
Abstract
Paclitaxel is one of the most frequently used chemotherapeutic agents for the treatment of breast cancer patients. Using a candidate gene approach, we hypothesized that polymorphisms in genes relevant to the metabolism and transport of paclitaxel are associated with treatment efficacy and toxicity. Patient and tumor characteristics and treatment outcomes were collected prospectively for breast cancer patients treated with paclitaxel-containing regimens in the neoadjuvant setting. Treatment response was measured before and after each phase of treatment by clinical tumor measurement and categorized according to RECIST criteria, while toxicity data were collected from physician notes. The primary endpoint was achievement of clinical complete response (cCR) and secondary endpoints included clinical response rate (complete response + partial response) and grade 3+ peripheral neuropathy. The genotypes and haplotypes assessed were CYP1B1*3, CYP2C8*3, CYP3A4*1B/CYP3A5*3C, and ABCB1*2. A total of 111 patients were included in this study. Overall, cCR was 30.1 % to the paclitaxel component. CYP2C8*3 carriers (23/111, 20.7 %) had higher rates of cCR (55 % vs. 23 %; OR = 3.92 [95 % CI: 1.46–10.48], corrected p = 0.046). In the secondary toxicity analysis, we observed a trend toward greater risk of severe neuropathy (22 % vs. 8 %; OR = 3.13 [95 % CI: 0.89–11.01], uncorrected p = 0.075) in subjects carrying the CYP2C8*3 variant. Other polymorphisms interrogated were not significantly associated with response or toxicity. Patients carrying CYP2C8*3 are more likely to achieve clinical complete response from neoadjuvant paclitaxel treatment, but may also be at increased risk of experiencing severe peripheral neurotoxicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Interpatient variability in toxicity and response are important problems in the use of cancer chemotherapy. For example, paclitaxel, one of the most commonly used therapies for breast cancer and other cancers, has interpatient variability of 19–26 % in (unbound) drug clearance [1], causes grade 3 or higher neuropathy and neutropenia in 5–8 % and 2–4 % of patients, respectively [2] and a response rate as first line, single-agent treatment in metastatic breast cancer of 20–30 % [3]. Studying host factors responsible for the variability in chemotherapeutic outcomes and developing strategies to individualize therapy to maximize response and minimize toxicity are active areas of research. Pharmacogenomics, study of the interplay of genetics and drug therapy outcomes, is one promising approach to achieving individualized therapy [4]. Genetic variation can influence therapy by a number of mechanisms. Variants in genes relevant to drug disposition or metabolism can modulate the patient’s exposure to the drug, whereas variation in genes that are involved in drug action can influence the patient’s sensitivity. For example, germline genetic polymorphisms have been discovered that increase the likelihood of a patient experiencing severe toxicity to irinotecan [5, 6] or modulate the optimal dose of a patient’s warfarin therapy [7].
A number of putative pharmacogenetic markers for paclitaxel outcomes, in breast cancer and in other solid tumors, have been evaluated [8–17]. Most of these studies focused on known mutations in biologically relevant candidate genes, such as CYP2C8, CYP3A4, and ABCB1, which code for the enzymes involved in paclitaxel metabolism and the transporters that influence paclitaxel disposition. More recent genome-wide association studies used an unbiased approach to examine the entire genome to address this question, and have reported intriguing candidate SNPs in genes not previously investigated [14, 15]. The clinical phenotypes most often studied were severe toxicities, such as neuropathy, or measures of survival. Some of these retrospective pharmacogenetic studies suggest that genetic variability may be associated with clinical outcome to paclitaxel therapy, and others do not. However, the toxicity endpoints are often confounded by prior or combination therapy, whereas the survival endpoint is confounded by a multitude of factors including ER status or tumor subtype [18] and stage at diagnosis [19]. To our knowledge, no published pharmacogenomic study has exclusively utilized a neoadjuvantly treated population, in which toxicity and tumor response to paclitaxel therapy can be assessed in the absence of these confounding factors.
In this study, we genotyped a cohort of patients treated with neoadjuvant paclitaxel for polymorphisms that have previously demonstrated significant associations with efficacy or toxicity. These subjects are uniquely informative because tumor response and toxicity data were collected exclusively for the taxane treatment phase. We hypothesized that polymorphisms in genes relevant to paclitaxel metabolism (CYP2C8 & CYP3A4/3A5), transport (ABCB1), or mechanism (CYP1B1) would influence the likelihood that a patient would respond to, or experience severe toxicity from, paclitaxel therapy.
