
Abstract
Tumorigenesis of the lung cancer is a multistep transformation involving genetic and epigenetic alterations. Aims To evaluate the general genetic pathways, their involvement, determining how they relate to the biological behavior of lung cancer, and their utility as diagnostic and therapeutic targets. Materials and method Online literature search through Google scholars, Pubmed, and hard copies, using keywords “biological alterations and lung carcinoma” yielded 9,380,000 publications. The 16 selected papers were used as a basis to formulate this report, and to evaluate the general pathways and involved genes, determining how they relate to the biological behaviour of lung cancer and their utility as diagnostic and therapeutic targets. Results and discussion Growth signals and lung cancer: In tumours cell, the activated oncogenes encode the molecules relating to the signalling growth factors either by direct initiation of the cell growths as an imitation of other growth factors or neutralization of the growth factor inhibitor. Conclusion Significant advances in molecular biologic research during recent decades have resulted in a substantial insight into critical signalling pathways and mediators contributing to lung cancer pathogenesis.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
The carcinogenesis of lung cancer is the multistep progress of transformation from normal epithelial cells of bronchi to cancer cells. Decades of research have contributed to our understanding of these processes, through which resulting DNA damage transforms normal lung epithelial cells into the lung cancer cells. The conclusion of these alterations leads to lung cancers, displaying every marks of lung malignancy, comprising self-sufficiency of growing signs, unresponsive to grow-inhibitory signs, avoidance of predetermined cell decease, unlimited proliferation ability, nonstop angiogenesis and tissue penetration and metastasis.
Clinically, lung cancer is a heterogeneous disease, biologically, histologically and molecularly. The molecular mutations lead to the deregulation of the critical genetic pathways, relating to cellular proliferation, differentiation, program cell death, invasion, migration and other progress harboring the malignant particularities [1]. Hanahan and Weinberg [2] suggested classification according to hallmarks of cancer: Unrestrained cell production; elusion of scheduled cell mortality; deficit of reliance on growing stimulatory indications; uninhibited replicative ability; unrestricted Angiogenesis; envasion and metastasis. This review presented the progress of each hallmark as expressed by clinical and genetic properties of carcinogenesis.
2 Materials and Method
An online literature search through Google scholars, PubMed, and hard copies, using keywords “biological alterations and lung carcinoma” yielded 9,380,000 publications. The 16 selected papers were used as a basis to formulate this report, and to evaluate the general pathways and involved genes, determining how they relate to the biological behavior of lung cancer and their utility as diagnostic and therapeutic targets.
3 Results and Discussion
3.1 Growth Signals and Lung Cancer
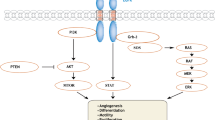
In tumors cell, the activated oncogenes encode the molecules relating to the signaling growth factors either by direct initiation of the cell growths as an imitation of other growth factors or neutralization of the growth factor inhibitor [3]. The tumor cell, losing the dependency on stimulatory growth signals from surrounding environment, have the ability of independent growth [4]. This phenotype of auto growth, by the influence of intracellular changes as molecular alterations and gene mutations, express the “self-supporting of growth factors,” then produce the necessary growth factors and receptors from “self-stimulatory autocrine signaling loop” leading to the uncontrolled proliferation [5].
In non-small cell lung cancer (NSCLC), these phenomena include the mutation of up regulating of some EPHB4 receptor tyrosine kinase (RTK), specifically epidermal growth factor receptor [EGFR (Erb1)] and other members of ErbB RTK family.
In SCLC, the overexpression of Insulin-like growth factor-1 (IGF-1) and its receptor, as well as its neuro-stimulating growth factors often observed.
