
Abstract
Mucins (MUC) protect epithelial barriers from environmental insult to maintain homeostasis. However, their aberrant overexpression and glycosylation in various malignancies facilitate oncogenic events from inception to metastasis. Mucin-associated sialyl-Tn (sTn) antigens bind to various receptors present on the dendritic cells (DCs), macrophages, and natural killer (NK) cells, resulting in overall immunosuppression by either receptor masking or inhibition of cytolytic activity. MUC1-mediated interaction of tumor cells with innate immune cells hampers cross-presentation of processed antigens on MHC class I molecules. MUC1 and MUC16 bind siglecs and mask Toll-like receptors (TLRs), respectively, on DCs promoting an immature DC phenotype that in turn reduces T cell effector functions. Mucins, such as MUC1, MUC2, MUC4, and MUC16, interact with or form aggregates with neutrophils, macrophages, and platelets, conferring protection to cancer cells during hematological dissemination and facilitate their spread and colonization to the metastatic sites. On the contrary, poor glycosylation of MUC1 and MUC4 at the tandem repeat region (TR) generates cancer-specific immunodominant epitopes. The presence of MUC16 neo-antigen-specific T cell clones and anti-MUC1 antibodies in cancer patients suggests that mucins can serve as potential targets for developing cancer therapeutics. The present review summarizes the molecular events involved in mucin-mediated immunomodulation, and metastasis, as well as the utility of mucins as targets for cancer immunotherapy and radioimmunotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Mucins (MUC) are glycosylated proteins, synthesized principally by epithelial cells and provide protection and lubrication to the epithelial surfaces [1]. The presence of highly O-glycosylated tandem repeat regions (TR) is a hallmark of mucins, consisting of proline, threonine, and serine (PTS) residues. Mucins are classified into two subfamilies, secreted and membrane-bound. The secreted mucin family comprises of MUC2, MUC5AC, MUC5B, MUC6, MUC7, MUC8, and MUC19. Membrane-bound mucins contain a transmembrane domain and include MUC1, MUC3A, MUC3B, MUC4, MUC11–13, MUC15–17, MUC20, MUC21, and MUC22 family members. Several structural attributes of mucins, such as extensive glycosylation and presence of growth factor-like C-terminal domains, modulate the microenvironment around the cell surface and mediate the interactions of mucins with other surface receptors [2,3,4,5,6,7,8]. Cellular transformation leads to the loss of polarity of the epithelial cells, which brings transmembrane mucins close to cell surface receptors that would otherwise be sequestered basolaterally in the polarized epithelium. In addition, altered mucins glycosylation patterns during malignant transformation enable their interaction with various receptors and thereby promote cancer cell differentiation, proliferation, invasion, and metastasis [9, 10].
Chronic inflammation is one of the risk factors for tumorigenesis. Mucins, such as MUC5B and MUC7, have been implicated in the chronic inflammatory state Toll-like receptor 4 (TLR4) recognition and accumulation of inflammatory cells [11]. Inflammation, in turn, regulates the expression of multiple mucins [12,13,14]. Under non-pathological conditions, mucins suppress inflammation by protecting epithelial cells from pathogens or injury. For instance, MUC2 maintains a suppressive environment in the intestinal lumen by establishing a mucosal barrier between epithelial lining and microflora. Similarly, MUC1 expression is upregulated during bacterial infection in the colon [15, 16]. Cancer cells exploit this immune-modulatory ability of mucins to evade immune surveillance. Mucins can interact with various inhibitory receptors like intercellular adhesion molecule-1 (ICAM-1) on T-cells, leading to their anergy and impaired antigen recognition, and with sialic-acid-binding immunoglobulin-like lectin receptors (siglecs) on antigen-presenting cells (APCs) [13, 17]. Aberrantly high levels of mucins on cancer cells create steric hindrance and mask the detection of tumor-associated antigens (TAAs) and prevent specific and non-specific lysis of the tumor cells by immune cells [13, 18,19,20]. However, the detection of circulating MUC1- and MUC16-specific antibodies in carcinoma patients suggests their immunogenic potential in stimulating antigen-specific humoral responses [21,22,23]. Apart from this, a recent study identified MUC16 as a hotspot with four-fold higher neoantigen frequency in the long-term survivors of pancreatic cancer as compared to the short-term survivors [24]. In under-glycosylated MUC1, the VNTR region with exposed cryptic peptide epitopes has been shown to elicit strong humoral- and cell-mediated immune responses [25, 26]. A study demonstrating the induction of strong antibody responses in mice using naked MUC4 peptides and Thomson-Friedenrich (T) and sialyl-Tn (sTn) glycopeptide antigens shows the significance of aberrantly glycosylated backbone as tumor-associated glycopeptide antigens [27]. These findings support multiple mechanisms by which mucins and their glycosylation states establish an immunosuppressive or immune stimulatory environment [26, 28,29,30,31]. This review summarizes the distinctive attributes of tumor-associated mucins in immune modulation during cancer progression and metastasis.
2 Immune modulation by aberrantly glycosylated mucins
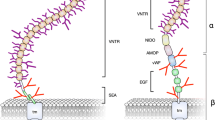
Aberrant overexpression and glycosylation patterns of tumor-associated mucins have been implicated in immunomodulation [32]. This immunomodulation can lead either to the activation of innate immune effector function and increased inflammatory stimuli or to the dampening of cytotoxic immune response and increased metastasis in a context-dependent manner [32, 33]. In this section, we discuss how tumor-associated mucins either activate or suppress the host–immune system depending upon the cellular crosstalk in the tumor microenvironment (TME). Figure 1 shows the immunomodulatory effects of tumor-associated mucins.

Immunomodulation by tumor-associated MUC. Tumor-associated mucins (MUC) promote an immunosuppressive microenvironment by either masking Toll-like receptors (TLRs) on antigen-presenting cells (APCs) or inhibiting synapse formation between cytolytic natural killer (NK) cells and cancer cells. Mucins increase immune tolerance by enhancing reprogramming toward a regulatory T cell (T-regs) phenotype and decreasing expression of activation markers on dendritic cells (DCs). Further, mucins activate tumor-specific humoral and cellular immune responses in the presence of adjuvants or TLR agonists. Antibody-dependent cellular cytotoxicity (ADCC), sialyl Tn antigen (sTn), cytotoxic T-lymphocytes (CTLs), interleukin (IL), transforming growth factor beta1 (TGF-β1), and cluster of differentiation (CDs)
2.1 Innate immune cell modulation
The formation of truncated O-linked glycans that lead to aberrant mucin glycosylation patterns results from alterations in the mucin core peptide, mucin subcellular localization or expression, and activity of various glycosyltransferases [32]. These changes in glycosylation result in the expression of unique carbohydrate epitopes, like sTn, fucosylated Lewisx/a, and T-antigen, on carcinoma-associated mucins. Galectin-1, a beta-galactoside-binding family protein, recognizes and binds specific cell membrane glycosylation patterns, triggering apoptosis of cancer specific effector T-cells [33]. Altered glycosylation patterns affect the binding of galectin-1 to mucins [34], which hampers cytotoxic T-lymphocyte (CTL) activation and results in immunosuppression [33]. In addition, mucins-associated sTn antigens, expressed in various adenocarcinomas, inhibit the cytotoxic activity of natural killer (NK) cells, and the effect is further enhanced in the presence of ammonium ions, which are known inhibitors of NK cell function [35]. NK cells are innate immune cells that exhibit cytolytic activity and kill cancer cells by the engagement of receptors present on their surface upon cancer cell encounter.
DCs bridge innate and adaptive innate and adaptive immune responses by regulated activation of antigen-specific T lymphocytes. Increased TLR expression on DCs and other APCs has been observed during infections and cancer [36, 37]. TLR activation and generation of mucin-specific immune responses (through MHC class I or MHC class II) may vary depending upon the extent and site of mucin glycosylation [38, 39]. Moreover, the formation of extracellular mucinous matrix by aberrantly glycosylated mucin masks various receptors on the cell surface, hence obstructing ligand–receptor or cellular interactions. For instance, membrane-bound MUC1 and MUC16 have been reported to suppress TLR-mediated innate immune activation at the ocular surface, inhibiting secretion of pro-inflammatory cytokines, such as IL-6, IL-8, and TNF-α, in human corneal epithelial cells [40].
The ovarian tumor marker, MUC16, downregulates the expression of CD16 and CD94/NKG2A on NK cells, so that NK cells are unable to use these receptors to bind and kill cancer cells [41]. Selective binding of MUC16 to CD16+CD56dim NK cells alters their phenotype to CD16−CD56br NK cells in ovarian cancer patients, resulting in reduced cytolytic activity and increased immune tolerance [42]. Inhibition of immune synapse formation between NK and cancer cells represents another effective MUC16-mediated immunosuppressive mechanism. MUC16 knockdown in mice demonstrated better synapse formation, thereby enhancing the NK cell-mediated cytolytic response [43].
