
Abstract
Stepwise progression of pulmonary adenocarcinoma is described from the viewpoint of both pathology and molecular biology. Pulmonary adenocarcinoma develops to invasive carcinoma through atypical adenomatous hyperplasia, adenocarcinoma in situ and minimally invasive adenocarcinoma. The Noguchi classification is well correlated with this sequential histological progression. On the other hand, in terms of molecular biology, p16 gene inactivation, EGFR mutation and KRAS mutation are early events, and tumors progress to invasive adenocarcinoma as a result of p53 mutation, loss of various chromosomes and other genetic abnormalities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Adenocarcinoma of the lung is the most frequent lung cancer occurring in Japan, and is increasing in the USA and most European countries [1–3]. Adenocarcinomas of the lung frequently reveal mixed histology (i.e., any combination of bronchioloalveolar (lepidic), papillary, acinar, or solid growth patterns), and are divided into several major subtypes (Table 1) [4, 5]. Among the four histologic subtypes, bronchioloalveolar carcinoma is a special subtype of adenocarcinoma. It is referred to as adenocarcinoma in situ (AIS) totally consisting of a lepidic growth pattern (formerly called a BAC pattern) and shows growth of neoplastic cells along pre-existing alveolar structures (lepidic growth) without evidence of stromal, vascular, or pleural invasion. AIS are subdivided into non-mucinous and mucinous variants. Non-mucinous-type AIS is the major form, and positive for thyroid transcription factor (TTF)-1 antigen. Therefore, it is included in terminal respiratory unit (TRU)-type adenocarcinoma [6]. On the other hand, it is still debatable whether mucinous-type AIS can be included in AIS. On thin-slice computed tomography (CT) scan examination, non-mucinous-type AIS reveales localized ground glass opacity (GGO). The 5-year survival of patients with localized resected AIS is 100%.
2 Atypical adenomatous hyperplasia
Atypical adenomatous hyperplasia (AAH) is a localized proliferation of mildly to moderately atypical cells lining involved alveoli and, sometimes, respiratory bronchioles, resulting in focal lesions in the peripheral alveolated lung (Fig. 1a,b). Initially, AAH had been considered a reactive, and not neoplastic, lesion. However, several studies subsequently proved that AAH is a pure neoplastic lesion and may be a precursor of peripheral-type adenocarcinoma. For example, Nakayama et al. demonstrated that some AAHs contain aneuploid stem lines [7]. AAH is usually less than 5 mm in diameter and lacks any underlying interstitial inflammation or fibrosis. AAH shows positivity for TTF-1 antigen and is a preinvasive lesion of peripheral-type adenocarcinoma, especially the TRU type. AAH is found by thin-slice CT scan examination and shows characteristic GGO, similar to AIS. Usually, AAH is a solitary lesion but cases of multiple AAH are detected occasionally [8].

Atypical adenomatous hyperplasia (A, ×20; B, ×200)
3 Adenocarcinoma in situ and minimally invasive adenocarcinoma
Adenocarcinoma with pure lepidic growth is a special subtype, because it mimics AAH, which is a preinvasive form of adenocarcinoma and has an extremely favorable prognosis. Therefore, it can be termed “adenocarcinoma in situ” (Fig. 2a,b). Minimally invasive adenocarcinoma (MIA) is included in adenocarcinoma with a predominantly lepidic growth pattern (Fig. 3a,b). MIA is most often non-mucinous, but rarely, mucinous-type adenocarcinoma can also be included. MIA is, by definition, solitary, and discrete. MIA criteria can be applied in the setting of multiple tumors only if the other tumors are regarded as synchronous primaries rather than intrapulmonary metastasis. The proportion of the lepidic growth pattern in MIA is closely associated with patient outcome. On the other hand, MIA contains a very limited invasive area, usually less than 5 mm in diameter. If the tumor is larger than 2 cm, diagnosis should be done with caution, and the tumor needs to be extensively sampled, especially the solid component. On thin-slice CT examination, MIA reveals a partly solid appearance. The 5-year survival of patients with localized resected MIA is more than 95%.