3.1.1 Over-Expression of Epidermal Growth Factor and Signaling in NSCLC
Overexpression of EGFR was observed in 50–90% of NSCLC and especially in SCC, but rare in SCLC. Entire EGFRs comprise an extracellular ligand-binding area, a trans membrane area, and a cytoplasmic protein tyrosine kinase field. Binding of the ligand epidermal growth factor leads to receptor homo or heterodimerization with another member of EGFR family and activation of TK domain through the phosphorylation of thymidine kinase (TK) residues [6].
Signal transduction stimulated by EGFR occurs through PI3 K/AKT/mTOR →→ Inhibits the regulator of the cell cycle (glycogen synthase kinase—GSK-3 and pro-apoptotic protein BAD. RAS/RAF/MAPK →→ cell proliferation, and JAK/STAT/PLC-γ signaling pathways →→ affecting the cell motility, migration, and invasion of the cancer cell.
EGFR involves the regulation of numerous oncogenic functions such as proliferation, survival, differentiation, neoangiogenesis, invasion, and metastasis. Also, it relates to the constitutive TK activation and oncogenic transformation of pulmonary epithelial cell in vitro.
EGFR activates other pathways leading to effects antiapoptotic, invasion, metastasis and proliferation, cross-act among pathways. A mutant EGFR—called IIIv EGFR found in different malignancies, about 16% in NSCLC. The EGFRvIII analysis assay is capable of detecting the EGFRvIII mutation. The mutant receptor is incapable of binding any known ligand. The pro-tumorigenic effects of EGFRvIII seem to rely directly on its ability to signal. EGFRvIII activates several downstream pathways; EGFRvIII is the most common mutation in glioblastoma multiforme (GBM), occurring in 25–64% of these tumors. It is also found in 20–36% of breast cancers and about 33% of head and neck squamous cell carcinoma (HNSCC) patients. Some trials report that this kind of mutant EGFR was present in about 5% of NSCLC, compared with wild-type EGFR, EGFRvIII appears to be relatively resistant to treatment with conventional anti-EGFR agents [7].
In NSCLC, EGFR mutations occur in the first four exons of the intracellular tyrosine kinase domain: most commonly exon 19 in-frame deletions (# 45%), of which there are over 20 variants, the universal being delE746-A750. The next most familiar EGFR are missense mutations, particularly L858R (# 40%) in exon 21. Another insertion mutation of EGFR in exon 20 (5–10%) of which there are many variants often resistant to EGFR-TKIs [8].
Secondary mutations in EGFR develop or clonally selected in patients that develop resistance to EGFR-TKIs. The most typical being the T790M activating point mutation in exon 20 which substitutes a bulkier methionine for threonine that interferes with binding of reversible TKIs. T790M found in about 50% of tumors from patients who develop acquired TKI resistance [9].
Another member of ErbB family is Her2/neu is overexpressed about 30% in NSCLC. Until now, there is not yet an adequately identified ligand for Her2 but can form heterodimers with other ligand-bound members of the receptor family. Oncogenic driver mutations identified in non-small cell lung cancer (NSCLC) have triggered the development of drugs capable of interfering in intracellular signaling pathways involved in tumorigenesis. Tyrosine kinase inhibitors, such as erlotinib or gefitinib, have demonstrated promising results in patients with advanced NSCLC that harbor EGFR mutations. Human epidermal growth factor 2 (HER2) mutations in NSCLC, described exclusively in adenocarcinoma histology, are present in approximately 4% of this subset of lung cancer patients, suggesting that thousands of patients per year may benefit from targeted therapy. Therefore, it can conclude that systematic genotypic testing in this subgroup of NSCLC patients should include detection of HER2 mutations.
3.1.2 Overexpression of Other Growth Factor Receptor and Ligand
The expression level of mitogen IGF-1 is elevated in the majority of SCLC, resulting in the formation of a “self-stimulatory autocrine signaling loop” involving IGF-1 receptor, which is commonly co-expressed in this malignancy. IGF signaling proceeds through binding of IGF ligands to cell surface RTK. The biologic activity of signaling system modulated by binding of IGF protein, which present in extracellular fluid, and serum to the IGF ligands. As for EGFR, activated IGF-1R signaling is complex but primarily occurs through the Ras/Raf/EEK and Pi3 K/AKT pathways. A correlation between significantly elevated IGF-1 serum level and lung cancer risk has been reported [10].