Materials and methods
Patients and treatments
Relevant candidate SNPs were evaluated in a cohort of patients treated between 2005 and 2009 and derived from the University of North Carolina Lineberger Comprehensive Cancer Center (UNC LCCC) Breast Cancer Neoadjuvant Database, which includes prospective annotation of clinical data, treatment details, toxicity, and outcome. Eligible women received neoadjuvant paclitaxel-containing regimens and enrolled in both the UNC neoadjuvant database and a concurrent IRB approved clinical trial that collected genomic DNA from all newly diagnosed patients. All patients received paclitaxel (T) treatment guided by standard neoadjuvant protocols that had a defined and conventional treatment dose, schedule, and duration. In most cases, patients received neoadjuvant doxorubicin and cyclophosphamide (AC) either before or after the paclitaxel; however, the tumors were measured before and after each phase of therapy so that clinical response to the anthracycline component and the taxane component could be separately identified. Some patients with HER2 overexpressing tumors received trastuzumab concurrent with the paclitaxel. Tumor size was clinically measured by the patient’s medical oncologist, and percent change in tumor size was calculated from these measurements. Clinical response was defined as complete response (cCR; 100 % reduction in tumor size), partial response (cPR; 30–99 % reduction), stable disease (cSD; 29 % reduction–20 % enlargement of tumor), or progressive disease (cPD; >20 % enlargement) according to RECIST criteria [20]. Patients who achieved complete response to AC before the start of taxane therapy were excluded from the efficacy analysis because they were not evaluable for response to taxane treatment. Pathological response, which could not be evaluated between treatment regimens, was not used as an endpoint in this study because of the inability to separate the confounding effects of other chemotherapy treatments. Toxicities were evaluated during paclitaxel treatment, recorded prospectively, and coded by NCI CTC AE V4.0 based on the physician’s description [21]. Patients who were treated at outside institutions did not have toxicity data and were excluded from that part of the analysis. All patients signed informed consent to participate and agreed to allow DNA to be collected for additional pharmacogenetic studies. The study protocol was approved by the UNC Institutional Review Board.
SNP genotyping
A 30 mL blood sample was collected from each patient at the time of study enrollment. DNA used for genotyping was extracted by the UNC Biospecimen Processing Facility and plated at 5 ng/uL. Target gene region amplification was carried out by PCR in a 20 μL reaction including 2 μL genomic DNA and polymorphisms were genotyped on a Pyromark Pyrosequencer Q96 MD as previously described [22, 23]. The polymorphisms genotyped were as follows: CYP1B1*3 (rs1056836, 4326C>G), CYP2C8*3 (rs11572080, 416G>A and rs10509681, 1196A>G), CYP3A4*1B (rs2740574, -392A>G), CYP3A5*3C (rs776746, 6986A>G), and ABCB1*2 (rs1045642, 3435C>T, rs2032582, 2677G>T/A, and rs1128503, 1236C>T). Genotyping was carried out blinded to clinical data with negative controls included in each, run and at least 5 % of samples were repeated for quality control to ensure accuracy of assay results. Any assay with call rate or concordance with repeated samples <95 % was excluded from analysis. Additional details for gene variants including PCR and pyrosequencing primers are included in Supplementary Table 1.
CYP3A4*1B/CYP3A5*3C and ABCB1 1236C > T, 2677G > T/A, and 3435C > T were included in haplotype analyses. Haplotypes were inferred using PHASE Version 2 [24, 25] for polymorphisms with LD > 0.7. Any subject with missing genotype information at any locus of the haplotype was considered to have an unknown haplotype and excluded from analysis. CYP3A4/3A5 haplotypes were grouped according to Baker et al. (*1: CYP3A4*1A/CYP3A5*3C, *2: CYP3A4*1B/CYP3A5*1A, *3: CYP3A4*1A/CYP3A5*1A, and *4: CYP3A4*1B/CYP3A5*3C) [26]. After ABCB1 haplotype inference (wild-type: C-G-C, Variant: T–T(A)-T, mixed: other), each patient was assigned a diplotype number (1–5) in order of increasing genetic variation as described in Sissung et al. [27].
Statistical analysis
Genotype calls were assessed for concordance with Hardy–Weinberg Equilibrium (HWE) using a Pearson’s chi-square test with df = 1. Assays with HWE p value < 0.05 in the cohort were then tested in the Caucasian subcohort as population admixture violates key assumptions of HWE [28]. Each genotype or haplotype was individually tested for an association with efficacy or toxicity using logistic regression modeling. In the haplotype analysis, the “variant” group was defined by grouping diplotypes. For CYP3A4/3A5, any individual carrying the *2 haplotype (CYP3A4*1B/CYP3A5*1A) was considered a variant carrier. For ABCB1, any individual with diplotype 4 or 5 was considered a variant carrier and was compared with non-carrier diplotypes 1–3. For the genotype analyses, variant carriers were compared with homozygotic wild-type individuals (dominant genetic model). The primary efficacy endpoint was clinical complete response (cCR) to taxane therapy (yes vs. no). The primary toxicity endpoint was any grade 3 or higher adverse event during taxane therapy (yes vs. no). Secondary endpoints of efficacy and toxicity were clinical response rate (cRR; cCR + cPR = cRR) and grade 3 or higher neuropathy during paclitaxel therapy, respectively. Following univariate testing, additional covariates for efficacy (estrogen receptor (ER) status, tumor grade, concurrent trastuzumab treatment, whether paclitaxel treatment was preceded by other chemotherapy phase) were included in a multivariable model to adjust for their prognostic importance. Backward selection was used to eliminate covariates that did not significantly contribute to the model using AIC as a selection criterion. Self-reported race was used as stratification factor for significant associations to account for racial heterogeneity in the cohort. To correct the primary efficacy analysis for multiple comparisons, the p values were multiplied by 7, the number of independent statistical associations performed (a Bonferroni correction for multiple comparisons) so that the p value to be compared with the standard significance threshold of α = 0.05 is valid. P values of all secondary and sub-analyses are uncorrected as these are exploratory in nature and should be interpreted as such. All statistical analyses were performed in R Statistical Software, version 2.13.0 (R Development Core Team, Vienna, Austria).