2.2 Adaptive immune cell modulation
When the ability of mucins to elicit adaptive immune response was tested using glycosylated and non-glycosylated OVA-MUC1 fusion peptides, GalNac O-glycosylation resulted in increased helper T-cell immune response and production of MUC1-specific antibodies through MHC class II presentation in HLA-A2 transgenic mice. However, the MHC class I presentation and CTL response were significantly impaired under similar settings [38]. The extent of adaptive immune responses against mucin antigens is influenced by the expression of specific proteins or enzymes that regulate the processing of cancer-associated mucins. Heat-shock protein 70 (HSP70), which is highly expressed in cancer cells, affects the antigenic processing of MUC1 peptides depending upon the extent and position of glycosylation [44]. In addition, the presence of of cathepsins in low-density low-density endosomes has been shown to catalyze site-specific MUC1 proteolysis and production of MHC class II-restricted peptides in DCs [31, 45]. The TR region of MUC1 contains three predominant cleavage sites: Thr3-Ser4, Gly13-Ser14, and His20-Gly1. However, O-linked glycosylation at either Thr3 or Ser4 sites renders MUC1 glycoforms unavailable for proteolytic processing by DCs, probably due to cleavage site masking [31]. Besides the O-glycosylation position, the number of GalNAc sugars linked to a particular residue in the TR region also modulates the CTL response. The addition of a GalNAc sugar to the central P5-Thr of MUC1 TR (MUC1–8-5GalNAc) enables the peptide to anchor more tightly into the groove, leading to its high affinity binding to MHC class I H-2Kb and generation of a stronger CTL response in mice challenged with MUC1-expressing tumors [46]. In contrast, the addition of a second saccharide (Gal-GalNAc) to the same P5-Thr residue completely abrogates the immunogenicity of the processed peptide by inhibiting its binding to the MHC class I molecule [47]. Human cathepsin-L and immuno-proteasomes are involved in generation of these immunogenic single GalNAc and non-immunogenic double GalNAc-linked tumor-associated MUC1 octameric or decameric glycopeptides [SAPDT (GalNAc) RPAPG] [47]. Apart from their processing and presentation by MHC molecules, mucins, due to their multivalent nature, directly activate CTLs. The cytotoxic T-cell line, W.D., established from a pancreatic cancer patient, has been shown to recognize tumor-associated mucins independent of MHC in pancreatic and breast cancer cell lines [28]. Together, these studies demonstrate that the aberrant expression and glycosylation of cancer-associated mucins modulate both innate and adaptive immune effector functions, enabling cancer cell survival.
3 Molecular basis of immune cell modulation by tumor-associated mucins
Immunosuppression is a state of blunted immune response upon antigen challenge due to activation of various anti-inflammatory mediators and regulatory T-cells (Tregs). Likewise, mucus-forming mucins not only provide a physical barrier but also dampen the immunogenicity of gut antigens by discharging tolerogenic signals [48]. Cancer cells release MUC1-associated sTn antigens which bind Siglec-9 expressed on immature DCs and cause elevated interleukin-10 (IL-10) and reduced IL-12 production. This limits the ability of DCs to activate a Th1 response [49, 50]. Similarly, treatment of immature human monocyte-derived DCs with recombinant MUC1-sialylated core 1 (ST) oligosaccharides promotes an IL-10hiIL-12lo DC phenotype, with increased CD1a and CD206 and reduced expression of CD40/CD80, MHC class I, MHC class II, and CD83 differentiation markers [51]. The DCs in MUC1 knockout mice express higher levels of co-stimulatory molecules, CD40, CD80, and CD86, and secrete higher amounts of pro-inflammatory cytokines, such as TNF-α and VEGF, compared to DCs from MUC1 wild-type animals, leading to better stimulation of allogeneic naïve T-cells [52]. Glycosylated MUC1 purified from ascitic fluid of pancreatic and breast cancer patients remains confined to the early endosomes due to poor processing and presentation, limiting DCs to the immature phenotype [45]. DCs also express MUC1 on their surface [53] which interferes with TLR activation and downstream signaling events. Thus, deletion of the muc1 gene produces strong TLR4- and TLR5-mediated DC responses [52]. Similarly, MUC2 promotes a tolerogenic DC phenotype by markedly reducing pro-inflammatory cytokines and increasing the production of IL-10 and TGFβ1, resulting in an increased Treg population. MUC2 binds to galectin-3 on the DC surface, favoring the galectin-3-Dectin-1-FcγRIIB receptor complex, resulting in the activation of β-catenin and in turn inhibition of NF-κB activation [48]. In contrast to the tolerogenic MUC1-siglec-9 interactions, MUC2-siglec-3 binding ameliorates apoptosis of human monocyte-derived DCs in culture, which can be rescued by the treatment of cells with anti-siglec-3 monoclonal antibody (mAb) or recombinant siglec-3 [50]. Apart from DC inactivation, MUC4 expression on pancreatic cancer cells mediates Fas-independent apoptosis of CTLs, resulting in dampening of effector immune function [54].
Contrarily, MUC1 has been shown to elicit specific CTLs [55, 56] and T-helper immune responses in various epithelial malignancies [57]. The presence of antibodies against MUC1 in carcinoma patients at the time of diagnosis strongly indicates the generation of a humoral immune response against MUC1 [22, 23]. Reduced glycosylation of MUC1 at its TR and generation of immunodominant epitopes, such as PPAHGVT, RPAPGS, and PDTRP [58, 59], contribute to strong humoral and cell-mediated immune responses [25, 26]. An improved antibody response has been observed in the case of peptide modifications with α-GalNAc, due to saccharide-mediated structural changes in the MUC1 peptide backbone [29, 60, 61]. Increased tumor-specific anti-MUC1 IgG2 subclass antibody titers correlated with improved overall survival in breast cancer patients, supporting the potential of MUC1-directed immunotherapy [62]. MUC1-mediated suppression of human T-cell responses can be reverted by the presence of IL-2 and anti-CD28 monoclonal antibodies [13]. In addition to the promising therapeutic utility of anti-MUC1 antibodies, MUC1-mediated CTL activation is important for anti-tumor response in advanced stages of the disease.
4 Mucin-based immune modulation facilitating metastasis
Under malignant conditions, mucins mediate interaction of cancer cells with leukocytes in the tumor microenvironment and facilitate the colonization of disseminated cells at distant sites. Aberrant glycosylation leads to the expression of T, sTn, sLea, and sLex structures on tumor-associated mucins, which contribute to the metastatic ability of several tumor types. These structures, present on mucins, such as MUC1, MUC2, MUC4, and MUC16, act as ligands for various selectins. Leukocytes and platelets express selectins on their surface for adhesion [63]. Mucins, being carriers for selectin ligands, form aggregates with these cells and promote metastasis [5, 64, 65]. MUC4, by its bulky glycosylated extracellular region, protects the disseminated tumor cells from immune recognition by masking various immunogenic cell surface antigens [5]. Leukocytes, especially neutrophils, help these tumor cells colonize at metastatic sites in colon and breast cancer by forming extracellular nets [66]. A recent study demonstrated that MUC4 further enhances the survival and extravasation of the disseminated tumor cells by physically interacting with platelets and immune cells, such as macrophages and hematopoietic progenitors [67]. Loss of MUC2 expression is observed in colon cancer and is associated with higher metastasis. Loss of MUC2 stimulates increased IL-6 production by tumor-associated macrophages, which in turn leads to increased STAT3 signaling and epithelial-to-mesenchymal transition in colon cancer cells [68]. MUC5AC leads to the suppression of anti-tumor function of neutrophils, promoting enhanced in vivo tumor growth and metastasis. Further, IL-8 from cancer cells is also responsible for neutrophil migration. MUC5AC silencing significantly increases IL-8 production and neutrophil activation in pancreatic cancer cells [69]. MUC16 increases metastasis by altering E-cadherin, N-cadherin, and vimentin expression in ovarian cancer cells [70]. MUC16 is involved in decreasing the expression of NK cell stimulating protein CD16, leading to reduced NK cell activity [41, 42]. NK cells have been implicated in reducing metastasis and their reduced activation might contribute to increased metastasis by MUC16. MUC16 also inhibits formation of immune synapses between tumor and NK cells. This immune protection provided by MUC16 might facilitate survival of ovarian cancer cells from cytotoxicity and promote peritoneal metastasis [43]. Conversely, a recent study showed the selective loss of MUC16 neo-antigen reactive T-cell clones in patients with pancreatic cancer during metastatic progression, resulting in reduced intratumoral and circulating T cell reactivity [24]. Overexpression of MUC1 in various malignancies is associated with increased tumor cell invasion and metastasis. MUC1, despite being efficiently taken up by the DCs, was not transported to late endosomes for degradation and was lodged in early endosomes. However, the long-term MUC1 retention in early endosomes did not affect the processing and presentation ability of DCs for other antigens, consistent with the generation of tumor-associated MUC1-specific tolerance in metastatic patients [45]. In addition, MUC1 has been shown to cooperate with the NF-κB signaling pathway and enhance MMP-2 and MMP-9 activities, thereby increasing cancer cell invasion and metastasis [71].
5 Targeting immunomodulatory effects of mucins
5.1 Antibody-based approach
Altered glycosylation of mucins generates new epitopes that form the basis of monoclonal antibody (mAb) based diagnosis and targeted therapy. Various mAbs have been generated against these altered mucins, which have shown potential in diagnostics and therapeutics development in multiple cancers. With the advent of new antibody engineering techniques, the focus has shifted toward the generation of genetically engineered antibody fragments with increased binding affinity and tumor localization abilities [72]. Furthermore, these high-affinity mAbs have been successfully used in immunotherapy and radioimmuno-diagnostics for several malignancies [73,74,75].
5.1.1 Antibodies targeting immune response
Murine anti-MUC16 (CA125) mAb-B43.13 (Oregovomab) was used to detect recurrence in ovarian cancer patients. Later, this antibody was observed to form strong immune complexes with circulating CA125 within 30 min of administration. Subsequently, mAb-B43.13 injection reinforced the production of more CA125-specific antibodies directed to different epitopes by induction of strong antigen-specific humoral and cellular immune responses. Furthermore, the generation of CA125-specific B- and T-cell responses after treatment with mAb-B43.13 improved survival in ovarian cancer patients [76, 77]. However, in clinical trials, oregovomab as a mono-immunotherapy did not achieve improved outcomes in patients with advanced stage ovarian cancer [78]. Alternatively, an anti-idiotypic monoclonal antibody (abagovomab) against oregovomab, mirroring CA125 in phase I or II clinical trial developed anti-anti-idiotypic antibodies (Ab3) and MUC16 antibodies and the patients with Ab3 showed better survival. Further, the administration of CA125 fusion protein with interleukin 6 increased the production of Ab3 through CA125-specific B-cells [79].