RTK c-kit and its ligand SCF (stem cell factor) is another receptor/ligand system, upregulated in more than 80% of SCLC tumors. A study of c-kit expression in SCLC patients identified c-kit as a marker for increased survival.
c-MET is another RTK often overexpressed in SCLC. Signaling through this receptor system has been reported to be associated with tumor growth and metastasis. In contrast to the c-Kit SCF system, the c-MET ligand hepatocyte growth factor (HGF) is rarely co-expressed with the receptor in SCLC.
3.1.3 Activating Ras Mutations
As mentioned, mutation of the intracellular membrane-associated signaling mediator Ras with a high overrepresentation of mutations in the Kras gene detected in up to 30% of NSCLC, but rarely in SCLC. Ras protein becomes activated by the binding of guanine triphosphate (GTP), allowing for transmission of growth stimulatory signals to the cell nucleus.
Downregulation of Ras signaling occurs by hydrolysis of GTP to GDP, mediated by GTPase activating protein (GAP).
In NSCLC and other malignancies, activating point mutations in the K-ras gene result in resistance to GAP activities, thereby trapping the Ras protein in a constitutively active state, capable of continuous growth promoting signaling [11].
3.1.4 Overexpression of Neuropeptides
Highly elevated expression of different neuropeptides is a hallmark of SCLC, and many of these markers have also detected in some (mainly poorly differentiated) NSCLC tumors. Neuropeptides exert their effect via binding to seven transmembrane G-protein coupled receptors, resulting in activation of various downstream signaling pathways including PLC. Gastrin-releasing peptide (GRP) signaling via the GRP-receptor has become one of the most intensively studied neuropeptide signaling pathways in SCLC. Other neuropeptides highly expressed in lung cancer include bradykinin, neuron-specific enolase, and l-Dopa decarboxylase.
In recent years, the expression of neuroendocrine transcription factor Achaete-scute homolog-1 (ASH-1) in SCLC has gained increased attention. ASH-1 usually is expressed in neuronal progenitor cell during early fetal development of various tissues including the central nervous system and the lung. The expression is virtually absent in normal adult organism but reactivated and highly expressed in SCLC and In other lung tumors with a neuroendocrine phenotype.
3.1.5 Amplification of Myc Gene
Myc is a member of a proto-oncogene family comprising N-myc, c-myc, and L-myc, commonly amplified in NSCLC and SCLC, resulting in overexpression of transcription factor Myc. Although the contribution of myc-amplification to the pathogenesis of lung cancer remains to be elucidated; a recent study has pointed out to a role of myc to promote cell cycle progression. Also, myc in combining with loss of TSG such as Rb has been shown to significantly contribute to decreased cell cycle arrest and deregulate tumor growth.
3.1.6 Anaplastic Lymphoma Kinase (ALK)
Rearrangement of RTK ALK is most common in the fusion of the intracellular kinase domain with the amino-terminal end of Echinoderm Microtubule-associated protein-Like 4 (EML4) occur in a subset of lung cancers. The rearrangement result from a short inversion in chromosome 2p whereby in the best-known variant, intron 13 of EML4 id fused to intron 19 of ALK. Numerous variants of EML4-ALK fusions have identified due to different lengths of EML4, the commonest being exons 1–13 of EML4 joining to exon 20–29 of ALK. More recently, various partner gene has identified in a small subset of ALK rearrangements including KIF5B (kinase family member 5b), TFG (TRK-fuse gene) and KLC-1 (kinesin light chain1). Activation of ALK is linked to cell proliferation and inhibition of apoptosis mediated through RAS/RAF/MAPK1, PI3K/AKT, and JAK3-STAT3 signaling pathways.