Results
Patient population
A total of 111 patients neoadjuvantly treated with paclitaxel-containing regimens were eligible for analysis. After excluding subjects missing efficacy or toxicity data, 103 subjects were included in the efficacy and 109 subjects in the toxicity analysis. Demographic data including patient, treatment, and tumor characteristics for the whole population are presented in Table 1.
Allele frequencies
The two highly linked SNPs in CYP2C8*3 (rs11572080, 416G>A and rs10509681, 1196A>G) were completely concordant in this population. CYP3A4 and CYP3A5 were out of HWE before accounting for race, as expected given the large difference in allele frequencies among Caucasian and African-American individuals [29], but no significant deviations were seen in stratified samples. Allele frequencies in Caucasian subjects for all variants were consistent with those reported in The International HapMap Project [30] or the NCBI EntrezSNP database [31] and replicated samples were 100 % concordant with the original genotype calls; thus, no assays were excluded from analysis (Supplementary Table 2). As expected, significant LD was seen between CYP3A4*1B and CYP3A5*3C (r = 0.93) and the three polymorphisms in ABCB1 (r > 0.7), which were then grouped into haplotypes as planned (Supplementary Table 3).
Response by genotype
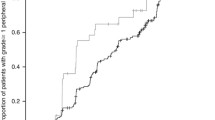
Clinical complete response to paclitaxel for the efficacy cohort was 30.1 % and the mean change in tumor size was a 49 % reduction (Table 2). Response by genotype is presented in Table 3, demonstrating significance only for CYP2C8; the odds ratio for an individual carrying CYP2C8*3 to achieve clinical complete response was 3.92 with a 95 % confidence interval of 1.46–10.48 (corrected p = 0.046) (Fig. 1). Of the 22 subjects carrying the CYP2C8*3 variant, 12 achieved clinical complete response (55 %) as compared with only 19 out of the 81 wild-type subjects (23 %). To ascertain whether this association was independent of other prognostic factors, a multivariable model that included tumor grade, ER status, concomitant trastuzumab, and whether paclitaxel was preceded by another phase of chemotherapy was tested. After backward elimination of covariates that were not significant, the final model included tumor grade and whether paclitaxel was preceded by another phase of chemotherapy. After controlling for these prognostic factors, the association of CYP2C8*3 status remained significant in the final model (uncorrected p = 0.003, corrected p = 0.022), while the other covariates were not significantly associated with achievement of clinical complete response (Table 4). Next, the association of CYP2C8*3 and clinical complete response was stratified by race to ensure that racial heterogeneity was not falsely inflating our results. In the self-reported Caucasian subjects, the magnitude of effect was marginally greater and the significance similar to that seen in the entire efficacy cohort (OR = 5.31, 95 % CI: 1.59–17.67, corrected p = 0.049). Only two non-Caucasians carried the *3 variant in our cohort so the association in non-Caucasians was not analyzed.

Percentage of patients carrying CYP2C8*3 vs. CYP2C8*1 wild-type homozygotes achieving clinical complete response (a: left) or experiencing severe peripheral neuropathy (b: right). Patients carrying CYP2C8*3 were more likely to achieve clinical complete response (OR = 3.92, 95 % CI: 1.46–10.48, corrected p = 0.046). There was a trend toward greater risk of severe neuropathy in patients carrying the *3 variant, though it did not achieve statistical significance (OR = 3.13, 95 % CI: 0.89–11.01, uncorrected p = 0.075)
To evaluate the robustness of our finding, a secondary efficacy analysis was carried out with clinical response rate (cRR = cCR + cPR). The clinical response rate in the cohort was 63 % (65/103). In the CYP2C8*3 carriers, the response rate was 82 % (18/22) versus 58 % (47/81) in the CYP2C8 wild-type subjects. In the univariate logistic regression model, this association showed a strong trend in the same direction with an odds ratio of 3.16 (95 % CI: 0.98-10.19, uncorrected p = 0.054), supporting our primary findings.
To examine whether, as hypothesized, this finding was specific to paclitaxel, we then tested the association between CYP2C8*3 status and clinical complete response to the doxorubicin–cyclophosphamide (AC) phase of therapy in patients who received the combination. Out of 100 subjects who received the AC combination who had evaluable response, the rate of clinical complete response was not significantly different between *3 carriers (2/22 = 9.0 %) and wild-type homozygotes (8/78 = 10.3 %) (OR = 1.14, 95 % CI: 0.62–15.96, uncorrected p = 0.872).
Toxicity by genotype
Of the 109 subjects included in the toxicity analysis, 34 experienced at least one grade 3 or higher toxicity (31.2 %) (Table 5); however, none of the genetic markers were associated with this cumulative endpoint (data not shown). Analysis of the secondary toxicity endpoint, grade 3 or higher peripheral neuropathy, revealed a trend toward increased neuropathy in subjects carrying the CYP2C8*3 variant (5/23 = 22 %) versus wild-type individuals (7/86 = 8 %) (OR = 3.13, 95 % CI: 0.89–11.01, p = 0.075) (Fig. 1, Supplementary Table 4).
