
Abstract
The authors review how cancer cells may cooperate in metastasis by means of microenvironmental changes. The main mechanisms underlying this cooperation are clustered migration of cancer cells, extracellular matrix degradation, paracrine loops of released signaling factors and/or induction of adhesion molecules on stromal cells. Another critical factor could be temporal cooperation: successive waves of cancer cells may induce progressive conditioning of the microenvironment. The “class action” of cancer cells against the microenvironment involves successive steps of the metastatic process: invasion of the primary tumor microenvironment, collective migration through the extracellular matrix, blood vessel disruption, vascular or lymphatic tumor emboli, establishment of a premetastatic niche by secreted factors and endothelial precursor recruitment, induction of cell adhesion molecule expression in endothelial cells, extravasation, micrometastasis dormancy and establishment of a new growth in distant sites. As a result, after completion of the metastatic process, the series of microenvironmental changes from the primary tumor to the metastatic site may promote colonization of metastases by nonmetastatic cancer cells of the primary tumor.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The metastatic process is a cellular marathon which combines both random and non-random selections of cancer cells. Random selection roughly corresponds to the mechanistic and “passive” aspects of the metastatic process (access to blood vessels, blood flow pressure, passive trapping of cancer cells in capillaries, etc.) [1], whereas non-random selection is mostly based on the molecular determinants displayed (or not displayed) by cancer cells [2]. These molecular determinants (e.g. E-Cadherin expression, Nm23 suppression, etc.) are required to proceed through the highly selective, and putatively “active” steps of the metastatic process, such as escape from anoikis, homing in a preferential host organ, extravasation and start of a new growth in secondary sites [3, 4]. According to the metastatic switch paradigm, their expression is restricted to a small subpopulation of cells which pre-exists within a parental neoplasm [5]. In this model, the lack of any of the required molecular determinants would prevent tumor cells from developing into metastases [6]. Globally, the success rate of the metastatic process is very low, less than 0.1% for each circulating cancer cell, justifying the term “metastatic inefficiency” [7]. However, cancer cells which fail to metastasize may facilitate the establishment of metastasis by other cells. A community effect (or “class action”) of cancer cells may be responsible for favorable conditioning of the host microenvironment, facilitating the final establishment of metastases.
2 General mechanisms of cancer cell cooperation
The main mechanisms used by cancer cells to cooperate have been largely described, although their cooperative potential has not been elucidated. Direct signaling via adhesion molecules between cancer cells and surrounding non-neoplastic cells have been described [8–9], but most of their interactions are mediated by secreted chemokines, together with other secreted proteins (e.g. proteases) [10]. The action of secreted factors on the microenvironment may also facilitate the survival and progression of other tumor subclones. It has been postulated that two adjacent tumor cells may overcome certain host defences and protect each other by means of diffusible products [11]. Another mechanism is the formation of cancer cell clusters: “autologous” intercellular junctions may cluster heterogeneous subclones in tumor emboli or in invasion through a basement membrane or endothelium [12–13]. The time dimension must also be taken into account: due to the genetic instability of the primary tumor, the tumor microenvironment is exposed to successive tumor subclones that may exhibit different phenotypes [14]. Once a potential metastatic subclone has undergone a metastatic switch, it may take advantage of the prior conditioning of the microenvironment induced by other cancer cells.
This review details the three main steps of the hematogenous metastatic process at which a community effect, or “class action”, can occur: invasion and migration through the extracellular matrix, pre-metastatic niche conditioning, final growth of macrometastasis and the late colonization process of metastases.
3 Invasion and migration through the extracellular matrix
During the metastatic process, invasion of the extracellular matrix (ECM) and migration of cancer cells occur during primary tumor growth and after arrest of cancer cells in the endothelium of the host organ [15]. Invasion and migration properties are closely coordinated, and both require morphologic changes of the cancer cell: formation of pseudopodia at the leading edge, release and activation of extracellular matrix proteases at the invasive front, cell adhesion to proteolysed ECM and cellular movement by detachment at the cell rear [16]. The loss of epithelioid polarization and acquisition of an invasive phenotype are mostly acquired via epithelial–mesenchymal transition (EMT) [17]. However, focused analyses on the invasive front of primary tumors revealed two phenomena which allow cooperation between heterogeneous cancer cells: the ability of cancer cells to migrate depends on ECM stiffness and their ability to degrade ECM components by proteolysis [18–19].