ALK rearrangement has identified in approximately 4% of unselected NSCLC. ALK inhibition with TKI Crizotinib produce a profound response, drug resistance develops with evidence of secondary ALK point mutations.
3.1.7 ROS1 Proto-Oncogene
Ros1 is a proto-oncogene located on chromosome 6q22, which encodes a transmembrane RTK that has high homology with ALK in its protein kinase domain. ROS1 activation leads to signaling through the PI3K/AKT/mTOR, STAT3 and RAS/MAPK/ERK pathways [4]. ROS1 rearrangements appear to be more common in patients who are younger, never smoker or of Asian Ethnicity similar to ALK rearrangement. Furthermore, there is in vitro and early clinical evidence that lung cancers with ROS1 rearrangement are sensitive to kinase inhibitors including inhibitor Crizotinib.
3.1.8 RET
RET is located on chromosome 10q11.2 and encodes a receptor tyrosine kinase in neural crest development. Alteration of the crest has long been known to play a role in papillary and medullary thyroid carcinoma. But it was not until recently that activation of RET through chromosomal rearrangement has identified in a small proportion of lung cancer. Similar to ALK and ROS1, rearrangements of RET also appear to be associated with ADC from never smoker [4, 5].
3.1.9 Experimental Therapeutic Targeting of Growth Factors and Oncogenes
Some different growth factors and oncogenes of importance for lung cancer biology have presented. A research area of the significant locus in recent years has been the therapeutic targeting of EGFR in NSCLC.
There are two global strategies: one is to prevent binding of the ligand through blocking the ligand-binding site (belonging to this group Cetuximab is a humanized mAB anti EGFR). The other is to directly inhibit receptor signaling by blocking activities of cytoplasmic tyrosine kinase domain (the two most clinical advanced are erlotinib and gefitinib).
Recently, some others newer agents: Afatinib (GilotrifR—Chemocare); Crizotinib (XalkoriR—Pfizer) for uses of EML4-ALK, ROS1, RET; Ceritinib (ZykadiaR—Novartis) used for NSCLC patient who has developed resistance to crizotinib.
3.2 Aberrant Anti-growth Signaling
In contrast to an oncogene, TSG act to prevent and control cell growth, often via tight control of cell cycle progression. Full inhibition of tumor suppressors activity often requires inactivation of both alleles of a TSG in the cancer cell. This dual inactivation is frequently accomplished by a two-step process, involving a chromosomal translocation or deletion resulting from loss of heterozygosity (LOH), followed by inactivating point mutation of the remaining allele. In lung cancer cells, LOH of distinct chromosomal regions frequently detected, and many of these regions harbor genes encoding central tumor suppressor, known or speculated to be involved in cancer pathogenesis.
3.2.1 TP53 Gene Mutations
The TP53 gene located within a region of chromosome 17 (17p13), which is mutated or altered in the majority of lung cancer with a specific high prevalence in SCLC or SCC. The type of TP53 alterations observed in lung cancer range from gross chromosomal changes such as LOH, homozygous deletion, and DNA rearrangements, to local point mutations, all of which contribute to TP53 malfunction or inactivation.
The cellular enzyme MDM2 play an important role in the downregulation of p53. It serves as a p53 binding partner, which facilitate the attachment of ubiquitin tags to p53, thereby targeting it for degradation. MDM2-p53 interaction generates an oscillating feedback loop of p53 and MDM2 degradation and synthesis within the cell.
Active p53 regulates transcription of some genes involved in cell cycle control, resulting in cell cycle arrest, allowing for repair of damaged DNA by the cellular repair machinery. Activation of p53 also induces apoptosis via activation of some apoptotic mediator (including BAX) and inhibits blood vessel formation by activation of genes encoding antiangiogenic factors. P53 mutations suggest a poorer prognostic in NSCLC and SCLC as well.