Discussion
This study investigated pharmacogenetic predictors of breast cancer treatment outcomes following neoadjuvant paclitaxel. By measuring the tumor before and after each phase of sequential chemotherapy, and collecting toxicity during each phase separately, we were able to isolate the taxane-specific outcomes from the sequential therapy. We employed a candidate polymorphism replicate strategy based on reported associations with clinical outcomes in previous pharmacogenetic studies in taxane-treated cancer patients. These candidate genes covered the major metabolic pathways of paclitaxel, CYP3A4/3A5 and CYP2C8, the efflux transporter ABCB1, and CYP1B1 that has been shown to influence taxane treatment efficacy.
Our results indicate that patients carrying the CYP2C8*3 polymorphism are more likely to achieve clinical complete response than patients homozygous for the wild-type isozyme. This finding is supported by the strong trend in the same direction for clinical response rate. The association with tumor response remained significant after adjustment for covariates and stratification by self-reported race. There was no association between CYP2C8 genotype and clinical complete response to the AC phase of sequential therapy, dismissing the possibility that patients carrying CYP2C8*3 had more chemosensitive tumors. Thus, there seems to be a true pharmacogenetic association between the CYP2C8*3 polymorphism and clinical response to neoadjuvant paclitaxel therapy.
Clinical response, instead of the more accepted pathological response, which has prognostic implications for future survival [32–34], was selected because of the collection of tumor size data between phases of sequential therapy, which enables us to isolate the response to the paclitaxel phase of treatment from that of the other administered therapy. Although clinical measurement is not a component of RECIST classification, that methodology was designed with radiographic measurements in the metastatic setting in mind. Conversely, pathologic response, a more conventional endpoint for neoadjuvant studies, cannot differentiate among drugs given preoperatively so would have introduced considerable noise from the inclusion of anthracycline and antimetabolite effect in the efficacy estimates. Furthermore, the ability to use response to the AC component of therapy as an internal control for the specificity of the findings for paclitaxel would have been lost. There is a documented relationship between clinical measurement and pathologic response [35] supporting the use of easily obtained serial clinical measurements in the palpable lesions relevant in this setting. The reported relationship between clinical and pathological response indicates that our finding may have an important influence on survival; however, it is essential that these findings are confirmed with pathological or radiographic tumor measurements before and after taxane therapy in independent patient cohorts.
Two previous groups have reported that patients carrying CYP2C8*3 are at a higher risk of paclitaxel-induced peripheral neuropathy [10, 11], but this finding was not observed in other studies [16, 36, 37]. All of these studies primarily included patients who were on combination therapy or who had been previously treated with chemotherapy. Our results, in previously untreated patients not receiving combination therapy with other neurotoxins, are consistent with those of Leskela et al. [11] and Green et al. [10] and suggest that there may be a true association between CYP2C8*3 and risk of peripheral neuropathy. The difference between our study and that of Leskela et al. [11] is the event rate. Their study included grade 2+ neurotoxicity, a substantially more common phenotype than our grade 3+ endpoint, but one that does not require a change in therapy, unlike higher grades of neuropathy as were measured in this study. Additionally, they used a cumulative dose-to-event analysis, which is consistent with the cumulative nature of neurotoxicity. This was not feasible in our study given the relatively small number of patients (12) who experienced grade 3 or higher neuropathy. In fact, it is important to note that a general limitation of the current study is the modest sample size.
The paclitaxel parent compound, not its metabolites, is thought to be responsible for the drug’s efficacy and toxicity [38]. Paclitaxel clinical outcomes are related to the amount of time the total drug concentration remains above a threshold level [39, 40] and the cumulative exposure may determine the extent of neuropathy development [10, 41]. The CYP2C8*3 variant has diminished in vitro metabolic activity for paclitaxel [42–45] and carriers of this variant have decreased clearance of free paclitaxel, and a corresponding increase in drug exposure [46]. These findings provide a rational mechanism for the increased paclitaxel treatment response and toxicity seen in CYP2C8*3 carriers in this study.
It is not possible to distinguish the influence of each of the two non-synonymous polymorphisms in CYP2C8*3 in this population because of the complete concordance, and it will be difficult to do this in any clinical study based on their high linkage disequilibrium. However, in vitro data suggest that the causative SNP is the K399R variant (rs10509681) that, unlike the R139K variant, has diminished paclitaxel metabolic activity when each is tested in isolation [42, 47]. If this were true, then it would be important for future researchers to focus their analyses specifically on the K399R variant that is sometimes present in patients without the R139K variant [48].