Experimental and theoretical models have shown that the primary invading cancer cells are highly selected in terms of their phenotype and correspond to a few tumor clones exhibiting aggressive traits [20]. Their migration through the ECM is accompanied by the formation of migration tracks signaled by cell membrane material, such as integrins, released by migrating cancer cells during their rear detachment [21–22]. The signaling role of this cellular debris and their ability to slow matrix remodeling have not been clearly evaluated. However, by creating a tunnel of least resistance within the ECM and reshaping the collagen fibers at the border of the tunnel, primary migrating cancer cells may create migrating pathways for other cancer cells [23]. It has also been reported that collective cell movement represents an efficient dissemination strategy. This collective migration of cancer cells exhibits an invasive front composed of clustered promigratory, beta-1 integrin-expressing cancer cells (described as “guiding” cells) and different cellular phenotypes at the rear end of the cell cluster [24–25]. Together with other hypotheses, the collective migration of cancer cells may explain why metastases of epithelial cancers still display epithelial markers and do not exhibit a mesenchymal phenotype [26]: EMT may concern only the first guiding cancer cells.
The late step of the migratory pathway within the microenvironment of the primary tumor is intravasation, i.e release of cancer cells into blood or lymph [27]. Some studies have demonstrated the active involvement of specific molecular determinants, such as adhesion molecules or chemokines [28–30], while others have reported the importance of passive, unregulated mechanisms of cancer cell release into lymph or blood vessels [1]. In clinical studies reporting the existence of circulating cancer cells in disseminated breast cancers, the number of circulating cancer cells appeared to be at least partially linked to disease progression [31] after an initial biological regulation [32]. These clinical observations are not in favor of a tight regulation of the intravasation process throughout tumor growth. To explain how circulating cancer cells may be a “biological staging beyond tumor burden,” we propose that early intravasating cancer cells require specific molecular determinants, and that subsequent cancer cells may take advantage of an altered endothelium to passively extravasate.
4 Premetastatic niche conditioning
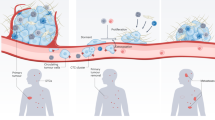
Circulating cancer cells are released into the blood by nonmetastatic primary tumors, as documented by many clinical studies [33]. Many biological studies, including those using in vivo videomicroscopy, have shown that the vast majority of these circulating cells cannot form metastases or micrometastases [34–36]. This has been described by the well known term “metastatic inefficiency” [37]. As these cells do not directly form macrometastases, no study has specifically reported the microenvironmental changes induced by these “inefficient” cancer cells. Concerning modification of the extracellular matrix by early migrating cancer cells, we can postulate that the host-organ microenvironment may be conditioned by certain circulating cells to promote the establishment of metastasis by other cancer cells (Fig. 1).

Premetastatic niche conditioning. Possible cooperations between successive waves of homogeneous or heterogeneous circulating cancer cells
A simple experiment in a human colorectal model of metastasis reported that E Selectin expression by endothelial cells mediated the arrest of cancer cells in the liver [38]. After injection of cancer cells into the portal vasculature, E Selectin was strongly upregulated in the liver, thereby facilitating the arrest of further incoming cancer cells [39]. More recently, E Selectin expression by sinusoidal endothelial cells was shown to be only part of the proinflammatory response of the host-organ microenvironment to arrested cancer cells: release of TNF-alpha by Kupffer cells, and P-Selectin, VCAM-1, and ICAM-1 expression by sinusoidal endothelial cells [40–41]. This process is one of the first steps leading to the creation of a favorable metastatic niche. Other alterations of the endothelial microenvironment can also upregulate the metastatic process: expression of integrin adhesion molecules in cancer cells and the endothelium, matrix metalloproteinases, and chemotactic factors that promote the attachment of tumor cells to the vessel wall and/or transvascular penetration [42–43]. Not surprisingly, together with intravascular tumor emboli of the primary tumor [44], prometastatic intravascular “homotypic” adhesive interactions between circulating cancer cells have also been reported at the site of primary attachment to the endothelium [12, 45]. These two kinds of cellular cluster may also promote cooperation against the host-organ microenvironment.