Adenocarcinoma in situ (formaly known as bronchioloalveolar carcinoma; type A, Noguchi classification; A, ×20; B, ×200)

Minimally invasive adenocarcinoma (A, ×20; B, ×200)
4 Noguchi classification and biology
Many peripheral type adenocarcinomas contain an area of lepidic growth in the early phase, such as AAH, AIS, and MIA. Early stage adenocarcinomas of the lung are classified into two groups [9] (Table 2), one containing lepidic growth and the other showing destructive growth of pre-existing alveolar structures. Both of these in turn are subdivided into three histological types as detailed below. This classification is closely associated with stepwise progression of peripheral type adenocarcinoma and is known as the “Noguchi Classification”.
-
Type A: Localized bronchioloalveolar carcinoma
-
Tumors of this type are solitary and show totally lepidic growth with minimal or mild thickening of the alveolar septa (Fig. 2). The tumors lack fibrotic foci and the individual tumor cells resemble Clara cells, type II pneumocytes, or goblet cells. Sometimes it is difficult to discriminate type A adenocarcinoma from AAH. On the basis of several features, such as cell stratification and nuclear and cell structure, they can be differentiated histologically (Table 2) [8]. Therefore, type A adenocarcinoma is identical with AIS.
-
Type B: Localized bronchioloalveolar carcinoma with foci of alveolar structural collapse
-
The overall microscopic appearance of these tumors is similar to that of type A, showing a lepidic growth pattern (Fig. 4a,b). However, the tumors contain fibrotic foci due to alveolar collapse (benign scarring). The fibrosis in this tumor is due to alveolar collapse without cellular growth, and is regarded as benign fibrosis. Type B adenocarcinoma is also included in AIS category.
Fig. 4 
Adenocarcinoma in situ with alveolar collapse (type B, Noguchi classification; A, ×20; B, ×200)
-
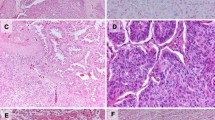
Type C: Localized bronchioloalveolar carcinoma with foci of active fibroblastic proliferation
-
This type constitutes the largest group of ealy stage adenocarcinomas. The tumors also show a lepidic growth pattern, but foci of active fibroblastic proliferation are detectable (Fig. 5a,b). In these foci, the proliferating fibroblasts and endothelial cells of small vessels contain prominent nuclei and nucleoli [10]. MIA is included in type C adenocarcinomas [11–14].
Fig. 5 
Adenocarcinoma with focus of fiborblastic proliferation (type C, Noguchi classification; A, ×20; B, ×200)
-
Type D, E, and F: (Adenocarcinomas showing non-lepidic growth)
-
Type D adenocarcinomas show largely solid growth, and papillary and acinar growth patterns are minor components (Fig. 6a,b). Histologically, their classification is almost the same as that of “solid adenocarcinoma with mucin production” in the World Health Organization (WHO) classification (third edition) [4]. Type E adenocarcinoma consists of acinar, tubular and cribriform structures, and tumor cells with a signet ring appearance may be present. This type is classified as “acinar adenocarcinoma” in the WHO classification (third edition). This specific type of adenocarcinoma is thought to originate from, or differentiate toward, bronchial gland cells. Type F adenocarcinomas show papillary growth. However, they do not grow by replacing the alveolar lining cells, but instead show expansive and destructive growth. This type is classified as “papillary adenocarcinoma” in the WHO classification (third edition) [4].
Fig. 6 
Solid adenocarcinoma (type D, Noguchi classification; A, ×20; B, ×200)



The Noguchi classification is well correlated with thin-slice CT findings and patient outcome. Type A and B tumors have an extremely favorable prognosis, and patients have a 5-year survival rate of 100% [9]. These types can be regarded as AIS. Type C tumors are similar to types A and B and show lepidic growth of alveolar lining cells, but the 5-year survival rate averages 75%. Type C adenocarcinoma contains MIA. Compared to lepidic growth-type adenocarcinomas, types D, E, and F tumors have a relatively poor prognosis, and the 5-year survival rate of patients with type D tumors is 50%. On the other hand, a histological lepidic growth pattern corresponds to the ground glass opacity feature evident by thin-slice CT. Therefore, types A, B, and C tumors and other non-lepidic-type tumors can be distinguished on the basis of thin-slice CT findings. These histological and radiological findings suggest malignant progression of peripheral-type adenocarcinomas from AAH, types A and B, to type C (Fig. 7). On the basis of these clinicopathological findings, type A and B adenocarcinomas with an extremely favorable prognosis are candidates for limited surgery such as wedge resection or segmentectomy, or careful observation alone.