These patients were treated according to standard neoadjuvant protocols, which specify an appropriate treatment dose, schedule, and duration. Recent data demonstrate that the 3-weekly regimen received by 20 % of these patients is inferior to the weekly or every 2-week regimen [49]. In follow-up analyses, multivariable models that included treatment schedule were analyzed to see if schedule had a significant influence on the achievement of clinical complete response, which it did not (uncorrected p = 0.100, data not shown). The predefined treatment duration also ensures that the assessment of response and neuropathy is not confounded by dramatic differences in cumulative paclitaxel received. Only 12 patients discontinued paclitaxel before receiving the full course of therapy, 11 for toxicity, and 1 because of disease progression during treatment. Indeed, carriers of the CYP2C8*3 variant received a similar number of cycles (median = 4) and weeks of therapy (mean = 9.99 vs. 9.60) compared with wild-type patients so it is unlikely that differences in response or neurotoxicity are attributable to differences in cumulative paclitaxel administered.
Numerous groups have investigated polymorphisms in other genes relevant to paclitaxel exposure or mechanism. The polymorphisms in CYP3A4 and CYP3A5 have been independently interrogated and typically do not show associations with outcome [13, 36, 50], though associations have been reported [51]. However, recent data suggest that looking at the CYP3A4*1B/3A5*1A variants as a high metabolic activity haplotype may be a superior strategy [26]. We were unable to identify a statistically significant association with paclitaxel treatment outcomes for either the CYP3A4*1B variant alone or the two variants in combination.
The ABCB1 variants have also been the focus of a number of retrospective pharmacogenetic studies, with inconsistent results. Variants at the 3435 position have been associated with shorter overall survival and worse progression free survival in paclitaxel-treated cancer patients [8, 9]. Variants at the 3435 and 2677 position have been implicated in higher risk of paclitaxel-induced neutropenia [17] and docetaxel treatment outcomes [27]. In our study, the variants of ABCB1 tested individually or in haplotypes did not have a statistically significant effect on paclitaxel treatment outcome.
CYP1B1 is not involved in taxane metabolism [52], yet an association between taxane efficacy and the CYP1B1*3 variant has been repeatedly demonstrated [13, 53–55]. In vitro studies by Sissung et al. [27] reveal that CYP1B1*3 enhances estrogen metabolism to compounds that antagonize the mechanism of action of docetaxel and paclitaxel, and covalently bind docetaxel, providing two plausible mechanisms for the decreased efficacy seen in patients with this genotype [53]. We found no evidence of a link between CYP1B1 genotype and paclitaxel efficacy.
In conclusion, we report evidence that CYP2C8*3 carriers are more likely to achieve complete clinical response to neoadjuvant paclitaxel. This association was independent of other important clinical covariates. The odds of an individual who carried the *3 variant achieving clinical complete response were nearly four times higher than those for an individual carrying two wild-type alleles. Additionally, our results support the previously reported possibility that individuals carrying this variant are at increased risk of experiencing paclitaxel related neuropathy. Our data suggest a potential biomarker for identifying patients before treatment who are more likely to benefit from therapy, but may be at an increased risk of experiencing certain adverse events. The results of this small study warrant further investigation of this association in larger neoadjuvantly treated patient cohorts, and if confirmed, may prompt studies of dose individualization based on host genotype.
References
Henningsson A, Sparreboom A, Sandström M, Freijs A, Larsson R, Bergh J, Nygren P, Karlsson MO (2003) Population pharmacokinetic modelling of unbound and total plasma concentrations of paclitaxel in cancer patients. Eur J Cancer 39(8):1105–1114
Sparano JA, Wang M, Martino S, Jones V, Perez EA, Saphner T, Wolff AC, Sledge GW, Wood WC, Davidson NE (2008) Weekly paclitaxel in the adjuvant treatment of breast cancer. N Engl J Med 358(16):1663–1671
Di Leo A, Gomez HL, Aziz Z, Zvirbule Z, Bines J, Arbushites MC, Guerrera SF, Koehler M, Oliva C, Stein SH, Williams LS, Dering J, Finn RS, Press MF (2008) Phase III, double-blind, randomized study comparing lapatinib plus paclitaxel with placebo plus paclitaxel as first-line treatment for metastatic breast cancer. J Clin Oncol 26(34):5544–5552