Similarly, a MUC1-specific murine antibody, known as mAb-HMFG1, was generated against the PDTR epitope in the extracellular TR region of MUC1 [80]. This antibody was humanized (huHMFG1) and clinically tested for the treatment of breast cancer [81]. The phase II clinical trials of huHMFG1 (AS1402) on breast cancer patients were discontinued due to worse response rates for early disease progression and its poor efficacy [82]. Moreover, the phase III clinical trials following treatment with radiolabeled HMFG1 in ovarian cancer patients failed to reach endpoints of increased time to relapse or patient survival [83]. Pankomab, also known as GT-MAB 2.5-GEX, targets a carbohydrate-induced, conformational tumor-associated epitope present on MUC1 (TA-MUC1). This unique feature of Pankomab enables it to distinguish between tumor-specific MUC1 expression and physiological MUC1 expression [84]. Major properties of this antibody include the following: (a) rapid internalization; (b) toxin-mediated elimination of tumor cells; and (c) strong induction of an ADCC response. Additionally, Pankomab does not bind to peripheral mononuclear cells and exhibits high tumor specificity and affinity [85]. As the work on Pankomab progressed, a humanized version of Pankomab (hPankomab) was developed for potential clinical application. The safety and tolerability of PankoMab-GEX was tested in phase I clinical trials in patients with advanced metastatic carcinomas. It was safely tolerated and showed anti-tumor response for advanced disease. Out of 60 evaluable patients, one ovarian cancer patient showed complete response and 19 patients were confirmed for the stable disease. Based on these findings, PankoMab-GEX was selected for pre-clinical efficacy in phase II trials [86]. In addition, immunohistochemistry studies of hPankomab, which analyzed 137 clinical samples from various carcinomas and non-epithelial malignancies, revealed the highest reactivity with carcinomas originating from glandular or squamous epithelium. Moderate reactivity was also observed in hepatocellular carcinomas but not with sarcomas [87].
Anti-mucin mAbs have also been exploited for targeted delivery of drug conjugates. Of the two anti-MUC16 mAbs, 3A5 and 11D10, the former was found to target the TR of MUC16, showing comparatively greater reactivity and more effective delivery of the microtubule-disrupting cytotoxic drug Monomethyl auristatin E (MMAE) to cancer cells [88]. Phase I clinical trial of a 3A-MMAE conjugate DMUC5754A in unresectable ovarian and pancreatic cancer patients revealed minimal cytotoxicity and anti-tumor activity for MUC16-expressing tumors. In addition to murine and humanized antibodies, NPC-1C, a chimeric IgG1 mAb, recognizes a tumor-associated MUC5AC antigen expressed in colorectal and pancreatic cancer tissues. Reactivity tests of NPC-1C against human tumor cells positively stained 52–94% of colorectal and pancreatic cancer cell lines, 43% of colon and 48% of pancreatic cancer tissues with no reactivity observed against normal colon or pancreatic tissues [89].
5.1.2 Mucin-based radioimmunotherapy
Radioimmunotherapy (RIT) is a promising approach for antigen-specific targeting and delivery of radiation into tumors with the help of native antibodies or their immunologically active bonsai fragments (Fig. 2). Successful RIT relies on both the expression of target antigens within the tumor and the epitope accessibility to the antibodies [72]. The radiolabeled antibodies target primary tumors, disseminated circulating cancer cells, newly developing metastatic niches, and residual malignant cells in antigen-specific manner. Successful clinical testing of RIT in lymphomas and leukemia with radiolabeled antibodies against CD20, CD33, CD37, C45, and HLA-DR and additional cell markers opened avenues for other malignancies, including solid tumors [90]. Cancer-specific expression of mucins and the epitope multiplicity in TRs make mucins appropriate targets for RIT development [79, 91]. Mucin-based RIT began with optimization and development of mAbs against epithelial mucin [92], including mucin expressed on ovarian cancer [79, 93] and breast cancer [94]. Anti-MUC1 antibodies have been used against several epitopes with variable success rates in tumor regression and survival [95,96,97,98,99,100,101,102,103]. RIT-based clinical trials using anti-MUC1 are listed in Table 1. Peterson et al. developed anti-mucin mAbs that recognized the overlapping epitopes in the TR of breast cancer mucin. One of these mAbs, Mc3, when conjugated with 90Y, significantly increased complete response rates in a breast cancer xenograft model [109]. Additionally, another monoclonal antibody PAM4, was evaluated in pancreatic cancer preclinical and clinical studies for both targeting as well as therapeutic efficacy using different radioisotopes [103, 110, 111]. Earlier this antibody was claimed to be MUC1-specific. Subsequent studies suspected MUC4 as its target antigen but a recent study by Gold et al. showed MUC5AC as a specific mucin to which PAM4 reacts [108]. In addition, studies in xenograft models and pancreatic cancer patients showed that, when combined with gemcitabine, 90Y-PAM4-based RIT prolonged survival and increased tumor regression [112,113,114]. Interestingly, gemcitabine treatment was reported to radiosensitize pancreatic tumors and enhance the therapeutic response to radiation [114]. Further, 90Y-humanized PAM4-tetraxetan (clivatuzumab tetraxetan) with concurrent low-dose gemcitabine showed clinical feasibility in pancreatic cancer patients with metastasis [107].

Stages for ADCC, ADC or radiolabeled anti-mucin antibody mediated targeting during cancer progression. Different stages (1-5) during cancer progression and metastasis that are vulnerable to targeting by mucin specific antibody/active fragment(s). ADCC, ADC, and RIT are all dependent on antibody specificity and antigen expression in a particular cancer type. In addition, RIT is shown to be suitable for targeting post resection residual disease (6). Abbreviations: Circulatory tumor cells (CTCs); disseminated tumor cells (DTC); monoclonal antibodies (mAbs). Organ structures are adapted from ChemDraw drawing software
Previously, combined therapeutic effects of RIT with IFN-γ in colon cancer [115] and with cisplatin in ovarian cancer [116] were observed in murine models. In addition, radiolabeled antibodies used in combination with chemotherapeutic agents either demonstrated tumoricidal effects or facilitated better penetration and distribution of radio-conjugated antibodies inside tumors, and improved therapeutic responses in multiple cancer types [117,118,119]. Several mucins, like MUC4, MUC5AC, MUC5B, MUC16 and MUC17, have also been shown to be differentially expressed in various cancers and, therefore, hold potential as targets for RIT. For instance, MUC4 expression significantly correlates with pancreatic cancer progression, while remaining absent in normal pancreas, and MUC4 can be selectively targeted because of the availability of specific antibodies against the TR and non-TRs [120,121,122]. However, antibodies generated against synthetic mucin peptide epitopes are sometimes unable to recognize the native epitope due to differential glycosylation of mucins. Further, the high molecular weight of antibodies and host compatibility are among the major challenges in development of successful RIT. Therefore, antibody engineering to develop immunologically active recombinant Ab fragments, as well as humanization of antibodies derived from different hosts, are important for the clinical success of RIT. Engineered antibodies show better therapeutic utility due to reduced molecular size, better pharmacokinetics, and lower immunogenicity [123,124,125]. In summary, overexpressed mucins are suitable for antibody-mediated targeting, and development of mucin-targeting RIT approaches might significantly enhance existing therapeutic regimens. These new mucin-based strategies are expected to overcome some of the challenges associated with cancer progression, metastasis, and post-surgical recurrence.
5.2 Mucin peptide-based vaccines
Ever since the clinical safety of MUC1 peptide vaccines was demonstrated [126], a number of clinical trials have tested synthetic MUC1 peptide vaccines. Potent anti-tumor vaccines necessitate the induction of strong CD8+ and CD4+ T cell responses and the generation of long lasting immunological memory. A vaccine consisting of 5 repeat sequences of MUC1 peptide fused with mannan polysaccharide was clinically tested in 25 patients with a variety of cancers. Despite eliciting MUC1-specific antibodies in 13 patients, the vaccine was unable to induce significant CTL responses [55]. Similarly, a clinical trial of MUC1 peptides in combination with various adjuvant therapies failed to promote T-cell activation in spite of generating high titers of MUC1 antibodies [127, 128]. Phase II studies of immunization with MUC1 peptide demonstrated increased survival in patients with non-small cell lung cancer (NSCLC). Liposomal trafficking enhanced antigen uptake and presentation of the MUC1 peptide by APCs, leading to the activation of MUC1-specific T-cells [129]. BLP25 is a liposomal MUC1 vaccine (L-BLP25) consisting of a 25-mer MUC1 sequence (STAPPAHGVTSAPDTRPAPGSTAPP), BL25 lipopeptide, TLR4 agonist monophosphoryl lipid A (MPLA) and three lipid adjuvants [130]. BLP25 stimulated peripheral blood lymphocytes, resulting in strong CTL responses in a phase III clinical trial for NSCLC patients [131]. TG4010, a recombinant virus vaccine encoding MUC1 and IL-2, induced reasonable MUC1-specific CD4+ and CD8+ T cell activity in a phase II clinical trial for metastatic renal clear-cell carcinoma [132]. Furthermore, TG4010 did not cause significant adverse events, while eliciting promising clinical responses when tested in patients with various cancers [132,133,134], including lung cancer [135]. When given in combination with chemotherapy, synergistic enhancement of the TG4010 efficacy resulted in better patient survival [135, 136]. MUC1-derived glycopeptides containing TAAs coupled with bovine serum albumin have also been evaluated as an anticancer vaccine [137]. Tripartite vaccine candidates consisting of MUC1-derived glycopeptides, a promiscuous T-helper epitope, and a TLR2 agonist elicited high-titer IgG antibodies recognizing cancer cells expressing those glycopeptides in vivo [138]. Further, these tripartite vaccine candidates induced a robust CTL response when lymph node CD8+ T cells were incubated with peptide-pulsed DCs. Moreover, in a humanized mouse model for mammary cancer, engineered tripartite vaccines generated an antibody-dependent, cell-mediated cytotoxic (ADCC) response against tumor-associated MUC1 antigens [139]. Apart from MUC1, a study with MUC4-positive HCT-116 colorectal cancer cells showed in vitro induction of a MUC4 peptide-specific CTL response using candidate HLA-A*0201-binding peptides, establishing the suitability of a MUC4 peptide (LLGVGTFVV) in cancer immunotherapy [140].
