of N, which is very similar to the theoretical values of 18.11 and 2.11%, respectively, calculated according to the proposed formula. This result once again strongly demonstrates that our proposed molecular formula is rational.

TG curve for L-Mn-PMoV
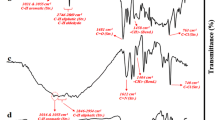
FT-IR spectra for various samples are presented in Fig. 2. PMoV gave four characteristic infrared bands for Keggin structure at 1,063, 962, 866 and 789 cm−1, assigned to P-Oa (central oxygen), M-Ob-M (corner-sharing oxygen), M-Oc-M (edge-sharing oxygen), and M-Od (terminal oxygen), respectively [19]. The bands from 1,100 to 1,600 cm−1 are the feature of the L-Mn complex [30], and the peak at 1,647 cm−1 is assigned to the stretching vibration of C=N in L ligand. The band at 1,615 cm−1 is indicative of the presence of water molecules contained in the solid. For L-Mn-PMoV, the characteristic bands for L-Mn and PMoV still appeared in its spectrum, suggesting that the structures of L-Mn and PMoV essentially remained well in L-Mn-PMoV. However, in the curve for L-Mn-PMoV, a clear red shift by about 6 cm−1 for the M-Od band and a blue shift by about 13 cm−1 for the C=N band were observed compared to its parent samples. This observation evidences the coordinate linkage between the terminal oxygen (M-Od) of the PMoV and the central Mn ions, through which the vibration strength of C=N and M-Od might be altered.

FT-IR spectra for various catalysts: (a) L-Mn; (b) PMoV; (c) L-Mn-PMoV; (d) L-Mn-PMoV#
UV–Vis spectra for various catalysts were recorded using DMSO as the solvent, as illustrated in Fig. 3. PMoV displays two absorbance bands (260 and 307 nm), which are attributed to the charge transition from the ligand oxygen to octahedrally coordinated metals [31]. L-Mn-PMoV also showed two absorbance bands at 261 and 314 nm, but interestingly, the later shifted to the higher position by 7 nm in comparison with the pure PMoV. It is suggested that this shift must be resulted from the strong chemical interaction between L-Mn and PMoV, providing another proof for the coordinate structure of L-Mn-PMoV.

UV–Vis spectra for various catalysts: (a) PMoV; (b) L-Mn-PMoV; (c) L-Mn-PMoV#
On the other hand, the other sample L-Mn-PMoV# gave almost the identical characterization information as compared to L-Mn-PMoV, as demonstrated in elemental analyses (Table 1), FT-IR spectra (Fig. 2), and UV–Vis results (Fig. 3). This implies that they have exactly the same composition and structure. One thus can suppose that although 1:1 M ratio of L-Mn to PMoV was used in the preparation of L-Mn-PMoV#, only a half amount of L-Mn got the chance to react with PMoV, generating the same coordinate structure as L-Mn-PMoV. This result confirms the formula [{Mn(C20H22N2O2)(Cl)}2(H4PMo11VO40)] for both of samples.
3.2 Catalytic Performance
Various catalysts for hydroxylation of benzene to phenol were compared under the same reaction conditions in Table 1, with molecular oxygen as the oxidant, ascorbic acid as the co-reductant, and aqueous solution of acetonitrile as the solvent. It can be seen that no reaction took place without using a catalyst and a reducing agent. After ascorbic acid was added into the reaction system as a reducing agent, a low yield of phenol 2.6% was detected. L-Mn itself was obviously not active for this reaction due to the observation of a very similar low yield of phenol 2.5%. Interestingly, upon attaching L-Mn to PMoV, the resulting hybrid catalyst L-Mn-PMoV exhibited a rather high yield of 12.2%, which is clearly higher than that of the pure PMoV (8.4%). The V-containing heteropolyacid PMoV is a well-known redox catalyst due to the co-action of the substituted vanadium with the framework molybdenum [32]. The V4+ species in PMoV could activate dioxygen as active species for accelerating the oxidation process [33]. For the current newly prepared L-Mn-PMoV catalyst, it is suggested that L-Mn acts as a modifier to create an electron donor-accepter system via the coordinate bond between the Mn center and the terminal oxygen atom of PMoV [23]. Consequently, the low oxidation state V4+ in PMoV may be stabilized in this system, and thus the oxidative activity is improved. Furthermore, the hybrid catalyst most possibly facilitates the structural stableness of PMoV against the decomposition of V out of the framework during the reaction [24]. In addition, L-Mn-PMoV#, being identical to L-Mn-PMoV in composition and structure, also showed almost equivalent catalytic performance to L-Mn-PMoV.