McLeod HL, Evans WE (2001) Pharmacogenomics: unlocking the human genome for better drug therapy. Annu Rev Pharmacol Toxicol 41:101–121
Hoskins JM, Goldberg RM, Qu P, Ibrahim JG, McLeod HL (2007) UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J Natl Cancer Inst 99(17):1290–1295
Hu Z, Yu Q, Zhao Y (2010) Dose-dependent association between UGT1A1∗28 polymorphism and irinotecan-induced diarrhoea: a meta-analysis. Eur J Cancer 46(10):1856–1865
Klein TE, Altman RB, Eriksson N, Gage BF, Kimmel SE, International Warfarin Pharmacogenetics Consortium et al (2009) Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med 360(8):753–764
Chang H, Rha SY, Jeung HC, Im CK, Noh SH, Kim JJ, Chung HC (2010) Association of the ABCB1 3435C>T polymorphism and treatment outcomes in advanced gastric cancer patients treated with paclitaxel-based chemotherapy. Oncol Rep 23(1):271–278
Chang H, Rha SY, Jeung H-, Im C-, Ahn JB, Kwon WS, Yoo NC, Roh JK, Chung HC (2009) Association of the ABCB1 gene polymorphisms 2677G>T/A and 3435C>T with clinical outcomes of paclitaxel monotherapy in metastatic breast cancer patients. Ann Oncol 20(2):272–277
Green H, Soderkvist P, Rosenberg P, Horvath G, Peterson C (2006) Mdr-1 single nucleotide polymorphisms in ovarian cancer tissue: G2677T/A correlates with response to paclitaxel chemotherapy. Clin Cancer Res 12(3):854–859
Leskela S, Jara C, Leandro-Garcia L, Martinez A, Garcia-Donas J, Hernando S, Hurtado A, Vicario JCC, Montero-Conde C, Landa I, Lopez-Jimenez E, Cascon A, Milne RL, Robeldo M, Rodriguez-Antona C (2011) Polymorphisms in cytochromes P450 2C8 and 3A5 are associated with paclitaxel neurotoxicity. Pharmacogenomics J 11(2):121–129
Petros WP, Hopkins PJ, Spruill S, Broadwater G, Vredenburgh JJ, Colvin OM, Peters WP, Jones RB, Hall J, Marks JR (2005) Associations between drug metabolism genotype, chemotherapy pharmacokinetics, and overall survival in patients with breast cancer. J Clin Oncol 23(25):6117–6125
Marsh S, Somlo G, Li X, Frankel P, King CR, Shannon WD, McLeod HL, Synold TW (2007) Pharmacogenetic analysis of paclitaxel transport and metabolism genes in breast cancer. Pharmacogenomics J 7(5):362–365
Kroetz DL, Baldwin RM, Owzar K, Jiang C, Zembutsu H, Kubo M, Nakamura Y, Shulman LN, Ratain MJ, Cancer and Leukemia Group B. (2010) Inherited genetic variation in EPHA5, FGD4, and NRDG1 and paclitaxel (P)-induced peripheral neuropathy (PN): results from a genome-wide association study (GWAS) in CALGB 40101. ASCO Meeting Abstracts. 2010 June 14;28(15 Suppl):3021
Schneider BP, Li L, Miller K, Flockhart D, Radovich M, Hancock BA, Kassem N, Forourd T, Koller DL, Badve SS, Li Z, Partridge AH, O’Neill AM, Sparano JA, Dang CT, Northfelt DW, Smith ML, Railey E, Sledge GW (2011) Genetic associations with taxane-induced neuropathy by a genome-wide association study (GWAS) in E5103. ASCO Meeting Abstracts. 2011 June 09;29(15_suppl):1000
Rizzo R, Spaggiari F, Indelli M, Lelli G, Baricordi O, Rimessi P, Ferlini A (2010) Association of CYP1B1 with hypersensitivity induced by taxane therapy in breast cancer patients. Breast Cancer Res Treat 124(2):593–598
Sissung TM, Mross K, Steinberg SM, Behringer D, Figg WD, Sparreboom A, Mielke S (2006) Association of ABCB1 genotypes with paclitaxel-mediated peripheral neuropathy and neutropenia. Eur J Cancer 42(17):2893–2896
Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lenning PE, Borresen-Dale A (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98(19):10869–10874
Eley JW, Hill HA, Chen VW, Austin DF, Wesley MN, Muss HB, Greenberg RS, Coates RJ, Correa P, Redmond CK (1994) Racial differences in survival from breast cancer. Results of the National Cancer Institute Black/White Cancer Survival Study. JAMA 272(12):947–954
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247
Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. U.S. Department of Health and Human Services, National Institutes of Health, National Cancer Institute; 2010 June 14, 2010
Marsh S, King CR, Garsa AA, McLeod HL (2005) Pyrosequencing of clinically relevant polymorphisms. Methods Mol Biol 311:97–114
Rose CM, Marsh S, Ameyaw MM, McLeod HL (2003) Pharmacogenetic analysis of clinically relevant genetic polymorphisms. Methods Mol Med 85:225–237
Stephens M, Donnelly P (2003) A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73(5):1162–1169
Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68(4):978–989
Baker SD, Verweij J, Cusatis GA, van Schaik R, Marsh S, Orwick SJ, Franke RM, Hu S, Schuetz EG, Lamba V, Messersmith WA, Wolff AC, Carducci MA, Sparreboom A (2008) Pharmacogenetic pathway analysis of docetaxel elimination. Clin Pharmacol Ther 85(2):155–163