5.3 Cell-based approaches
In contrast to vaccination, adoptive immune cell therapy (AT) is another immunotherapeutic approach, which specifically exploits the MUC1-induced T-cell response. AT involves autologous transplantation of ex vivo activated T-cells stimulated by MUC1 peptide-pulsed DCs and macrophages. Despite several initial failures, adoptive transfer of MUC1-specific CD4+ T-cells secreting IFN-γ and IL-10 showed the appearance of a memory cell phenotype, and enhanced T-cell survival, effectively increasing patient survival in ovarian cancer. In addition, the ratio of natural and inducible T-regs after immunization was responsible for the long-term anticancer activity and immune memory response [141]. Multiple studies have evaluated the potential of adoptive transfer of MUC1-pulsed DCs in clinical trials. Twelve pancreatic and biliary cancer patients after resection were immunized with MUC1 peptide-loaded DCs, and 4 out of the 12 patients survived without disease recurrence [142]. Administration of autologous DCs transfected with MUC1 cDNA increased IFN-γ-secreting CTLs two- to ten-folds in 10 pancreatic, breast, and papillary cancer patients in a phase I/II clinical trial [143]. Adoptive transfer therapy by co-administration of autologous 100-mer TR MUC1 peptide-loaded DCs and CTLs in twenty pancreatic cancer patients with unresectable or recurrent tumors resulted in a complete response in one patient with multiple lung metastasis while 5 patients showed stable disease with mean survival of 9.8 months [144]. Genetically engineered T-cells expressing mucin-specific chimeric antigen receptors (CARs) have also been exploited as therapy. Anti-cancer-associated MUC1 Tn-specific (Anti-Tn-MUC1) CAR T-cells show potential target specific cytotoxicity and growth inhibitory effect on T cell leukemia and pancreatic cancer xenograft models []. Apart from MUC-1, tumor-associated glycoprotein (TAG)-72-specific CAR T-cells were tested in phase I clinical trials in patients with metastatic colorectal cancer. The study demonstrated the safety of CART72 cells in the patients and use of fully human CAR constructs for better efficacy [145].
6 Conclusion
Mucins expressed by epithelial cells are involved in a plethora of biological activities under normal and pathological conditions. Among others, immunomodulation by mucins is involved in various inflammatory and cancerous conditions. Aberrant glycosylation and extensive splicing of carcinoma mucins result in the generation of cancer-specific B- and T-cell epitopes, providing opportunities for vaccine design. Further, the presence of neo-antigen reactive CTL clones responsible for long-term survival in cancer patients underscores the protective role of anti-mucin adaptive immunity. However, immunosuppression by multiple mechanisms and stearic hindrance caused by mucins shielding cancer-associated epitopes facilitate tumor progression and dissemination and highlight their dichotomous role in cancer biology. The attributes of mucins, like extensive glycosylation, difficulty in their purification, secretion, or cleavage of immunogenic epitopes, lack of antigen-specific neutralizing antibodies, and the unavailability of appropriate genetically engineered mouse models, pose a major challenge in understanding their role in immune modulation. The success in generation of MUC1 and MUC4 knockout murine models, humanization of mucin-specific antibodies, discovery of immunogenic potential of MUC16-specific neo-antigenic T-cell clones in long-term patient survival, and clinical trials using MUC1-specific chimeric antigen receptor T-cell (CAR-T cell)-based therapeutic modalities are among the major advances in establishing the therapeutic suitability of mucins in cancer. Along these lines, cell-surface targeting by antibody–drug conjugate using humanized mAb 3D1 against non-shed membrane-retained MUC1 C-terminal subunit conjugated to MMAE demonstrated the efficacy of this antibody in successful delivery of payload to the cancer cell both in vitro and in vivo [146]. Recently developed high-throughput technologies, such as single-cell RNA sequencing, analysis of gene expression-specific molecular signatures associated with anti-tumor immune response, whole-exome sequencing, and in silico neo-antigen prediction, will provide more in-depth understanding regarding the appropriate utilization of mucins in immunotherapy. This will also provide better understanding regarding highly immunogenic mucin-specific epitopes that can be utilized as vaccines either in conjunction with checkpoint inhibitors or CAR-T cell therapy.
References
Kaur, S., Kumar, S., Momi, N., Sasson, A. R., & Batra, S. K. (2013). Mucins in pancreatic cancer and its microenvironment. Nature Reviews. Gastroenterology & Hepatology, 10(10), 607–620. https://doi.org/10.1038/nrgastro.2013.120.
Moniaux, N., Andrianifahanana, M., Brand, R. E., & Batra, S. K. (2004). Multiple roles of mucins in pancreatic cancer, a lethal and challenging malignancy. British Journal of Cancer, 91(9), 1633–1638. https://doi.org/10.1038/sj.bjc.6602163.
Lakshmanan, I., Rachagani, S., Hauke, R., Krishn, S. R., Paknikar, S., Seshacharyulu, P., et al. (2016). MUC5AC interactions with integrin beta4 enhances the migration of lung cancer cells through FAK signaling. Oncogene, 35(31), 4112–4121. https://doi.org/10.1038/onc.2015.478.
Lakshmanan, I., Seshacharyulu, P., Haridas, D., Rachagani, S., Gupta, S., Joshi, S., et al. (2015). Novel HER3/MUC4 oncogenic signaling aggravates the tumorigenic phenotypes of pancreatic cancer cells. Oncotarget, 6(25), 21085–21099. https://doi.org/10.18632/oncotarget.3912.
Chaturvedi, P., Singh, A. P., Chakraborty, S., Chauhan, S. C., Bafna, S., Meza, J. L., et al. (2008). MUC4 mucin interacts with and stabilizes the HER2 oncoprotein in human pancreatic cancer cells. Cancer Research, 68(7), 2065–2070. https://doi.org/10.1158/0008-5472.Can-07-6041.
Ramasamy, S., Duraisamy, S., Barbashov, S., Kawano, T., Kharbanda, S., & Kufe, D. (2007). The MUC1 and galectin-3 oncoproteins function in a microRNA-dependent regulatory loop. Molecular Cell, 27(6), 992–1004. https://doi.org/10.1016/j.molcel.2007.07.031.
Senapati, S., Chaturvedi, P., Chaney, W. G., Chakraborty, S., Gnanapragassam, V. S., Sasson, A. R., et al. (2011). Novel INTeraction of MUC4 and galectin: potential pathobiological implications for metastasis in lethal pancreatic cancer. Clinical Cancer Research, 17(2), 267–274. https://doi.org/10.1158/1078-0432.Ccr-10-1937.
Chen, S. H., Hung, W. C., Wang, P., Paul, C., & Konstantopoulos, K. (2013). Mesothelin binding to CA125/MUC16 promotes pancreatic cancer cell motility and invasion via MMP-7 activation. Scientific Reports, 3, 1870. https://doi.org/10.1038/srep01870.
Brockhausen, I. (1999). Pathways of O-glycan biosynthesis in cancer cells. Biochimica et Biophysica Acta, 1473(1), 67–95.
Pinho, S. S., & Reis, C. A. (2015). Glycosylation in cancer: mechanisms and clinical implications. Nature Reviews. Cancer, 15(9), 540–555. https://doi.org/10.1038/nrc3982.
Barrera, M. J., Aguilera, S., Veerman, E., Quest, A. F., Diaz-Jimenez, D., Urzua, U., et al. (2015). Salivary mucins induce a Toll-like receptor 4-mediated pro-inflammatory response in human submandibular salivary cells: are mucins involved in Sjogren's syndrome? Rheumatology (Oxford), 54(8), 1518–1527. https://doi.org/10.1093/rheumatology/kev026.
Hollingsworth, M. A., & Swanson, B. J. (2004). Mucins in cancer: protection and control of the cell surface. Nature Reviews. Cancer, 4(1), 45–60. https://doi.org/10.1038/nrc1251.
Agrawal, B., Krantz, M. J., Reddish, M. A., & Longenecker, B. M. (1998). Cancer-associated MUC1 mucin inhibits human T-cell proliferation, which is reversible by IL-2. Nature Medicine, 4(1), 43–49.
Van Seuningen, I., Pigny, P., Perrais, M., Porchet, N., & Aubert, J. P. (2001). Transcriptional regulation of the 11p15 mucin genes. Towards new biological tools in human therapy, in inflammatory diseases and cancer? Frontiers in Bioscience, 6, D1216–D1234.
McAuley, J. L., Linden, S. K., Png, C. W., King, R. M., Pennington, H. L., Gendler, S. J., et al. (2007). MUC1 cell surface mucin is a critical element of the mucosal barrier to infection. The Journal of Clinical Investigation, 117(8), 2313–2324. https://doi.org/10.1172/jci26705.
Linden, S. K., Florin, T. H., & McGuckin, M. A. (2008). Mucin dynamics in intestinal bacterial infection. PLoS One, 3(12), e3952. https://doi.org/10.1371/journal.pone.0003952.
Cornelissen, L. A., & Van Vliet, S. J. (2016). A bitter sweet symphony: immune responses to altered O-glycan epitopes in cancer. Biomolecules, 6(2). https://doi.org/10.3390/biom6020026.
Chauhan, S. C., Kumar, D., & Jaggi, M. (2009). Mucins in ovarian cancer diagnosis and therapy. Journal of Ovarian Research, 2, 21. https://doi.org/10.1186/1757-2215-2-21.
Komatsu, M., Yee, L., & Carraway, K. L. (1999). Overexpression of sialomucin complex, a rat homologue of MUC4, inhibits tumor killing by lymphokine-activated killer cells. Cancer Research, 59(9), 2229–2236.
van de Wiel-van Kemenade, E., Ligtenberg, M. J., de Boer, A. J., Buijs, F., Vos, H. L., Melief, C. J., et al. (1993). Episialin (MUC1) inhibits cytotoxic lymphocyte-target cell interaction. Journal of Immunology, 151(2), 767–776.
Marcos-Silva, L., Ricardo, S., Chen, K., Blixt, O., Arigi, E., Pereira, D., et al. (2015). A novel monoclonal antibody to a defined peptide epitope in MUC16. Glycobiology, 25(11), 1172–1182. https://doi.org/10.1093/glycob/cwv056.
von Mensdorff-Pouilly, S., Verstraeten, A. A., Kenemans, P., Snijdewint, F. G., Kok, A., Van Kamp, G. J., et al. (2000). Survival in early breast cancer patients is favorably influenced by a natural humoral immune response to polymorphic epithelial mucin. Journal of Clinical Oncology, 18(3), 574–583. https://doi.org/10.1200/jco.2000.18.3.574.