3.2.2 Mutated RB and P16INK4A
A central tumor is suppressing signaling cascade, frequently altered in human lung cancer, is the p16INK4A/CDK-cycling/Rb pathway. The retinoblastoma (Rb) tumor suppressor gene at 13q14 encodes a transcription factor involved in the regulation of G1 to S phase transition in the cell cycle. The tumor-suppressing activity of Rb depends on its level of phosphorylation. In its hypo-phosphorylated state, Rb binds to and inhibit the activity of different binding partners, including members of the E2F family of transcription factors. Upon phosphorylation of Rb, E2F is released and activated, resulting in transcription of the gene responsible for G1 to S phase cell cycle progression. The formation of these complexes inhibited by p16INK4A which thereby serves as a tumor suppressor upstream of Rb by indirect inhibiting its phosphorylation and thereby promoting Rb association with its binding partner.
3.2.3 Aberrant TGF-β Signaling
The transforming growth factor β (TGF-β) receptor system is also commonly altered in lung cancer. The effects of signaling by TGF-β mostly associated with inhibited cellular proliferation in many cell types. TGF-β signaling related to some cell functions, the best described of which relate to inhibition of the cell cycle. Growth inhibitory effects of TGF-β signaling have associated with inhibition of expression and assembly of some of cyclin/CDK components responsible for Rb activation.
3.2.4 Loss of Chromosome 3p and Related Genes
Probably the most frequent chromosomal abnormality in lung cancer is a loss of regions within chromosome 3p. LOH at chromosome 3p has been reported in 70–100% of all NSCLC and more than 90% of SCLC. Some genes within this region have suggested as putative tumor suppressors.
The loss of the fragile histidine triad (FHIT) gene located at position 3p14, 2 is frequent in lung cancer. Expression of FHIT protein in NSCLC cell line and mouse xenograft models have been showed to suppress tumor growth and induce apoptosis. And recently FHIT has been found to stabilize p53 presumably by interaction with MDM2 [10, 12].
RASSFIA is a different candidate tumor suppressor gene residing at chromosome 3p (position 3p21). This gene inactivated in virtually all SCLC and more than 60% NSCLC. RASSFIA has been found to reduce motility of NSCLC cell and increase cell adhesion, suggesting a role for RASSFIA in cell migration and metastasis.
Several other genes reside at the frequently deleted regions of chromosome 3p but much remains to be learned about the role of these genes in tumor suppression.
3.2.5 Experimental Treatment: The Reintroduction of Tumor Suppressors
Since the loss of activity of specific tumor suppressor pathways is a distinctive characteristic of human lung cancer, the reinstatement of tumor suppressor activity is an attractive strategy for the therapeutic intervention. For this purpose, replacement therapy by delivery of lost tumor suppressor genes to cancer has become increasingly attractive. Most reports of tumor suppressor replacement gene therapy involve reintroduction of TP53 in NSCLC, where some clinical trials have published. However, the limiting factor of gene therapy today remains poor delivery of the therapeutic gene to the cancer cells. The clinical study performed to date have all used modified viruses for gene delivery, but a significant drawback of using viruses for gene delivery is the induction of immune responses against the virus in the patients. Results in the production of antibodies, which target the virus for degradation and limit the efficiency of repeated treatment. Novel non-viral delivery vehicles developed. Which may in the future provide a potent alternative to viral gene therapy [11, 13].
3.3 Apoptosis in Lung Cancer
Apoptosis is a morphologically and biochemically distinct form of cell death that occurs under various physiologic and pathologic conditions triggered by extrinsic and intrinsic cellular and molecular damage. It characterized by the activation of a specific event of molecular processes followed by specific morphologic changes such as shrinkage of the cell, condensation of chromatin, and disintegration of the cell into small fragments [6, 16].