Stepwise progression of adenocarcinoma (type A-C)
5 Genetic alteration of peripheral type adenocarcinoma
Lung adenocarcinoma can be divided into at least three types genetically: the V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) type, the epidermal growth factor receptor (EGFR) type, and the non-EGFR/KRAS type, based on accumulated genetic alterations in the adenocarcinoma cells.
Ras belongs to the guanosine triphosphatase superfamily of proteins that cycle between active and inactive forms. In particular, KRAS point mutations are detected in many adenocarcinomas of various organs, such as the lung, pancreas, ovary, stomach, and colon. About 10-30% of pulmonary adenocarcinomas are reported to carry point mutation of the KRAS gene, but the frequency differs between Asian and Western countries [15]. Pulmonary adenocarcinomas containing KRAS mutation have certain cytological characteristics. Most of them are classified as goblet cell-type adenocarcinoma and are negative for TTF-1 [16].
Ligands of EGFR such as EGF, upon binding to EGFR, trigger receptor tyrosine kinase activation and a series of downstream signaling actions that mediate cellular proliferation, migration, invasion, and suppression of apoptosis [17]. In 2004, two research groups simultaneously reported that patients who were responsive to EGFR tyrosine kinase inhibitors such as gefitinib had somatic mutations in the kinase domain of the EGFR gene in their tumor cells [18, 19]. Subsequently, frequent EGFR mutations were reported in Asian patients, including Japanese, in comparison to Caucasians [20–22]. The incidence of EGFR mutation was significantly high in female patients with adenocarcinoma without a smoking history. EGFR mutation is frequently detected in lepidic-type adenocarcinoma and papillary adenocarcinoma [23]. Most of them are positive for TTF-1 antigen.
The p16 tumor suppressor gene is frequently inactivated in lung adenocarcinoma, most prominently through promoter methylation with loss of chromosome 9p21 and less frequently through point mutations or intragenic deletions. As the p16 gene is an important tumor suppressor gene, loss of function of p16 is significantly associated with the outcome of patients with adenocarcinoma [24]. Recently, Nakanishi et al. reported that p16 homologous deletion occurs early in the development of lung adenocarcnomas and with similar frequencies among the EGFR, KRAS, and non-EGFR/KRAS types. These results indicate that either EGFR or KRAS mutations occur prior to homologous deletion or methylation of the p16 gene during the progression of adenocarcinomas [25].
The P53 gene is a well-studied tumor-suppressor gene located on chromosome 17p13.1. Forty to fifty percent of human tumors contain mutations of this gene. The fact that p53 mutations are common in a variety of human malignancies suggests that p53 protein serves as a critical gatekeeper against the development of malignancy. It is localized to the nucleus, and wild-type p53 binds to DNA and stimulates transcription of several genes that mediate the two major effects of p53: cell-cycle arrest and apoptosis. About half of all pulmonary adenocarcinomas harbor mutated p53 [26]. There are no definite correlations between p53 mutation and EGFR mutation and/or KRAS mutation. In comparison with EGFR and KRAS abnormalities, p53 mutation is a late event in pulmonary adenocarcinogenesis [23].
In 2007, Soda et al. reported the transforming EML4-ALK fusion gene. Both EML4 and ALK are located on the short arm of chromosome 2 (2p21 and 2p23, respectively), but in opposite orientation [27]. This fusion gene is formed as a result of a small inversion with the short arm of chromosome 2 that joins intron 13 of EML4 to intron 19 of ALK. Inamura et al. examined many cases of adenocarcinoma for the fusion gene and found that 3.4% of them showed EML4-ALK fusion mRNA [28]. Cases harboring the EML4-ALK fusion gene showed papillary with bronchioloalveolar components or acinar adenocarcinoma. All of them expressed TTF-1 protein and are classified into the TRU-type adenocarcinoma subgroup. None of the fusion-positivsae cases demonstrated any mutations of EGFR or KRAS, suggesting a mutually exclusive relationship.