Blixt, O., Bueti, D., Burford, B., Allen, D., Julien, S., Hollingsworth, M., et al. (2011). Autoantibodies to aberrantly glycosylated MUC1 in early stage breast cancer are associated with a better prognosis. Breast Cancer Research, 13(2), R25. https://doi.org/10.1186/bcr2841.
Balachandran, V. P., Luksza, M., Zhao, J. N., Makarov, V., Moral, J. A., Remark, R., et al. (2017). Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature, 551(7681), 512–516. https://doi.org/10.1038/nature24462.
Lloyd, K. O., Burchell, J., Kudryashov, V., Yin, B. W., & Taylor-Papadimitriou, J. (1996). Comparison of O-linked carbohydrate chains in MUC-1 mucin from normal breast epithelial cell lines and breast carcinoma cell lines. Demonstration of simpler and fewer glycan chains in tumor cells. The Journal of Biological Chemistry, 271(52), 33325–33334.
Croce, M. V., Isla-Larrain, M. T., Capafons, A., Price, M. R., & Segal-Eiras, A. (2001). Humoral immune response induced by the protein core of MUC1 mucin in pregnant and healthy women. Breast Cancer Research and Treatment, 69(1), 1–11.
Cai, H., Palitzsch, B., Hartmann, S., Stergiou, N., Kunz, H., Schmitt, E., et al. (2015). Antibody induction directed against the tumor-associated MUC4 glycoprotein. Chembiochem, 16(6), 959–967. https://doi.org/10.1002/cbic.201402689.
Barnd, D. L., Lan, M. S., Metzgar, R. S., & Finn, O. J. (1989). Specific, major histocompatibility complex-unrestricted recognition of tumor-associated mucins by human cytotoxic T cells. Proceedings of the National Academy of Sciences of the United States of America, 86(18), 7159–7163.
Karsten, U., Serttas, N., Paulsen, H., Danielczyk, A., & Goletz, S. (2004). Binding patterns of DTR-specific antibodies reveal a glycosylation-conditioned tumor-specific epitope of the epithelial mucin (MUC1). Glycobiology, 14(8), 681–692. https://doi.org/10.1093/glycob/cwh090.
Grinstead, J. S., Schuman, J. T., & Campbell, A. P. (2003). Epitope mapping of antigenic MUC1 peptides to breast cancer antibody fragment B27.29: a heteronuclear NMR study. Biochemistry, 42(48), 14293–14305. https://doi.org/10.1021/bi0301237.
Hanisch, F. G., Schwientek, T., Von Bergwelt-Baildon, M. S., Schultze, J. L., & Finn, O. (2003). O-linked glycans control glycoprotein processing by antigen-presenting cells: a biochemical approach to the molecular aspects of MUC1 processing by dendritic cells. European Journal of Immunology, 33(12), 3242–3254. https://doi.org/10.1002/eji.200324189.
Anandkumar, A., & Devaraj, H. (2013). Tumour immunomodulation: mucins in resistance to initiation and maturation of immune response against tumours. Scandinavian Journal of Immunology, 78(1), 1–7. https://doi.org/10.1111/sji.12019.
Hauselmann, I., & Borsig, L. (2014). Altered tumor-cell glycosylation promotes metastasis. Frontiers in Oncology, 4, 28. https://doi.org/10.3389/fonc.2014.00028.
Cousin, J. M., & Cloninger, M. J. (2016). The role of galectin-1 in cancer progression, and synthetic multivalent systems for the study of galectin-1. International Journal of Molecular Sciences, 17(9). https://doi.org/10.3390/ijms17091566.
Ogata, S., Maimonis, P. J., & Itzkowitz, S. H. (1992). Mucins bearing the cancer-associated sialosyl-Tn antigen mediate inhibition of natural killer cell cytotoxicity. Cancer Research, 52(17), 4741–4746.
Rakoff-Nahoum, S., & Medzhitov, R. (2009). Toll-like receptors and cancer. Nature Reviews. Cancer, 9(1), 57–63. https://doi.org/10.1038/nrc2541.
Schmidt, C. (2006). Immune system’s Toll-like receptors have good opportunity for cancer treatment. Journal of the National Cancer Institute, 98(9), 574–575. https://doi.org/10.1093/jnci/djj198.
Madsen, C. B., Petersen, C., Lavrsen, K., Harndahl, M., Buus, S., Clausen, H., et al. (2012). Cancer associated aberrant protein O-glycosylation can modify antigen processing and immune response. PLoS One, 7(11), e50139. https://doi.org/10.1371/journal.pone.0050139.
Tarang, S., Kumar, S., & Batra, S. K. (2012). Mucins and toll-like receptors: kith and kin in infection and cancer. Cancer Letters, 321(2), 110–119. https://doi.org/10.1016/j.canlet.2012.01.040.
Menon, B. B., Kaiser-Marko, C., Spurr-Michaud, S., Tisdale, A. S., & Gipson, I. K. (2015). Suppression of Toll-like receptor-mediated innate immune responses at the ocular surface by the membrane-associated mucins MUC1 and MUC16. Mucosal Immunology, 8(5), 1000–1008. https://doi.org/10.1038/mi.2014.127.
Patankar, M. S., Jing, Y., Morrison, J. C., Belisle, J. A., Lattanzio, F. A., Deng, Y., et al. (2005). Potent suppression of natural killer cell response mediated by the ovarian tumor marker CA125. Gynecologic Oncology, 99(3), 704–713. https://doi.org/10.1016/j.ygyno.2005.07.030.
Belisle, J. A., Gubbels, J. A., Raphael, C. A., Migneault, M., Rancourt, C., Connor, J. P., et al. (2007). Peritoneal natural killer cells from epithelial ovarian cancer patients show an altered phenotype and bind to the tumour marker MUC16 (CA125). Immunology, 122(3), 418–429. https://doi.org/10.1111/j.1365-2567.2007.02660.x.
Gubbels, J. A., Felder, M., Horibata, S., Belisle, J. A., Kapur, A., Holden, H., et al. (2010). MUC16 provides immune protection by inhibiting synapse formation between NK and ovarian tumor cells. Molecular Cancer, 9, 11. https://doi.org/10.1186/1476-4598-9-11.
Schreiber, J., Stahn, R., Schenk, J. A., Karsten, U., & Pecher, G. (2000). Binding of tumor antigen mucin (MUC1) derived peptides to the heat shock protein DnaK. Anticancer Research, 20(5a), 3093–3098.
Hiltbold, E. M., Vlad, A. M., Ciborowski, P., Watkins, S. C., & Finn, O. J. (2000). The mechanism of unresponsiveness to circulating tumor antigen MUC1 is a block in intracellular sorting and processing by dendritic cells. Journal of Immunology, 165(7), 3730–3741.
Apostolopoulos, V., Yuriev, E., Ramsland, P. A., Halton, J., Osinski, C., Li, W., et al. (2003). A glycopeptide in complex with MHC class I uses the GalNAc residue as an anchor. Proceedings of the National Academy of Sciences of the United States of America, 100(25), 15029–15034. https://doi.org/10.1073/pnas.2432220100.
Ninkovic, T., Kinarsky, L., Engelmann, K., Pisarev, V., Sherman, S., Finn, O. J., et al. (2009). Identification of O-glycosylated decapeptides within the MUC1 repeat domain as potential MHC class I (A2) binding epitopes. Molecular Immunology, 47(1), 131–140. https://doi.org/10.1016/j.molimm.2008.09.032.
Shan, M., Gentile, M., Yeiser, J. R., Walland, A. C., Bornstein, V. U., Chen, K., et al. (2013). Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science, 342(6157), 447–453. https://doi.org/10.1126/science.1237910.
Monti, P., Leone, B. E., Zerbi, A., Balzano, G., Cainarca, S., Sordi, V., et al. (2004). Tumor-derived MUC1 mucins interact with differentiating monocytes and induce IL-10highIL-12low regulatory dendritic cell. Journal of Immunology, 172(12), 7341–7349.
Ohta, M., Ishida, A., Toda, M., Akita, K., Inoue, M., Yamashita, K., et al. (2010). Immunomodulation of monocyte-derived dendritic cells through ligation of tumor-produced mucins to Siglec-9. Biochemical and Biophysical Research Communications, 402(4), 663–669. https://doi.org/10.1016/j.bbrc.2010.10.079.
Rughetti, A., Pellicciotta, I., Biffoni, M., Backstrom, M., Link, T., Bennet, E. P., et al. (2005). Recombinant tumor-associated MUC1 glycoprotein impairs the differentiation and function of dendritic cells. Journal of Immunology, 174(12), 7764–7772.
Williams, M. A., Bauer, S., Lu, W., Guo, J., Walter, S., Bushnell, T. P., et al. (2010). Deletion of the mucin-like molecule muc1 enhances dendritic cell activation in response to toll-like receptor ligands. Journal of Innate Immunity, 2(2), 123–143. https://doi.org/10.1159/000254790.
Wykes, M., MacDonald, K. P., Tran, M., Quin, R. J., Xing, P. X., Gendler, S. J., et al. (2002). MUC1 epithelial mucin (CD227) is expressed by activated dendritic cells. Journal of Leukocyte Biology, 72(4), 692–701.
Zhu, Y., Zhang, J. J., Liang, W. B., Zhu, R., Wang, B., Miao, Y., et al. (2014). Pancreatic cancer counterattack: MUC4 mediates Fas-independent apoptosis of antigen-specific cytotoxic T lymphocyte. Oncology Reports, 31(4), 1768–1776. https://doi.org/10.3892/or.2014.3016.
Karanikas, V., Hwang, L. A., Pearson, J., Ong, C. S., Apostolopoulos, V., Vaughan, H., et al. (1997). Antibody and T cell responses of patients with adenocarcinoma immunized with mannan-MUC1 fusion protein. The Journal of Clinical Investigation, 100(11), 2783–2792. https://doi.org/10.1172/jci119825.