In addition to activation of the endothelium and clustering of cancer cells, a primary tumor may also trigger the recruitment of bone-marrow derived cells at future metastatic sites. It has been reported that the secretion of inflammatory chemokines, induced by the primary tumor, attracts both cancer cells and MAC1+ myeloid cells in the premetastatic lung [46]. Moreover, VEGFR1+/VLA-4+ bone marrow-derived hematopoietic progenitor cells may form a premetastatic niche in future host organs, and their recruitment is mediated by signaling factors secreted by cancer cells [47–48]. However, it has not been reported whether or not proliferation at the metastatic site is restricted to the cancer cells which were initially responsible for the recruitment of metastasis-facilitating bone marrow cells. Importantly, in the reported experiments, the metastatic pattern (i.e. preferential homing of metastasizing cells) of injected tumor cells depended on the conditioned microenvironment, but not on their own intrinsic metastatic pattern. In the absence of supplementary experiments, it can be hypothesized that chemokine-secreting subclones of the primary tumor are responsible for initiation of the premetastatic niche, but that the resulting conditioned microenvironment may also be a niche for other tumor subclones.
5 Final growth and colonization of macrometastases
The early growth and regulation of micrometastatic cancer cells within a host organ remain unclear. Many studies have reported that bone marrow micrometastases (BM MM) are a strong prognostic factor for metastatic relapse of early breast cancers [49–50], in accordance with our results [51]. After successful dissemination, isolated cancer cells appear to undergo a dormancy phase which could last several years, before some of them grow into macrometastases [52]. Strikingly, BM MM have almost completed the metastatic process but still remain genetically and phenotypically heterogeneous [53–55]. In the breast cancer adjuvant setting, 40 months after completion of treatment, the detection of BM MM and circulating cancer cells were not correlated in patients, and only BM MM had a significant impact on survival. Although circulating cancer cells had no prognostic significance in the overall population, their detection resulted in an especially poor prognosis for the few patients who also exhibited BM MM [56]. It can be hypothesized that circulating cancer cells might form macrometastases when the local microenvironment has been favorably conditioned by other cancer cells (namely BM MM), but this hypothesis needs to be further investigated.
The late growth of metastases, after the start of secondary proliferation by metastasizing cancer cells, has been studied in our laboratory. Although the underlying molecular determinants have not been determined, we demonstrated colonization of metastases by nonmetastatic circulating cancer cells [57]. These types of tumor subpopulation interactions in metastasis were also indirectly reported in a murine model [58]. We concluded that the late part of the metastatic process creates a favorable microenvironment for the arrest and growth of other tumor subclones. This cooperative process could also explain why primary tumors and macrometastases may exhibit a similar molecular profile after clonal initiation of metastases [59–60].
6 Conclusion
We have reviewed the main steps of the metastatic process in which cooperation of cancer cells progressively creates a conditioned microenvironment, and its potential mechanisms. The cooperation between cancer cells may have been underestimated by the use of highly selected cell lines injected intravenously to mice. It is almost impossible at the present time, for technical reasons and due to genetic instability, to distinguish all of the genetically and phenotypically different subclones in a primary tumor and to follow them in the course of the metastatic process. However, this class action type of process might also exist in many other hallmarks of cancer, such as angiogenesis or immunity escape. If confirmed by further experiments, this cooperation may change our understanding of the metastatic process.
References
Bockhorn, M., Jain, R. K., & Munn, L. L. (2007). Active versus passive mechanisms in metastasis: Do cancer cells crawl into vessels, or are they pushed? Lancet Oncology, 8, 444–448.
Hoon, D. S., Kitago, M., Kim, J., Mori, T., Piris, A., Szyfelbein, K., et al. (2006). Molecular mechanisms of metastasis. Cancer and Metastasis Reviews, 25, 203–220.
Chambers, A. F., Groom, A. C., & MacDonald, I. C. (2002). Dissemination and growth of cancer cells in metastatic sites. Nature Reviews. Cancer, 2, 563–572.
Steeg, P. S. (2006). Tumor metastasis: Mechanistic insights and clinical challenges. Nature Medicine, 12, 895–904.
Fidler, I. J. (2002). Critical determinants of metastasis. Seminars in Cancer Biology, 12, 89–96.