To optimize the reaction conditions when using L-Mn-PMoV as the catalyst, various parameters including the concentration of acetonitrle, reaction temperature, reaction pressure, amount of ascorbic acid, amount of catalyst, and reaction time were investigated, respectively, as shown in Fig. 4. It can be seen that all of them influenced drastically the yield of phenol. As expected, the yield of phenol increased with increasing the reaction temperature (Fig. 4b), reaction pressure of molecular oxygen (Fig. 4c), amount of catalyst (Fig. 4e), and reaction time (Fig. 4f), respectively. However, the further increase of these parameters caused lower yields of phenol most probably due to the deep oxidation of the produced phenol. Because no catechol, hydroquinone and benzoquinone were found in the product mixture, the coke and/or CO2 (CO) that cannot be detected by the GC technique may be the over-oxidation products.

Influence of various reaction conditions on the yield of phenol in hydroxylation of benzene by molecular oxygen over the L-Mn-PMoV catalyst. a Influence of concentration of acetonitrile. b Influence of reaction temperature. c Influence of reaction pressure. d Influence of amount of ascorbic acid. e Influence of amount of catalyst. f Influence of reaction time
When using the aqueous solution of acetonitrile as the solvent, a biphasic reaction system was observed: organic phase (benzene and CH3CN) and aqueous phase (H2O and CH3CN). Benzene and the hybrid catalyst tend to be dissolved in the organic phase, while product phenol in the aqueous phase. During the reaction, the produced phenol was transferred from the organic phase to aqueous phase, which is in favor of the increase of the phenol yield and also restrains the further oxidation of phenol. Thus, 50 vol% concentration of acetonitrile can be a good compromise between the dissolving of benzene and L-Mn-PMoV in organic phase and the separating of the produced phenol into aqueous phase, which proved to be true in Fig. 4a.
A reducing agent is indispensible in this reaction, and ascorbic acid, an often used reducing agent, is employed in our case. The phenol yield increased with raising the amount of ascorbic acid, and the yield of phenol attained its highest value when the ascorbic acid dosage was beyond 1.0 g (Fig. 4d). It is inferred that ascorbic acid plays a key role in reducing high valance vanadium (V5+) of the hybrid catalyst to its low valance state (V4+) [33].
From Fig. 4, one can conclude the optimal reaction conditions: 2 mL benzene, 25 mL aqueous solution of acetonitrile (50 vol%), 0.1 g (0.038 mmol) catalyst, 1.0 g ascorbic acid, 2.0 MPa O2, 373 K, and 10 h. In this case, a very high phenol yield of 14.4% was obtained.
4 Conclusions
In this study, we prepared a novel organic–inorganic hybrid catalyst L-Mn-PMoV by coordinately linking the schiff base manganese complex (L-Mn) with the Keggin-strutured molybdovanadophosphoric acid (PMoV). In the direct hydroxylation of benzene to phenol by molecular oxygen using ascorbic acid as a reducing regent, this hybrid catalyst exhibits a very high phenol yield of 14.4% under the optimal reaction conditions: 2 mL benzene, 25 mL aqueous solution of acetonitrile (50 vol%), 0.1 g (0.038 mmol) catalyst, 1.0 g ascorbic acid, 2.0 MPa O2, 373 K, and 10 h. It is proposed that the coordinate bond between the Mn center and the terminal oxygen atom of PMoV results in an electron donor-accepter system, in which the low oxidation state V4+ in PMoV may be stabilized, and thus the catalytic performance is improved. Another interpretation for the high yield of phenol is that the hybrid catalyst is able to hinder the decomposition of V out of the PMoV framework during the reaction.