Ioannides, C. G., Fisk, B., Jerome, K. R., Irimura, T., Wharton, J. T., & Finn, O. J. (1993). Cytotoxic T cells from ovarian malignant tumors can recognize polymorphic epithelial mucin core peptides. Journal of Immunology, 151(7), 3693–3703.
Barratt-Boyes, S. M., Vlad, A., & Finn, O. J. (1999). Immunization of chimpanzees with tumor antigen MUC1 mucin tandem repeat peptide elicits both helper and cytotoxic T-cell responses. Clinical Cancer Research, 5(7), 1918–1924.
Graves, C. R., Robertson, J. F., Murray, A., Price, M. R., & Chapman, C. J. (2005). Malignancy-induced autoimmunity to MUC1: initial antibody characterization. The Journal of Peptide Research, 66(6), 357–363. https://doi.org/10.1111/j.1399-3011.2005.00297.x.
von Mensdorff-Pouilly, S., Petrakou, E., Kenemans, P., van Uffelen, K., Verstraeten, A. A., Snijdewint, F. G., et al. (2000). Reactivity of natural and induced human antibodies to MUC1 mucin with MUC1 peptides and n-acetylgalactosamine (GalNAc) peptides. International Journal of Cancer, 86(5), 702–712.
Coltart, D. M., Royyuru, A. K., Williams, L. J., Glunz, P. W., Sames, D., Kuduk, S. D., et al. (2002). Principles of mucin architecture: structural studies on synthetic glycopeptides bearing clustered mono-, di-, tri-, and hexasaccharide glycodomains. Journal of the American Chemical Society, 124(33), 9833–9844.
Dziadek, S., Griesinger, C., Kunz, H., & Reinscheid, U. M. (2006). Synthesis and structural model of an alpha(2,6)-sialyl-t glycosylated MUC1 eicosapeptide under physiological conditions. Chemistry, 12(19), 4981–4993. https://doi.org/10.1002/chem.200600144.
Fremd, C., Stefanovic, S., Beckhove, P., Pritsch, M., Lim, H., Wallwiener, M., et al. (2016). Mucin 1-specific B cell immune responses and their impact on overall survival in breast cancer patients. Oncoimmunology, 5(1), e1057387. https://doi.org/10.1080/2162402x.2015.1057387.
McEver, R. P. (2015). Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovascular Research, 107(3), 331–339. https://doi.org/10.1093/cvr/cvv154.
Baldus, S. E., Monig, S. P., Hanisch, F. G., Zirbes, T. K., Flucke, U., Oelert, S., et al. (2002). Comparative evaluation of the prognostic value of MUC1, MUC2, sialyl-Lewis(a) and sialyl-Lewis(x) antigens in colorectal adenocarcinoma. Histopathology, 40(5), 440–449.
Chen, S. H., Dallas, M. R., Balzer, E. M., & Konstantopoulos, K. (2012). Mucin 16 is a functional selectin ligand on pancreatic cancer cells. The FASEB Journal, 26(3), 1349–1359. https://doi.org/10.1096/fj.11-195669.
Park, J., Wysocki, R. W., Amoozgar, Z., Maiorino, L., Fein, M. R., Jorns, J., et al. (2016). Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Science Translational Medicine, 8(361), 361ra138. https://doi.org/10.1126/scitranslmed.aag1711.
Rowson-Hodel, A. R., Wald, J. H., Hatakeyama, J., O'Neal, W. K., Stonebraker, J. R., VanderVorst, K., et al. (2018). Membrane mucin Muc4 promotes blood cell association with tumor cells and mediates efficient metastasis in a mouse model of breast cancer. Oncogene, 37(2), 197–207. https://doi.org/10.1038/onc.2017.327.
Hsu, H. P., Lai, M. D., Lee, J. C., Yen, M. C., Weng, T. Y., Chen, W. C., et al. (2017). Mucin 2 silencing promotes colon cancer metastasis through interleukin-6 signaling. Scientific Reports, 7(1), 5823. https://doi.org/10.1038/s41598-017-04952-7.
Hoshi, H., Sawada, T., Uchida, M., Iijima, H., Kimura, K., Hirakawa, K., et al. (2013). MUC5AC protects pancreatic cancer cells from TRAIL-induced death pathways. International Journal of Oncology, 42(3), 887–893. https://doi.org/10.3892/ijo.2013.1760.
Theriault, C., Pinard, M., Comamala, M., Migneault, M., Beaudin, J., Matte, I., et al. (2011). MUC16 (CA125) regulates epithelial ovarian cancer cell growth, tumorigenesis and metastasis. Gynecologic Oncology, 121(3), 434–443. https://doi.org/10.1016/j.ygyno.2011.02.020.
Mori, Y., Akita, K., Tanida, S., Ishida, A., Toda, M., Inoue, M., et al. (2014). MUC1 protein induces urokinase-type plasminogen activator (uPA) by forming a complex with NF-kappaB p65 transcription factor and binding to the uPA promoter, leading to enhanced invasiveness of cancer cells. The Journal of Biological Chemistry, 289(51), 35193–35204. https://doi.org/10.1074/jbc.M114.586461.
Wittel, U. A., Goel, A., Varshney, G. C., & Batra, S. K. (2001). Mucin antibodies—new tools in diagnosis and therapy of cancer. Frontiers in Bioscience, 6, D1296–D1310.
Hughes, O. D., Perkins, A. C., Frier, M., Wastie, M. L., Denton, G., Price, M. R., et al. (2001). Imaging for staging bladder cancer: a clinical study of intravenous 111indium-labelled anti-MUC1 mucin monoclonal antibody C595. BJU International, 87(1), 39–46.
Hughes, O. D., Bishop, M. C., Perkins, A. C., Frier, M., Price, M. R., Denton, G., et al. (1997). Preclinical evaluation of copper-67 labelled anti-MUC1 mucin antibody C595 for therapeutic use in bladder cancer. European Journal of Nuclear Medicine, 24(4), 439–443.
Hughes, O. D., Bishop, M. C., Perkins, A. C., Wastie, M. L., Denton, G., Price, M. R., et al. (2000). Targeting superficial bladder cancer by the intravesical administration of copper-67-labeled anti-MUC1 mucin monoclonal antibody C595. Journal of Clinical Oncology, 18(2), 363–370. https://doi.org/10.1200/jco.2000.18.2.363.
Noujaim, A. A., Schultes, B. C., Baum, R. P., & Madiyalakan, R. (2001). Induction of CA125-specific B and T cell responses in patients injected with MAb-B43.13—evidence for antibody-mediated antigen-processing and presentation of CA125 in vivo. Cancer Biotherapy & Radiopharmaceuticals, 16(3), 187–203. https://doi.org/10.1089/10849780152389384.
Ehlen, T. G., Hoskins, P. J., Miller, D., Whiteside, T. L., Nicodemus, C. F., Schultes, B. C., et al. (2005). A pilot phase 2 study of oregovomab murine monoclonal antibody to CA125 as an immunotherapeutic agent for recurrent ovarian cancer. International Journal of Gynecological Cancer, 15(6), 1023–1034. https://doi.org/10.1111/j.1525-1438.2005.00483.x.
Berek, J., Taylor, P., McGuire, W., Smith, L. M., Schultes, B., & Nicodemus, C. F. (2009). Oregovomab maintenance monoimmunotherapy does not improve outcomes in advanced ovarian cancer. J Clin Oncol, 27(3), 418-425, https://doi.org/10.1200/jco.2008.17.8400.
Singh, A. P., Senapati, S., Ponnusamy, M. P., Jain, M., Lele, S. M., Davis, J. S., et al. (2008). Clinical potential of mucins in diagnosis, prognosis, and therapy of ovarian cancer. The Lancet Oncology, 9(11), 1076–1085. https://doi.org/10.1016/s1470-2045(08)70277-8.
Burchell, J., Gendler, S., Taylor-Papadimitriou, J., Girling, A., Lewis, A., Millis, R., et al. (1987). Development and characterization of breast cancer reactive monoclonal antibodies directed to the core protein of the human milk mucin. Cancer Research, 47(20), 5476–5482.
Verhoeyen, M. E., Saunders, J. A., Price, M. R., Marugg, J. D., Briggs, S., Broderick, E. L., et al. (1993). Construction of a reshaped HMFG1 antibody and comparison of its fine specificity with that of the parent mouse antibody. Immunology, 78(3), 364–370.
Ibrahim, N. K., Yariz, K. O., Bondarenko, I., Manikhas, A., Semiglazov, V., Alyasova, A., et al. (2011). Randomized phase II trial of letrozole plus anti-MUC1 antibody AS1402 in hormone receptor-positive locally advanced or metastatic breast cancer. Clinical Cancer Research, 17(21), 6822–6830. https://doi.org/10.1158/1078-0432.Ccr-11-1151.
Song, H., & Sgouros, G. (2011). Radioimmunotherapy of solid tumors: searching for the right target. Current Drug Delivery, 8(1), 26–44.
Dian, D., Lenhard, M., Mayr, D., Heublein, S., Karsten, U., Goletz, S., et al. (2013). Staining of MUC1 in ovarian cancer tissues with PankoMab-GEX detecting the tumour-associated epitope, TA-MUC1, as compared to antibodies HMFG-1 and 115D8. Histology and Histopathology, 28(2), 239–244. https://doi.org/10.14670/hh-28.239.
Danielczyk, A., Stahn, R., Faulstich, D., Loffler, A., Marten, A., Karsten, U., et al. (2006). PankoMab: a potent new generation anti-tumour MUC1 antibody. Cancer Immunology, Immunotherapy, 55(11), 1337–1347. https://doi.org/10.1007/s00262-006-0135-9.
Fiedler, W., DeDosso, S., Cresta, S., Weidmann, J., Tessari, A., Salzberg, M., et al. (2016). A phase I study of PankoMab-GEX, a humanised glyco-optimised monoclonal antibody to a novel tumour-specific MUC1 glycopeptide epitope in patients with advanced carcinomas. European Journal of Cancer, 63, 55–63. https://doi.org/10.1016/j.ejca.2016.05.003.