Apoptosis activated by a family of intracellular cysteine proteases called caspases. They are synthesized as zymogens and activated by proteolytic cleavage. They divided into two distinct classes, initiator caspase, which include caspase P8, P9 and P10; and effector caspase, which provides for P3, P6, and P7. There are two separate pathways of caspase activation: one starts with binding of an extracellular ligand to its cell surface receptor. The ligands are TNF, FasL, and Trail, and their respective receptors are TNFRI, FAS, and DR4 and DR5. The other caspase activation pathway starts with the release of cytochrome C from the intermembrane space of mitochondria. Two proapoptotic family members, BAX and BAK, appear to facilitate cytochrome C release by participating in the formation of a pore that releases mitochondrial intermembrane space protein. Once discharge, cytochrome C fixes to apoptotic protease activating factor 1 (APAF-1) fastening to procaspase 9 establishing a multiprotein compound, named an apoptosome. Inhibitor of apoptosis protein (IAP) binds and inhibits apoptosome related caspases. The most widely studied IAP is survivin, which elevated in the majority of NSCLC and it has shown that absence of its expression might associate with improved prognosis. Activators and inhibitors are influenced by several other proteins including p53, Rb, PTEN, Raf-ERK, PI3K-PKB and Hsp70 [5].
Another member of apoptosis in lung cancer: programmed cell death 1 (PD-1) is also CD279, a cell surface receptor encoded by PDCD-1 gene, member of the superfamily of immunity. It expressed on the surface of T and preB cell, NSCLC and SCC cells. PD-1 binds to two ligands: PD-L1 and PD-L2, member of family B7-homolog and encoded by CD 247. PD-1 and its ligand regulate downstream immune system [12].
3.3.1 Targeting Apoptotic Pathway
Treatment with TNF has undertaken. However, due to pronounced general toxicities, its potential as a therapeutic drug is limited; Recently, Trail agonist have approved for clinical trials, but no data are presently available [7].
Small molecule inhibitor of Bcl-2 have been developed and are at the moment being tested in preclinical trial and phase I test.; antisense constructs against survivin have been produced and test in phase I clinical trial. In addition, adenovirus-based gene therapy is under development.
Inhibition of PD-1 leads to promote an immune system for the treatment of non-squamous NSCLC. Nivolumab (Opdivo—BMS; FDA approved 12/2014; Pembrolizumab (Keytruda—MERCK) target PD-1; FDA approved 9/2014 for NSCLC and HCC; other agents PALIVIZUMAB—BMS; MPDL—Roche also target PD-1, on the study of phase III [14].
3.4 Angiogenesis
Good vascularity is vital for cell task and existence in each matter as oxygen and nutrients delivered by the vessels. The growth of new blood vessel, called the process of angiogenesis, is a typical physiologic process-taking place under angiogenesis, which under these conditions, is transitory and carefully regulated. Similarly, tumors must develop angiogenic ability to progress. This ability appears by activating the angiogenetic switch. Once cancer has activated its angiogenic switch, it becomes able to grow. Many different pro and anti-angiogenic factor have identified. The angiogenesis instigating signs are demonstrated by vascular endothelial growth factors (VEGF)/vascular permeability factor (VPF) and acidic (FGF1) and basic fibroblast growth factors (FGF2), which completely attach to transmembrane tyrosine kinase receptor presented by endothelial cell. VEGF has been proven to be a important prognosticator of insignificant diagnosis in NSCLC.
3.4.1 Targeting Angiogenic Factors
Inhibition of angiogenesis is through anti-angiogenic and/or vascular targeting agents seem logical, as the new anti-cancer treatment strategy. There has been increasing attention focused on targeting VEGF and VEGFR. The recent clinical trial has shown that the anti-VEGF antibody bevacizumab, combined with standard first-line chemotherapy in NSCLC, provided a statistically and clinically significant survival advantage with tolerable toxicity. Also, more recently tested compounds characterized as an antivasculature agent have been shown to be effective against multiple targets Sunitinib, regorafenib, ramucirumab. The efficacy of such compounds currently investigated in a clinical trial for NSCLC.