Sissung TM, Baum CE, Deeken J, Price DK, Aragon-Ching J, Steinberg SM, Dahut W, Sparreboom A, Figg WD (2008) ABCB1 genetic variation influences the toxicity and clinical outcome of patients with androgen-independent prostate cancer treated with docetaxel. Clin Cancer Res 14(14):4543–4549
Barnholtz-Sloan JS, de Andrade M, Chakraborty R (2001) The impact of population admixture on traditional linkage analysis. Ethn Dis 11(3):519–531
Garsa A, McLeod H, Marsh S (2005) CYP3A4 and CYP3A5 genotyping by pyrosequencing. BMC Med Genet 6(1):19
International HapMap Consortium (2003) The international HapMap project. Nature 426(6968):789–796
NCBI (2010) NCBI entrez SNP database. Available from: http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp. Accessed Oct 05, 2008
Cance WG, Carey LA, Calvo BF, Sartor C, Sawyer L, Moore DT, Rosenman J, Ollila DW, Graham M (2002) Long-term outcome of neoadjuvant therapy for locally advanced breast carcinoma: effective clinical downstaging allows breast preservation and predicts outstanding local control and survival. Ann Surg 236(3):295–302 Discussion: 302–303
Carey LA, Metzger R, Dees EC, Collichio F, Sartor CI, Ollila DW, Klauber-DeMore N, Halle J, Sawyer L, Moore DT, Graham ML (2005) American Joint Committee on Cancer Tumor–Node–Metastasis stage after neoadjuvant chemotherapy and breast cancer outcome. J Natl Cancer Inst 97(15):1137–1142
Fisher B, Bryant J, Wolmark N, Mamounas E, Brown A, Fisher ER, Wickerham DL, Begovic M, DeCillis A, Robidoux A, Margolese RG, Cruz AB, Hoehn JL, Lees AW, Dimitrov NV, Bear HD (1998) Effect of preoperative chemotherapy on the outcome of women with operable breast cancer. J Clin Oncol 16(8):2672–2685
Zheng S, Zhang BL, Zhang RZ, Yang JL, Zou SM, Xue LY, Luo W, Yuan YL, Lu N (2010) Differences between clinical response and pathologic response of breast cancer after neoadjuvant chemotherapy. Zhonghua Bing Li Xue Za Zhi 39(11):734–738 in Chinese
Marsh S, Paul J, King CR, Gifford G, McLeod HL, Brown R (2007) Pharmacogenetic assessment of toxicity and outcome after platinum plus taxane chemotherapy in ovarian cancer: the Scottish randomised trial in ovarian cancer. J Clin Oncol 25(29):4528–4535
Bergmann TK, Gréen H, Brasch-Andersen C, Mirza M, Herrstedt J, Hølund B, du Bois A, Damkier P, Vach W, Brosen K, Peterson C (2011) Retrospective study of the impact of pharmacogenetic variants on paclitaxel toxicity and survival in patients with ovarian cancer. Eur J Clin Pharmacol 67(7):693–700
Sparreboom A, Huizing MT, Boesen JJB, Nooijen WJ, Beijnen JH, Tellingen OV (1995) Isolation, purification, and biological activity of mono- and dihydroxylated paclitaxel metabolites from human feces. Cancer Chemother Pharmacol 36(4):299–304
Mielke S, Sparreboom A, Steinberg SM, Gelderblom H, Unger C, Behringer D, Mross K (2005) Association of paclitaxel pharmacokinetics with the development of peripheral neuropathy in patients with advanced cancer. Clin Cancer Res 11(13):4843–4850
de Jonge ME, van den Bongard HJGD, Huitema ADR, Mathôt RAA, Rosing H, Baas P, van Zandwijk N, Beijnen JH, Schellens JHM (2004) Bayesian pharmacokinetically guided dosing of paclitaxel in patients with non-small cell lung cancer. Clin Cancer Res 10(7):2237–2244
van Gerven JMA, Moll JWB, van den Bent MJ, Bontenbal M, van der Burg MEL, Verweij J, Vecht CJ (1994) Paclitaxel (taxol) induces cumulative mild neurotoxicity. Eur J Cancer 30(8):1074–1077
Rowbotham SE, Boddy AV, Redfern CPF, Veal GJ, Daly AK (2010) Relevance of nonsynonymous CYP2C8 polymorphisms to 13-cis retinoic acid and paclitaxel hydroxylation. Drug Metab Dispos 38(8):1261–1266
Soyama A, Saito Y, Hanioka N, Murayama N, Nakajima O, Katori N, Ishida S, Kimie S, Ozawa S, Sawada J (2001) Non-synonymous single nucleotide alterations found in the CYP2C8 gene result in reduced in vitro paclitaxel metabolism. Biol Pharm Bull 24(12):1427–1430
Bahadur N, Leathart JBS, Mutch E, Steimel-Crespi D, Dunn SA, Gilissen R, Houdt JV, Hendrickx J, Mannens G, Bohets H, Williams FM, Armstrong M, Crespi CL, Daly AK (2002) CYP2C8 polymorphisms in caucasians and their relationship with paclitaxel 6α-hydroxylase activity in human liver microsomes. Biochem Pharmacol 64(11):1579–1589
Dai D, Zeldin DC, Blaisdell JA, Chanas B, Coulter SJ, Ghanayem BI, Goldstein JA (2001) Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics 11(7):597–607
Bergmann TK, Brasch-Andersen C, Green H, Mirza M, Pedersen RS, Nielsen F, Skougaard K, Wihl J, Keldsen N, Damkier P, Friberg LE, Peterson C, Vach W, Karlsson MO, Brosen K (2011) Impact of CYP2C8*3 on paclitaxel clearance: A population pharmacokinetic and pharmacogenomic study in 93 patients with ovarian cancer. Pharmacogenomics J 11(2):113–120
Gao Y, Liu D, Wang H, Zhu J, Chen C (2010) Functional characterization of five CYP2C8 variants and prediction of CYP2C8 genotype-dependent effects on in vitro and in vivo drug–drug interactions. Xenobiotica 40(7):467–475