Fan, X. N., Karsten, U., Goletz, S., & Cao, Y. (2010). Reactivity of a humanized antibody (hPankoMab) towards a tumor-related MUC1 epitope (TA-MUC1) with various human carcinomas. Pathology, Research and Practice, 206(8), 585–589. https://doi.org/10.1016/j.prp.2010.03.006.
Felder, M., Kapur, A., Gonzalez-Bosquet, J., Horibata, S., Heintz, J., Albrecht, R., et al. (2014). MUC16 (CA125): tumor biomarker to cancer therapy, a work in progress. Molecular Cancer, 13, 129. https://doi.org/10.1186/1476-4598-13-129.
Patel, S. P., Bristol, A., Saric, O., Wang, X. P., Dubeykovskiy, A., Arlen, P. M., et al. (2013). Anti-tumor activity of a novel monoclonal antibody, NPC-1C, optimized for recognition of tumor antigen MUC5AC variant in preclinical models. Cancer Immunology, Immunotherapy, 62(6), 1011–1019. https://doi.org/10.1007/s00262-013-1420-z.
Larson, S. M., Carrasquillo, J. A., Cheung, N. K., & Press, O. W. (2015). Radioimmunotherapy of human tumours. Nature Reviews. Cancer, 15(6), 347–360. https://doi.org/10.1038/nrc3925.
Kufe, D. W. (2009). Mucins in cancer: function, prognosis and therapy. Nature Reviews. Cancer, 9(12), 874–885. https://doi.org/10.1038/nrc2761.
Price, M. R., Sekowski, M., & Tendler, S. J. (1991). Purification of anti-epithelial mucin monoclonal antibodies by epitope affinity chromatography. Journal of Immunological Methods, 139(1), 83–90.
Buckman, R., De Angelis, C., Shaw, P., Covens, A., Osborne, R., Kerr, I., et al. (1992). Intraperitoneal therapy of malignant ascites associated with carcinoma of ovary and breast using radioiodinated monoclonal antibody 2G3. Gynecologic Oncology, 47(1), 102–109.
Peterson, J. A., & Ceriani, R. L. (1994). Breast mucin and associated antigens in diagnosis and therapy. Advances in Experimental Medicine and Biology, 353, 1–8.
Murray, A., Simms, M. S., Scholfield, D. P., Vincent, R. M., Denton, G., Bishop, M. C., et al. (2001). Production and characterization of 188Re-C595 antibody for radioimmunotherapy of transitional cell bladder cancer. Journal of Nuclear Medicine, 42(5), 726–732.
Supiot, S., Faivre-Chauvet, A., Couturier, O., Heymann, M. F., Robillard, N., Kraeber-Bodere, F., et al. (2002). Comparison of the biologic effects of MA5 and B-B4 monoclonal antibody labeled with iodine-131 and bismuth-213 on multiple myeloma. Cancer, 94(4 Suppl), 1202–1209.
Berger, M. A., Masters, G. R., Singleton, J., Scully, M. S., Grimm, L. G., Soltis, D. A., et al. (2005). Pharmacokinetics, biodistribution, and radioimmunotherapy with monoclonal antibody 776.1 in a murine model of human ovarian cancer. Cancer Biotherapy & Radiopharmaceuticals, 20(6), 589–602. https://doi.org/10.1089/cbr.2005.20.589.
Garkavij, M., Samarzija, M., Ewers, S. B., Jakopovic, M., Tezak, S., & Tennvall, J. (2005). Concurrent radiotherapy and tumor targeting with 111In-HMFG1-F(ab')2 in patients with MUC1-positive non-small cell lung cancer. Anticancer Research, 25(6c), 4663–4671.
Qu, C. F., Songl, Y. J., Rizvi, S. M., Li, Y., Smith, R., Perkins, A. C., et al. (2005). In vivo and in vitro inhibition of pancreatic cancer growth by targeted alpha therapy using 213Bi-CHX.A″-C595. Cancer Biology & Therapy, 4(8), 848–853.
Song, E. Y., Qu, C. F., Rizvi, S. M., Raja, C., Beretov, J., Morgenstern, A., et al. (2008). Bismuth-213 radioimmunotherapy with C595 anti-MUC1 monoclonal antibody in an ovarian cancer ascites model. Cancer Biology & Therapy, 7(1), 76–80.
Salouti, M., Babaei, M. H., Rajabi, H., & Rasaee, M. (2011). Preparation and biological evaluation of (177)Lu conjugated PR81 for radioimmunotherapy of breast cancer. Nuclear Medicine and Biology, 38(6), 849–855. https://doi.org/10.1016/j.nucmedbio.2011.02.009.
Cardillo, T. M., Ying, Z., & Gold, D. V. (2001). Therapeutic advantage of (90)yttrium- versus (131)iodine-labeled PAM4 antibody in experimental pancreatic cancer. Clinical Cancer Research, 7(10), 3186–3192.
Han, S., Jin, G., Wang, L., Li, M., He, C., Guo, X., et al. (2014). The role of PAM4 in the management of pancreatic cancer: diagnosis, radioimmunodetection, and radioimmunotherapy. Journal of Immunology Research, 2014, 268479. https://doi.org/10.1155/2014/268479.
Maraveyas, A., Snook, D., Hird, V., Kosmas, C., Meares, C. F., Lambert, H. E., et al. (1994). Pharmacokinetics and toxicity of an yttrium-90-CITC-DTPA-HMFG1 radioimmunoconjugate for intraperitoneal radioimmunotherapy of ovarian cancer. Cancer, 73(3 Suppl), 1067–1075.
Maraveyas, A., Stafford, N., Rowlinson-Busza, G., Stewart, J. S., & Epenetos, A. A. (1995). Pharmacokinetics, biodistribution, and dosimetry of specific and control radiolabeled monoclonal antibodies in patients with primary head and neck squamous cell carcinoma. Cancer Research, 55(5), 1060–1069.
Gulec, S. A., Cohen, S. J., Pennington, K. L., Zuckier, L. S., Hauke, R. J., Horne, H., et al. (2011). Treatment of advanced pancreatic carcinoma with 90Y-Clivatuzumab Tetraxetan: a phase I single-dose escalation trial. Clinical Cancer Research, 17(12), 4091–4100. https://doi.org/10.1158/1078-0432.CCR-10-2579.
Picozzi, V. J., Ramanathan, R. K., Lowery, M. A., Ocean, A. J., Mitchel, E. P., O'Neil, B. H., et al. (2015). (90)Y-clivatuzumab tetraxetan with or without low-dose gemcitabine: a phase Ib study in patients with metastatic pancreatic cancer after two or more prior therapies. European Journal of Cancer, 51(14), 1857–1864. https://doi.org/10.1016/j.ejca.2015.06.119.
Gold, D. V., Newsome, G., Liu, D., & Goldenberg, D. M. (2013). Mapping PAM4 (clivatuzumab), a monoclonal antibody in clinical trials for early detection and therapy of pancreatic ductal adenocarcinoma, to MUC5AC mucin. Molecular Cancer, 12(1), 143. https://doi.org/10.1186/1476-4598-12-143.
Peterson, J. A., Couto, J. R., Taylor, M. R., & Ceriani, R. L. (1995). Selection of tumor-specific epitopes on target antigens for radioimmunotherapy of breast cancer. Cancer Research, 55(23 Suppl), 5847s–5851s.
Mariani, G., Molea, N., Bacciardi, D., Boggi, U., Fornaciari, G., Campani, D., et al. (1995). Initial tumor targeting, biodistribution, and pharmacokinetic evaluation of the monoclonal antibody PAM4 in patients with pancreatic cancer. Cancer Research, 55(23 Suppl), 5911s–5915s.
Gold, D. V., Cardillo, T., Goldenberg, D. M., & Sharkey, R. M. (2001). Localization of pancreatic cancer with radiolabeled monoclonal antibody PAM4. Critical Reviews in Oncology/Hematology, 39(1–2), 147–154.
Cardillo, T. M., Blumenthal, R., Ying, Z., & Gold, D. V. (2002). Combined gemcitabine and radioimmunotherapy for the treatment of pancreatic cancer. International Journal of Cancer, 97(3), 386–392.
Gold, D. V., Schutsky, K., Modrak, D., & Cardillo, T. M. (2003). Low-dose radioimmunotherapy ((90)Y-PAM4) combined with gemcitabine for the treatment of experimental pancreatic cancer. Clinical Cancer Research, 9(10 Pt 2), 3929s–3937s.
Gold, D. V., Modrak, D. E., Schutsky, K., & Cardillo, T. M. (2004). Combined 90Yttrium-DOTA-labeled PAM4 antibody radioimmunotherapy and gemcitabine radiosensitization for the treatment of a human pancreatic cancer xenograft. International Journal of Cancer, 109(4), 618–626. https://doi.org/10.1002/ijc.20004.
Greiner, J. W., Ullmann, C. D., Nieroda, C., Qi, C. F., Eggensperger, D., Shimada, S., et al. (1993). Improved radioimmunotherapeutic efficacy of an anticarcinoma monoclonal antibody (131I-CC49) when given in combination with gamma-interferon. Cancer Research, 53(3), 600–608.
Molthoff, C. F., Pinedo, H. M., Schluper, H. M., Rutgers, D. H., & Boven, E. (1992). Comparison of 131I-labelled anti-episialin 139H2 with cisplatin, cyclophosphamide or external-beam radiation for anti-tumor efficacy in human ovarian cancer xenografts. International Journal of Cancer, 51(1), 108–115.
Jain, M., Chauhan, S. C., Singh, A. P., Venkatraman, G., Colcher, D., & Batra, S. K. (2005). Penetratin improves tumor retention of single-chain antibodies: a novel step toward optimization of radioimmunotherapy of solid tumors. Cancer Research, 65(17), 7840–7846. https://doi.org/10.1158/0008-5472.Can-05-0662.
Wittel, U. A., Jain, M., Goel, A., Baranowska-Kortylewicz, J., Kurizaki, T., Chauhan, S. C., et al. (2005). Engineering and characterization of a divalent single-chain Fv angiotensin II fusion construct of the monoclonal antibody CC49. Biochemical and Biophysical Research Communications, 329(1), 168–176. https://doi.org/10.1016/j.bbrc.2005.01.101.