3.5 Replicative Potential and Telomerase
After some division, the cells are predetermined to enter a crisis, a state characterized by extensive cell death and chromosomal aberrations. This phenomenon has been termed the mitotic clock and is part of the precise regulation of healthy cell growth. In contrast, cancer cells propagated in culture have an unlimited potential for continuous cell division, and said to be immortalized.
The molecular explanation for the mitotic clock resides in the chromosomal structure and mechanism of DNA replication. Upon cell division, the cell initiates DNA replication which proceeds to produce new leading and lagging strands from the DNA double helix. Since DNA replication can only proceed in one direction (3′–5′), only the leading strand of the double helix is continuously synthesized, whereas the new lagging strand assembled by ligation of smaller DNA fragments. The discontinuous replication of the lagging strand results in a gap at the 5′ end of the newly synthesized DNA strand, resulting in loss of chromosomal material during each mitotic cycle. Due to the continuous shortening of telomeric DNA following cell division, lack of telomere maintenance ultimately results in chromosomal degradation and end-to-end chromosome fusion. For the cells to overcome the limitation of telomere shortening, cancer cell activates a program for telomere maintenance, which is usually shut down in fully differentiated normal cells. Most frequently, this accomplished by activation of an enzyme complex known as telomerase, but a subset of cancer cell lacks telomerase activity and immortalized by a process known as alternative lengthening of telomerase (ALT).
3.5.1 Telomere Maintenance in Lung Cancer
The core telomerase enzyme comprises an RNA subunit (hTERC) which provides the template for synthesis of new telomeric DNA facilitated by the catalytic subunit human telomerase reverse transcriptase (hTERT). Some studies have shown that increased telomerase activity and increased level of hTERT mRNA are mainly found in patients with poorly differentiated tumor (such as SCLC) and advanced disease and correlate with poor survival, suggesting telomerase activity as an important prognostic marker for patients with lung cancer.
3.5.2 Experimental Therapeutic Targeting of Telomerase in Lung Cancer
Due to the central role of telomerase in the transformation of lung cancer cell, and the lack of telomere maintenance in normal tissue, blocking the activity of this enzyme appear an intriguing target for therapeutic intervention.
The compound GRN136L is a lipid-modified oligonucleotide, which binds to the hTERC subunit of telomerase with high affinity, thereby inhibiting reverse transcription by blocking access of hTERT to it RNA template. GRN 136L has recently been reported to successfully inhibit telomerase activity, leading to telomere shortening and resulting in decreased growth of adenocarcinoma cell in vitro and effective prevention of tumor metastasis in a xenograft mouse model [11].
3.6 Tissue Invasions and Metastasis
Tumor cell can produce some proteolytic enzyme, which can degrade these protein structures. Including matrix metalloproteinase (MMPs), collagenase, urokinase plasminogen activator (uPA), Plasmin, cathepsin, and others [6, 14]. MMPs are known to play a functional role in the metastatic spread of lung cancer. Different MMPs are active in various steps of the invasive and metastatic process, and a better understanding of the involvement of MMPs in the invasion and regulation of growth of both primary and metastatic tumors may help to implement these as anti-cancer therapy targets. Cathepsin has demonstrated to have a prognostic value in NSCLC and levels of the receptors for uPA and other components of the plasminogen activation system associated with survival in NSCLC.
3.6.1 Targeting Protease and the Metastatic Process
It expected that inhibition of the metastatic potential of the tumor by interaction with extracellular protein degradation could be an essential target, especially during early tumor development. Drug targeting MMPs have been in a clinical trial but have shown little or no activity in lung cancer. Inhibition of MMPs has primarily focused on targetting MMP-2 and -9. At the moment, new MMP inhibitors such as CP471.358 are being evaluated in Phase I and II studies in some malignancies including lung cancer [13, 15].