Fidler, I. J., & Kripke, M. L. (2003). Genomic analysis of primary tumors does not address the prevalence of metastatic cells in the population. Nature Genetics, 34, 23.
Weiss, L. (2000). Metastasis of cancer: A conceptual history from antiquity to the 1990s. Cancer and Metastasis Reviews, 19, 193–383.
Kapoor, P., Saunders, M. M., Li, Z., Zhou, Z., Sheaffer, N., Kunze, E. L., et al. (2004). Breast cancer metastatic potential: Correlation with increased heterotypic gap junctional intercellular communication between breast cancer cells and osteoblastic cells. International Journal of Cancer, 111, 693–697.
el-Sabban, M. E., & Pauli, B. U. (1994–1995). Adhesion-mediated gap junctional communication between lung-metastatic cancer cells and endothelium. Invasion Metastasis, 14, 164–176.
Opdenakker, G., & Van Damme, J. (2004). The countercurrent principle in invasion and metastasis of cancer cells. Recent insights on the roles of chemokines. International Journal of Developmental Biology, 48, 519–527.
Axelrod, R., Axelrod, D. E., & Pienta, K. J. (2006). Evolution of cooperation among tumor cells. Proceedings of the National Academy of Sciences of the United States of America, 103, 13474–13479.
Glinsky, V. V. (2006). Intravascular cell-to-cell adhesive interactions and bone metastasis. Cancer and Metastasis Reviews, 25, 531–540.
Kerbel, R. S. (1994–1995). Impact of multicellular resistance on the survival of solid tumors, including micrometastases. Invasion Metastasis, 14, 50–60.
Nicolson, G. L. (1984). Generation of phenotypic diversity and progression in metastatic tumor cells. Cancer and Metastasis Reviews, 3, 25–42.
Wittekind, C., & Neid, M. (2005). Cancer invasion and metastasis. Oncology, 69, 14–16.
Yamaguchi, H., Wyckoff, J., & Condeelis, J. (2005). Cell migration in tumors. Current Opinion in Cell Biology, 17, 559–564.
Thiery, J. P., & Sleeman, J. P. (2006). Complex networks orchestrate epithelial–mesenchymal transitions. Nature Reviews. Molecular Cell Biology, 7, 131–142.
Zaman, M. H., Trapani, L. M., Sieminski, A. L., Mackellar, D., Gong, H., Kamm, R. D., et al. (2006). Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell–matrix adhesion and proteolysis. Proceedings of the National Academy of Sciences of the United States of America, 103, 10889–10894.
Jodele, S., Blavier, L., Yoon, J. M., & DeClerck, Y. A. (2006). Modifying the soil to affect the seed: Role of stromal-derived matrix metalloproteinases in cancer progression. Cancer and Metastasis Reviews, 25, 35–43.
Anderson, A. R., Weaver, A. M., Cummings, P. T., & Quaranta, V. (2006). Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell, 27, 905–915.
Palecek, S. P., Huttenlocher, A., Horwitz, A. F., & Lauffenburger, D. A. (1998). Physical and biochemical regulation of integrin release during rear detachment of migrating cells. Journal of Cell Science, 111, 929–940.
Kirfel, G., Rigort, A., Borm, B., & Herzog, V. (2004). Cell migration: mechanisms of rear detachment and the formation of migration tracks. European Journal of Cell Biology, 83, 717–724.
Friedl, P., Maaser, K., Klein, C. E., Niggemann, B., Krohne, G., & Zänker, K. S. (1997). Migration of highly aggressive MV3 melanoma cells in 3-dimensional collagen lattices results in local matrix reorganization and shedding of alpha2 and beta1 integrins and CD44. Cancer Research, 57, 2061–2070.
Hegerfeldt, Y., Tusch, M., Bröcker, E. B., & Friedl, P. (2002). Collective cell movement in primary melanoma explants: plasticity of cell–cell interaction, beta1-integrin function, and migration strategies. Cancer Research, 62, 2125–2130.
Brakebusch, C., & Fässler, R. (2005). Beta 1 integrin function in vivo: adhesion, migration and more. Cancer and Metastasis Reviews, 24, 403–411.
Christiansen, J. J., & Rajasekaran, A. K. (2006). Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Research, 66, 8319–8326.