Jain, M., Venkatraman, G., & Batra, S. K. (2007). Cell-penetrating peptides and antibodies: a new direction for optimizing radioimmunotherapy. European Journal of Nuclear Medicine and Molecular Imaging, 34(7), 973–977. https://doi.org/10.1007/s00259-007-0395-4.
Moniaux, N., Varshney, G. C., Chauhan, S. C., Copin, M. C., Jain, M., Wittel, U. A., et al. (2004). Generation and characterization of anti-MUC4 monoclonal antibodies reactive with normal and cancer cells in humans. The Journal of Histochemistry and Cytochemistry, 52(2), 253–261. https://doi.org/10.1177/002215540405200213.
Jain, M., Venkatraman, G., Moniaux, N., Kaur, S., Kumar, S., Chakraborty, S., et al. (2011). Monoclonal antibodies recognizing the non-tandem repeat regions of the human mucin MUC4 in pancreatic cancer. PLoS One, 6(8), e23344. https://doi.org/10.1371/journal.pone.0023344.
Gautam, S. K., Kumar, S., Cannon, A., Hall, B., Bhatia, R., Nasser, M. W., et al. (2017). MUC4 mucin—a therapeutic target for pancreatic ductal adenocarcinoma. Expert Opinion on Therapeutic Targets, 21(7), 657–669. https://doi.org/10.1080/14728222.2017.1323880.
Batra, S. K., Jain, M., Wittel, U. A., Chauhan, S. C., & Colcher, D. (2002). Pharmacokinetics and biodistribution of genetically engineered antibodies. Current Opinion in Biotechnology, 13(6), 603–608.
Jain, M., & Batra, S. K. (2003). Genetically engineered antibody fragments and PET imaging: a new era of radioimmunodiagnosis. Journal of Nuclear Medicine, 44(12), 1970–1972.
Jain, M., Kamal, N., & Batra, S. K. (2007). Engineering antibodies for clinical applications. Trends in Biotechnology, 25(7), 307–316. https://doi.org/10.1016/j.tibtech.2007.05.001.
Goydos, J. S., Elder, E., Whiteside, T. L., Finn, O. J., & Lotze, M. T. (1996). A phase I trial of a synthetic mucin peptide vaccine. Induction of specific immune reactivity in patients with adenocarcinoma. The Journal of Surgical Research, 63(1), 298–304. https://doi.org/10.1006/jsre.1996.0264.
Gilewski, T., Adluri, S., Ragupathi, G., Zhang, S., Yao, T. J., Panageas, K., et al. (2000). Vaccination of high-risk breast cancer patients with mucin-1 (MUC1) keyhole limpet hemocyanin conjugate plus QS-21. Clinical Cancer Research, 6(5), 1693–1701.
Musselli, C., Ragupathi, G., Gilewski, T., Panageas, K. S., Spinat, Y., & Livingston, P. O. (2002). Reevaluation of the cellular immune response in breast cancer patients vaccinated with MUC1. International Journal of Cancer, 97(5), 660–667.
Sharma, S., Srivastava, M. K., Harris-White, M., Lee, J. M., & Dubinett, S. (2011). MUC1 peptide vaccine mediated antitumor activity in non-small cell lung cancer. Expert Opinion on Biological Therapy, 11(8), 987–990. https://doi.org/10.1517/14712598.2011.598146.
Sangha, R., & Butts, C. (2007). L-BLP25: a peptide vaccine strategy in non small cell lung cancer. Clinical Cancer Research, 13(15 Pt 2), s4652–s4654. https://doi.org/10.1158/1078-0432.Ccr-07-0213.
Xia, W., Wang, J., Xu, Y., Jiang, F., & Xu, L. (2014). L-BLP25 as a peptide vaccine therapy in non-small cell lung cancer: a review. Journal of Thoracic Disease, 6(10), 1513–1520. https://doi.org/10.3978/j.issn.2072-1439.2014.08.17.
Oudard, S., Rixe, O., Beuselinck, B., Linassier, C., Banu, E., Machiels, J. P., et al. (2011). A phase II study of the cancer vaccine TG4010 alone and in combination with cytokines in patients with metastatic renal clear-cell carcinoma: clinical and immunological findings. Cancer Immunology, Immunotherapy, 60(2), 261–271. https://doi.org/10.1007/s00262-010-0935-9.
Ramlau, R., Quoix, E., Rolski, J., Pless, M., Lena, H., Levy, E., et al. (2008). A phase II study of Tg4010 (Mva-Muc1-Il2) in association with chemotherapy in patients with stage III/IV non-small cell lung cancer. Journal of Thoracic Oncology, 3(7), 735–744. https://doi.org/10.1097/JTO.0b013e31817c6b4f.
Dreicer, R., Stadler, W. M., Ahmann, F. R., Whiteside, T., Bizouarne, N., Acres, B., et al. (2009). MVA-MUC1-IL2 vaccine immunotherapy (TG4010) improves PSA doubling time in patients with prostate cancer with biochemical failure. Investigational New Drugs, 27(4), 379–386. https://doi.org/10.1007/s10637-008-9187-3.
Quoix, E., Ramlau, R., Westeel, V., Papai, Z., Madroszyk, A., Riviere, A., et al. (2011). Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: a controlled phase 2B trial. The Lancet Oncology, 12(12), 1125–1133. https://doi.org/10.1016/s1470-2045(11)70259-5.
Quoix, E., Lena, H., Losonczy, G., Forget, F., Chouaid, C., Papai, Z., et al. (2016). TG4010 immunotherapy and first-line chemotherapy for advanced non-small-cell lung cancer (TIME): results from the phase 2b part of a randomised, double-blind, placebo-controlled, phase 2b/3 trial. The Lancet Oncology, 17(2), 212–223. https://doi.org/10.1016/s1470-2045(15)00483-0.
Dziadek, S., Kowalczyk, D., & Kunz, H. (2005). Synthetic vaccines consisting of tumor-associated MUC1 glycopeptide antigens and bovine serum albumin. Angewandte Chemie (International Ed. in English), 44(46), 7624–7630. https://doi.org/10.1002/anie.200501593.
Ingale, S., Wolfert, M. A., Gaekwad, J., Buskas, T., & Boons, G. J. (2007). Robust immune responses elicited by a fully synthetic three-component vaccine. Nature Chemical Biology, 3(10), 663–667. https://doi.org/10.1038/nchembio.2007.25.
Lakshminarayanan, V., Thompson, P., Wolfert, M. A., Buskas, T., Bradley, J. M., Pathangey, L. B., et al. (2012). Immune recognition of tumor-associated mucin MUC1 is achieved by a fully synthetic aberrantly glycosylated MUC1 tripartite vaccine. Proceedings of the National Academy of Sciences of the United States of America, 109(1), 261–266. https://doi.org/10.1073/pnas.1115166109.
Wu, J., Wei, J., Meng, K., Chen, J., Gao, W., Zhang, J., et al. (2009). Identification of an HLA-A*0201-restrictive CTL epitope from MUC4 for applicable vaccine therapy. Immunopharmacology and Immunotoxicology, 31(3), 468–476. https://doi.org/10.1080/08923970902795203.
Dobrzanski, M. J., Rewers-Felkins, K. A., Samad, K. A., Quinlin, I. S., Phillips, C. A., Robinson, W., et al. (2012). Immunotherapy with IL-10- and IFN-gamma-producing CD4 effector cells modulate “natural” and “inducible” CD4 TReg cell subpopulation levels: observations in four cases of patients with ovarian cancer. Cancer Immunology, Immunotherapy, 61(6), 839–854. https://doi.org/10.1007/s00262-011-1128-x.
Lepisto, A. J., Moser, A. J., Zeh, H., Lee, K., Bartlett, D., McKolanis, J. R., et al. (2008). A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Therapy, 6(B), 955–964.
Pecher, G., Haring, A., Kaiser, L., & Thiel, E. (2002). Mucin gene (MUC1) transfected dendritic cells as vaccine: results of a phase I/II clinical trial. Cancer Immunology, Immunotherapy, 51(11–12), 669–673. https://doi.org/10.1007/s00262-002-0317-z.
Kondo, H., Hazama, S., Kawaoka, T., Yoshino, S., Yoshida, S., Tokuno, K., et al. (2008). Adoptive immunotherapy for pancreatic cancer using MUC1 peptide-pulsed dendritic cells and activated T lymphocytes. Anticancer Research, 28(1b), 379–387.
Posey, A. D., Jr., Schwab, R. D., Boesteanu, A. C., Steentoft, C., Mandel, U., Engels, B., et al. (2016). Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity, 44(6), 1444–1454. https://doi.org/10.1016/j.immuni.2016.05.014.
Hege, K. M., Bergsland, E. K., Fisher, G. A., Nemunaitis, J. J., Warren, R. S., McArthur, J. G., et al. (2017). Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. Journal for ImmunoTherapy of Cancer, 5, 22. https://doi.org/10.1186/s40425-017-0222-9.
Panchamoorthy, G., Jin, C., Raina, D., Bharti, A., Yamamoto, M., Adeebge, D., et al. (2018). Targeting the human MUC1-C oncoprotein with an antibody-drug conjugate. JCI Insight, 3(12). https://doi.org/10.1172/jci.insight.99880.
Grant support
This work was supported by funding from the National Institutes of Health (PO1 CA 217798, P50 CA127297, UO1 CA210240, UO1 CA200466, UO 1 CA213862, R21 CA223429, F30 CA225117, R01 CA183459, RO1 CA 195586, RO1 CA206444, R21 AA 026428, and RO1 CA228524).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
SKB is one of the co-founders of Sanguine Diagnostics and Therapeutics, Inc. The other authors have no potential conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bhatia, R., Gautam, S.K., Cannon, A. et al. Cancer-associated mucins: role in immune modulation and metastasis. Cancer Metastasis Rev 38, 223–236 (2019). https://doi.org/10.1007/s10555-018-09775-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-018-09775-0