Similar to other types of lung carcinoma, adenocarcinoma carries various allelic losses at a number of chromosomal loci. Aoyagi et al. reported that accumulation of loss of heterozygosity in crucial chromosome regions occurred stepwise during sequential progression from type A/B to type C in the Noguchi classification [9]. Matsumoto et al. indicated that loss of the chromosome 13q13 region was evident in more than 50% of both early and advanced adenocarcinomas. Allelic imbalances in the chromosome 11p11-12p, 17p12-p13, and 18p11 regions are detected more frequently in advanced than in early adenocarcinomas. In particular, allelic imbalance at 8p21 was significantly more frequent at the advanced stage, and associated with poor outcome [23].
6 Multistep carcinogenesis
On the basis of histological, cytological, and molecular biological findings, common pulmonary adenocarcinogenesis is considered to be a multistep process as follows (Fig. 8).

Stepwise progression of adenocarcinoma (histology and molecular alterations)
Multi-potential epithelial cells located in TRUs acquire AAH as a result of exposure to various carcinogens. At this time, abnormality of the EGFR gene occurs in about 50% of cases of AAH. AAH progresses to AIS (pure bronchioloalveolar carcinoma) over a long period, and meanwhile, apoptotic function such as that mediated by the p16 gene, becomes perturbed and the expression of Bax inhibitor 1 [29]. The tumor cells also acquire several allelic inbalances such as 13q13. At this stage, the main genetic alterations are EGFR gene mutation and anti-apoptotic abnormality. Therefore, the tumor cells can survive, but their growth rate is still very low.
In the course of development to overt invasive adenocarcinoma, the tumor cells acquire various additional genetic abnormalities, such as p53 mutation and allelic imbalances at 17p12-p13, 18p11, and 11p11-p12. In particular, allelic imbalance at 8p21 is closely associated with poor prognosis [25]. Thus, adenocarcinoma of the lung develops after a series of well-defined pathological stages accompanied by a sequentially acquired series of molecular changes. It is now well established that this progression model is closely linked to patient prognosis.
References
Travis, W. D., Lubin, J., Ries, L., & Devesa, S. (1996). United States lung carcinoma incidence trends: declining for most histologic types among males, increasing among females. Cancer, 77, 2464–2470.
Valaitis, J., Warren, S., & Gamble, D. (1981). Increasing incidence of adenocarcinoma of the lung. Cancer, 47, 1042–1046.
Travis, W. D., Travis, L. B., & Devesa, S. S. (1995). Lung cancer. Cancer, 75, 191–202.
Travis, W. D., Colby, T. V., Corrin, B., et al. (1999). WHO histological typing of lung and pleural tumors (3rd ed.). Geneva: World Health Organization.
Travis, W. D., Brambilla, E. B., Muller-Hermelink, H. K., & Harris, C. C. (2004). WHO pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon: IARCPress.
Yatabe, Y., Mitsudomi, T., & Takahashi, T. (2002). TTF-1 expression in pulmonary adenocarcinomas. American Journal of Surgical Pathology, 26, 767–773.
Nakayama, H., Noguchi, M., Tsuchiya, R., Kodama, T., & Shimosato, Y. (1990). Clonal growth of atypical adenomatous hyperplasia of the lung: cytofluorometric analysis of nuclear DNA content. Modern Pathology, 3, 314–320.
Anami, Y., Matsuno, Y., Yamada, T., et al. (1998). A case of double primary adenocarcinoma of the lung with multiple atypical adenomatous hyperplasia. Pathology International, 48, 634–640.
Noguchi, M., Morikawa, A., Kawasaki, M., et al. (1995). Small adenocarcinoma of the lung. Histologic characteristics and prognosis. Cancer, 75, 2844–2852.
Nakamura, N., Iijima, T., Mase, K., et al. (2004). Phenotypic differences of proliferating fibroblasts in the stroma of lung adenocarcinoma and normal bronchus tissue. Cancer Science, 95, 226–232.