Woodhouse, E. C., Chuaqui, R. F., & Liotta, L. A. (1997). General mechanisms of metastasis. Cancer, 80, 1529–1537.
Gupta, A., Deshpande, C. G., & Badve, S. (2003). Role of E-cadherins in development of lymphatic tumor emboli. Cancer, 97, 2341–2347.
Galaup, A., Cazes, A., Le Jan, S., Philippe, J., Connault, E., Le Coz, E., et al. (2006). Angiopoietin-like 4 prevents metastasis through inhibition of vascular permeability and tumor cell motility and invasiveness. Proceedings of the National Academy of Sciences of the United States of America, 103, 18721–18726.
Siclari, V. A., Guise, T. A., & Chirgwin, J. M. (2006). Molecular interactions between breast cancer cells and the bone microenvironment drive skeletal metastases. Cancer and Metastasis Reviews, 25, 621–633.
Cristofanilli, M., Budd, G. T., Ellis, M. J., Stopeck, A., Matera, J., Miller, M. C., et al. (2004). Circulating tumor cells, disease progression, and survival in metastatic breast cancer. New England Journal of Medicine, 351, 781–791.
Cristofanilli, M., Broglio, K. R., Guarneri, V., Jackson, S., Fritsche, H. A., Islam, R., et al. (2007). Circulating tumor cells in metastatic breast cancer: Biologic staging beyond tumor burden. Clinical Breast Cancer, 7, 471–479.
Wiedswang, G., & Naume, B. (2007). Can detection of circulating tumor cells in peripheral blood provide prognostic data in breast cancer? Nature Clinical Practice. Oncology, 4, 154–155.
Luzzi, K. J., MacDonald, I. C., Schmidt, E. E., Kerkvliet, N., Morris, V. L., Chambers, A. F., et al. (1998). Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. American Journal of Pathology, 153, 865–873.
Cameron, M. D., Schmidt, E. E., Kerkvliet, N., Nadkarni, K. V., Morris, V. L., Groom, A. C., et al. (2000). Temporal progression of metastasis in lung: Cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Research, 60, 2541–2546.
Ito, S., Nakanishi, H., Ikehara, Y., Kato, T., Kasai, Y., Ito, K., et al. (2001). Real-time observation of micrometastasis formation in the living mouse liver using a green fluorescent protein gene-tagged rat tongue carcinoma cell line. International Journal of Cancer, 93, 212–217.
Weiss, L. (1996). Metastatic inefficiency: Intravascular and intraperitoneal implantation of cancer cells. Cancer Treatment and Research, 82, 1–11.
Khatib, A. M., Fallavollita, L., Wancewicz, E. V., Monia, B. P., & Brodt, P. (2002). Inhibition of hepatic endothelial E-selectin expression by C-raf antisense oligonucleotides blocks colorectal carcinoma liver metastasis. Cancer Research, 62, 5393–5398.
Khatib, A. M., Kontogiannea, M., Fallavollita, L., Jamison, B., Meterissian, S., & Brodt, P. (1999). Rapid induction of cytokine and E-selectin expression in the liver in response to metastatic tumor cells. Cancer Research, 59, 1356–1361.
Khatib, A. M., Auguste, P., Fallavollita, L., Wang, N., Samani, A., Kontogiannea, M., et al. (2005). Characterization of the host proinflammatory response to tumor cells during the initial stages of liver metastasis. American Journal of Pathology, 167, 749–759.
Auguste, P., Fallavollita, L., Wang, N., Burnier, J., Bikfalvi, A., & Brodt, P. (2007). The host inflammatory response promotes liver metastasis by increasing tumor cell arrest and extravasation. American Journal of Pathology, 170, 1781–1792.
Lafrenie, R., Shaughnessy, S. G., & Orr, F. W. (1992). Cancer cell interactions with injured or activated endothelium. Cancer and Metastasis Reviews, 11, 377–388.
Orr, F. W., Wang, H. H., Lafrenie, R. M., Scherbarth, S., & Nance, D. M. (2000). Interactions between cancer cells and the endothelium in metastasis. Journal of Pathology, 190, 310–329.
Ruiter, D. J., van Krieken, J. H., van Muijen, G. N., & de Waal, R. M. (2001). Tumour metastasis: Is tissue an issue? Lancet Oncology, 2, 109–112.