4 Conclusion
Significant advances in molecular biologic research during recent decades have resulted in a substantial insight into critical signaling pathways and mediators contributing to lung cancer pathogenesis. The identification of driver mutations in EGFR, ALK, and others heralded a new era of targeted therapy in lung adenocarcinoma, and advanced sequencing technologies are providing more sophisticated insights into the molecular alterations underlying lung cancer. These researchers have also identified a range of potentially targetable genetic modifications for lung cancer, have also encountered a troubling complexity and heterogeneity posing significant challenges for diagnosis as well as targeted therapy for lung cancer, a genuinely multi-mechanistic and heterogenous malignant disease.
References
Weinberg, R.A.: Multi-step Tumorigenesis. The biology of cancer, 2nd edn, pp. 31–70. Garland Science (2014)
Hanahan, D., Weinberg, R.A.: The hallmarks of cancer. The next generation. Cell 144(5), 646–674 (2011) (Elsevier. Inc)
Cagle, P., Allen, T., Beasley, M.B., et al.: Molecular pathology of lung cancer. Springer, New York (2012)
Bergethon, K., Shaw, A.T., Ou, S.H., et al.: ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 30, 863–870 (2012)
Thang, H.Q.: Basically molecular in Cancer. J. Med. HCM city 19(5–2015), 1–7 (2015)
Cooper, W.A., Lam, D.C.L., O’Toole, S.A., Minna, J.D.: Molecular biology of lung cancer. J. Thorac. Dis. 5, 1–22 (2013)
Thang, H.Q.: Molecular alterations in lung cancer. J. Med. HCM city 19(5–2015), 8–15 (2015)
Thang, H.Q.: Up dating biology, diagnosis, and treatment of lung cancer. Training documentations of Cantho Cancer Society 2014 (2014)
Larsen, Jill E., Minna, John D.: Molecular biology of lung cancer: clinical implications. Clin. Chest Med. 32(2011), 703–740 (2011)
Tan, W.W.: Nonsmall cell lung cancer treatment and management. Medscape Updated 17 Aug 2017, pp 01–40 (2017)
Zappa, C., Mousa, S.A.: Nonsmall cell lung cancer: current treatment and future advances. Transl. Lung Cancer Res. TLCR 5(3), 288–300 (2016)
Berinstein, N.L.: Biological therapy of cancer. In: Basic Science of Oncology, 4th edn, pp. 453–484. McGraw Hill, New York (2005)
Zarogoudilis, K., Zarogoudilis, P., Darwiche, K., Machairiolis, N., et al.: Treatment of NSCLC. J. Thorac. Dis. 5(4), 389–396 (2013)
Mackinnon, A.C., Kopatz, J., Sethi, T.: The molecular and cellular biology of lung cancer: Identifying novel therapeutic strategies. Br. Med. Bull. 95, 57–61 (2010)
Reck, M., Rabe, K.F.: Precision diagnosis and treatment for advanced NSCLC. Review article New England J. Med. 31 Aug 2017, pp. 849–861 (2017)
Shivapurkar, N., Reddy, J., Chaudhary, P.M.: Gazdar AF (2003) Apoptosis and lung cancer: a review. J. Cell. Biochem. 88, 885–898 (2003)
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this paper

Cite this paper
Huynh, T.Q., Tran, D.N., Chau, T.P., Nguyen, M.T., Doan, N. (2020). Biological Alterations of Lung Carcinoma. In: Van Toi , V., Le, T., Ngo, H., Nguyen, TH. (eds) 7th International Conference on the Development of Biomedical Engineering in Vietnam (BME7). BME 2018. IFMBE Proceedings, vol 69. Springer, Singapore. https://doi.org/10.1007/978-981-13-5859-3_102
Download citation
DOI: https://doi.org/10.1007/978-981-13-5859-3_102
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-5858-6
Online ISBN: 978-981-13-5859-3
eBook Packages: EngineeringEngineering (R0)