Minami, Y., Matsuno, Y., Iijima, T., et al. (2005). Prognostication of small-sized primary pulmonary adenocarcinomas by histopathological and karyometric analysis. Lung Cancer, 48, 339–348.
Suzuki, K., Yokose, T., Yoshida, J., et al. (2000). Prognostic significance of the size of central fibrosis in peripheral adenocarcinoma of the lung. Annals of Thoracic Surgery, 69, 893–897.
Yokose, T., Suzuki, K., Nagai, K., Nishiwaki, Y., Sasaki, S., & Ochiai, A. (2000). Favorable and unfavorable morphological prognostic factors in peripheral adenocarcinoma of the lung 3 cm or less in diameter. Lung Cancer, 29, 179–188.
Sakurai, H., Maeshima, A., Watanabe, S., et al. (2004). Grade of stromal invasion in small adenocarcinoma of the lung: histopathological minimal invasion and prognosis. American Journal of Surgical Pathology, 28, 198–206.
Sakamoto, H., Shimizu, J., Horio, Y., et al. (2007). Disproportionate representation of KRAS gene mutation in atypical adenomatous hyperplasia, but even distribution of EGFR gene mutation from preinvasive to invasive adenocarcinomas. Journal of Pathology, 212, 287–294.
Kobayashi, T., Tsuda, H., Noguchi, M., et al. (1990). Association of point mutation in c-Ki-ras oncogene in lung adenocarcinoma with particular reference to cytologic subtypes. Cancer, 66, 289–294.
Mok, T. S., Wu, Y. L., Yu, C. J., et al. (2009). Randomized, placebo-controlled, phase II study of sequential erlotinib and chemotherapy as first-line treatment for advanced non-small cell lung cancer. Journal of Clinical Oncology, 27, 5080–5087.
Paez, J. G., Janne, P. A., Lee, J. C., et al. (2004). EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science, 304, 1497–1500.
Lynch, T. J., Bell, D. W., Sordella, R., et al. (2004). Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small cell lung cancer to gefitinib. New England Journal of Medicine, 350, 2129–2139.
Takano, T., Ohe, Y., Sakamoto, H., et al. (2005). Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. Journal of Clinical Oncology, 23, 6829–6837.
Shigematsu, H., Lin, L., Takahashi, T., et al. (2005). Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. Journal of the National Cancer Institute, 97, 339–346.
Yatabe, Y., Takahashi, T., & Mitsudomi, T. (2008). Epidermal growth factor receptor gene amplification is acquired in association with tumor progression of EGFR-mutated lung cancer. Cancer Research, 68, 2106–2111.
Matsumoto, S., Iwakawa, R., Kohno, T., et al. (2006). Frequent EGFR mutations in noninvasive bronchioloalveolar carcinoma. International Journal of Cancer, 118, 2498–2504.
Tanaka, R., Wang, D., Morishita, Y., et al. (2005). Loss of function of p16 gene and prognosis of pulmonary adenocarcinoma. Cancer, 103, 608–615.
Nakanishi, H., Matsumoto, S., Iwakawa, R., et al. (2009). Whole genome comparison of allelic imbalance between noninvasive and invasive small-sized lung adenocarcinomas. Cancer Research, 69, 1615–1623.
Sato, K., Ueda, Y., Shikata, H., & Katsuda, S. (2006). Bronchioloalveolar carcinoma of mixed mucinous and nonmucinous type: immunohistochemical studies and mutation analysis of the p53 gene. Pathology, Research and Practice, 202, 751–756.
Soda, M., Choi, Y. L., Enomoto, M., et al. (2007). Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature, 448, 561–566.
Inamura, K., Takeuchi, K., Togashi, Y., et al. (2008). EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. Journal of Thoracic Oncology, 3, 13–17.
Tanaka, R., Ishiyama, T., Uchihara, T., et al. (2006). Expression of the Bax inhibitor-1 gene in pulmonary adenocarcinoma. Cancer, 106, 648–653.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Noguchi, M. Stepwise progression of pulmonary adenocarcinoma—clinical and molecular implications. Cancer Metastasis Rev 29, 15–21 (2010). https://doi.org/10.1007/s10555-010-9210-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-010-9210-y