Glinsky, V. V., Glinsky, G. V., Glinskii, O. V., Huxley, V. H., Turk, J. R., Mossine, V. V., et al. (2003). Intravascular metastatic cancer cell homotypic aggregation at the sites of primary attachment to the endothelium. Cancer Research, 63, 3805–3811.
Hiratsuka, S., Watanabe, A., Aburatani, H., & Maru, Y. (2006). Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nature Cell Biology, 8, 1369–1375.
Kaplan, R. N., Riba, R. D., Zacharoulis, S., Bramley, A. H., Vincent, L., Costa, C., et al. (2005). VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature, 438, 820–827.
Kaplan, R. N., Psaila, B., & Lyden, D. (2006). Bone marrow cells in the ‘pre-metastatic niche’: Within bone and beyond. Cancer and Metastasis Reviews, 25, 521–529.
Braun, S., Vogl, F. D., Naume, B., Janni, W., Osborne, M. P., Coombes, R. C., et al. (2005). A pooled analysis of bone marrow micrometastasis in breast cancer. New England Journal of Medicine, 353, 793–802.
Wiedswang, G., Borgen, E., Kåresen, R., Kvalheim, G., Nesland, J. M., Qvist, H., et al. (2003). Detection of isolated tumor cells in bone marrow is an independent prognostic factor in breast cancer. Journal of Clinical Oncology, 21, 3469–3478.
Bidard, F. C., Vincent-Salomon, A., Gomme, S., Thiery, J. P., Sigal-Zafrani, B., De Rycke, Y., et al. (2007). Bone marrow micrometastasis are a powerful prognostic factor in women with stage I to III breast cancer. (Abstract). AACR annual meeting.
Wiedswang, G., Borgen, E., Karesen, R., Qvist, H., Janbu, J., Kvalheim, G., et al. (2004). Isolated tumor cells in bone marrow three years after diagnosis in disease-free breast cancer patients predict unfavorable clinical outcome. Clinical Cancer Research, 10, 5342–5348.
Gangnus, R., Langer, S., Breit, E., Pantel, K., & Speicher, M. R. (2004). Genomic profiling of viable and proliferative micrometastatic cells from early-stage breast cancer patients. Clinical Cancer Research, 10, 3457–3464.
Braun, S., Hepp, F., Sommer, H. L., & Pantel, K. (1999). Tumor-antigen heterogeneity of disseminated breast cancer cells: Implications for immunotherapy of minimal residual disease. International Journal of Cancer, 84, 1–5.
Schmidt-Kittler, O., Ragg, T., Daskalakis, A., Granzow, M., Ahr, A., Blankenstein, T. J., et al. (2003). From latent disseminated cells to overt metastasis: Genetic analysis of systemic breast cancer progression. Proceedings of the National Academy of Sciences of the United States of America, 100, 7737–7742.
Wiedswang, G., Borgen, E., Schirmer, C., Kåresen, R., Kvalheim, G., Nesland, J. M., et al. (2006). Comparison of the clinical significance of occult tumor cells in blood and bone marrow in breast cancer. International Journal of Cancer, 118, 2013–1019.
Bidard, F. C., Auger, N., Rosty, C., Assayag, F., Di Santo, J. P., & Poupon, M. F. (2007). Specific colonization of metastases by non metastasizing circulating tumor cells. (Abstract). AACR annual meeting.
Miller, F. R. (1983). Tumor subpopulation interactions in metastasis. Invasion Metastasis, 3, 234–242.
Bernards, R., & Weinberg, R. A. (2002). A progression puzzle. Nature, 418, 823.
Inamura, K., Shimoji, T., Ninomiya, H., Hiramatsu, M., Okui, M., Satoh, Y., et al. (2007). A metastatic signature in entire lung adenocarcinomas irrespective of morphological heterogeneity. Human Pathology, 38, 702–709.
Acknowledgments
This work was funded by the Institut Curie and Inserm.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bidard, FC., Pierga, JY., Vincent-Salomon, A. et al. A “class action” against the microenvironment: do cancer cells cooperate in metastasis?. Cancer Metastasis Rev 27, 5–10 (2008). https://doi.org/10.1007/s10555-007-9103-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-007-9103-x