References
Kuznetsova NI, Kuznetsova LI, Likholobov VA, Pez GP (2005) Catal Today 99:193
Niwa S, Eswaramoorthy M, Nair J, Raj A, Itoh N, Shoji H, Namba T, Mizukami F (2002) Science 295:105
Sun KQ, Xia HA, Feng ZC, Santen RV, Hensen E, Li C (2008) J Catal 254:383
Balducci L, Bianchi D, Bortolo R, D’Aloisio R, Ricci M, Tassinari R, Ungarelli R (2003) Angew Chem Int Ed 42:4937
Liu YY, Murata K, Inaba M (2005) Catal Commun 6:679
Kanzaki H, Kitamura T, Hamada R, Nishiyama S, Tsuruya S (2004) J Mol Catal A Chem 208:203
Miyake T, Hamada M, Niwa H, Nishizuka M, Oguri M (2002) J Mol Catal A Chem 178:199
Itoh N, Niwa S, Mizukami F, Inoue T, Igarashi A, Namba T (2003) Catal Commun 4:243
Battistel E, Tassinari R, Fornaroli M, Bonoldi L (2003) J Mol Catal A Chem 202:107
Passoni LC, Cruz AT, Buffon R, Schuchardt U (1997) J Mol Catal A Chem 120:117
Sakamoto T, Takagaki T, Sakakura A, Obora Y, Sakaguchi S, Ishii Y (2008) J Mol Catal A Chem 288:19
Peng ZH (2004) Angew Chem Int Ed 43:930
Hu CW, Wang YH, Li YG, Wang EB (2001) Chem J Internet 3:22
Gouzerh P, Proust A (1998) Chem Rev 98:77
Arichi J, Eternot M, Louis B (2008) Catal Today 138:117
Chen JQ, Gao S, Xu J (2008) Catal Commun 9:728
Santos ICMS, Rebelo SLH, Balula MSS, Martins RRL, Pereira MMMS, Simões MMQ, Neves MGPMS, Cavaleiro JAS, Cavaleiro AMV (2005) J Mol Catal A Chem 231:35
Bar-Nahum I, Khenkin AM, Neumann R (2004) J Am Chem Soc 126:10236
Ge HQ, Leng Y, Zhou CJ, Wang J (2008) Catal Lett 124:324
Ge HQ, Leng Y, Zhang FM, Zhou CJ, Wang J (2008) Catal Lett 124:250
Leng Y, Ge HQ, Zhou CJ, Wang J (2008) Chem Eng J 145:335
Patel SA, Sinha S, Mishra AN, Kamath BV, Ram RN (2003) J Mol Catal A Chem 192:53
Bar-Nahum I, Cohen H, Neumann R (2003) Inorg Chem 42:3677
Mirkhani V, Moghadam M, Tangestaninejad S, Mohammadpoor-Baltork I, Rasouli N (2008) Catal Commun 9:2171
Bäckvall JE, Hopkins RB, Grennberg H, Mader MM, Awasthi AK (1990) J Am Chem Soc 112:5160
Li YW, Wang YH, Li YG, Wang EB (2009) Inorg Chem Commun 12:112
Leng Y, Wang J, Zhu DR, Ren XQ, Ge HQ, Shen L (2009) Angew Chem Int Ed 48:168
Aiken JD, Finke RG (1999) Chem Mater 11:1035
Yokoyama A, Kojima T, Ohkubo K, Fukuzumi S (2007) Chem Commun 39:3997
Pouralimardan O, Chamayou A-C, Janiak C, Hosseini-Monfared H (2007) Inorg Chim Acta 360:1599
Ilkenhans Th, Herzog B, Braun Th, Schlögl R (1995) J Catal 153:275
Nomia K, Yagishita K, Nemoto Y, Kamataki T (1997) J Mol Catal A Chem 126:43
Gao XH, Xu J (2006) Catal Lett 111:203
Acknowledgments
The authors thank the National Natural Science Foundation of China (Nos. 20476046 and 20776069).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhou, C., Wang, J., Leng, Y. et al. Hydroxylation of Benzene to Phenol by Molecular Oxygen over an Organic–Inorganic Hybrid Catalyst: Schiff Base Manganese Complex Attached to Molybdovanadophosphoric Heteropolyacid. Catal Lett 135, 120–125 (2010). https://doi.org/10.1007/s10562-010-0260-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-010-0260-0