Speed WC, Kang SP, Tuck DP, Harris LN, Kidd KK (2009) Global variation in CYP2C8–CYP2C9 functional haplotypes. Pharmacogenomics J 9(4):283–290
Seidman AD, Berry D, Cirrincione C, Harris L, Muss H, Marcom PK, Gipson G, Burstein H, Lake D, Shapiro CL, Ungaro P, Norton L, Winer E, Hudis C (2008) Randomized phase III trial of weekly compared with every-3-weeks paclitaxel for metastatic breast cancer, with trastuzumab for all HER-2 overexpressors and random assignment to trastuzumab or not in HER-2 nonoverexpressors: final results of cancer and leukemia group B protocol 9840. J Clin Oncol 26(10):1642–1649
Henningsson A, Marsh S, Loos WJ, Karlsson MO, Garsa A, Mross K, Mielke S, Vigano L, Locatelli A, Verweij J, Sparreboom A, McLeod HL (2005) Association of CYP2C8, CYP3A4, CYP3A5, and ABCB1 polymorphisms with the pharmacokinetics of paclitaxel. Clin Cancer Res 11(22):8097–8104
Gandara DR, Kawaguchi T, Crowley J, Moon J, Furuse K, Kawahara M, Teramukai S, Ohe Y, Kubota K, Williamsoon ST, Gautschi O, Lenz HJ, McLeod HL, Lara PN, Coltman CA, Fukuoka M, Saijo N, Fukushima M, Mack PC (2009) Japanese–US common-arm analysis of paclitaxel plus carboplatin in advanced non-small-cell lung cancer: a model for assessing population-related pharmacogenomics. J Clin Oncol 27(21):3540–3546
Bournique B, Lemarié A (2002) Docetaxel (taxotere) is not metabolized by recombinant human CYP1B1 in vitro, but acts as an effector of this isozyme. Drug Metab Dispos 30(11):1149–1152
Sissung TM, Danesi R, Price DK, Steinberg SM, de Wit R, Zahid M, Gaikwad N, Cavalieri E, Dahut WL, Sackett DL, Figg WD, Sparreboom A (2008) Association of the CYP1B1*3 allele with survival in patients with prostate cancer receiving docetaxel. Mol Cancer Ther 7(1):19–26
Pastina I, Giovannetti E, Chioni A, Sissung TM, Crea F, Orlandini C, Price DK, Cianci C, Figg WD, Ricci S, Danesi R (2010) Cytochrome 450 1B1 (CYP1B1) polymorphisms associated with response to docetaxel in castration-resistant prostate cancer (CRPC) patients. BMC Cancer 27(10):511
Figg WD, Li H, Sissung T, Retter A, Wu S, Gulley JL, Arlen P, Wright JJ, Parnes H, Fedenko K, Latham L, Steinberg SM, Jones E, Chen C, Dahut W (2007) Pre-clinical and clinical evaluation of estramustine, docetaxel and thalidomide combination in androgen-independent prostate cancer. BJU Int 99(5):1047–1055
Acknowledgments
We would like to thank Anne Misher for assistance with genotyping and Janelle Hoskins for guidance on choosing candidate SNPs. This study was supported by the Breast Cancer Research Foundation (LAC), Lineberger Comprehensive Cancer Center, a Clinical and Translational Science Award 5UL1RR025747-04, the National Institute of Health-National Institute of General Medical Sciences T32GM081057 and NIH SPORE in Breast Cancer 5P50CA058223. Daniel Hertz is an American Foundation for Pharmaceutical Education Pre-Doctoral Fellow in clinical pharmaceutical science. They have received research funding from pharmaceutical companies that are not related to the current work: Millennium Pharmaceuticals, Roche-Genentech, GlaxoSmithKline, and Novartis (ECD). This report has original material and there have been no other previous reports or publications to disclose.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
All experiments comply with current laws of the United States of America.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10549_2012_2054_MOESM1_ESM.docx
Supplementary Table 1: SNP Information: rsID, allelic position and change, resulting protein change, polymerase chain reaction annealing temperature and primers. (DOCX 12 kb)
10549_2012_2054_MOESM2_ESM.docx
Supplementary Table 2: Genotyping results: call rate, variant allele frequency, uncorrected p values of Hardy–Weinberg Equilibrium test in combined cohort and Caucasian subcohort, variant allele frequency in Caucasian subcohort and expected allele frequency in Caucasians from The International HapMap Project or dbSNP database. (DOCX 12 kb)
10549_2012_2054_MOESM3_ESM.docx
Supplementary Table 3: Haplotype Results: Complete breakdown of number of individuals who fall into each haplotype category as defined for CYP3A4/3A5 in Baker et al. [26] and ABCB1 in Sissung et al. [27] followed by the grouping for the association study. (CYP3A4/3A5 *2 carriers vs. other and ABCB1 Diplotype 1–3 vs. 4 and 5). (DOCX 11 kb)
10549_2012_2054_MOESM4_ESM.docx
Supplementary Table 4: Severe neuropathy (≥ grade 3) in toxicity cohort (N = 109) by genotype or haplotype, comparing variant carriers with wild-type homozygous patients. (DOCX 11 kb)
Rights and permissions
About this article
Cite this article
Hertz, D.L., Motsinger-Reif, A.A., Drobish, A. et al. CYP2C8*3 predicts benefit/risk profile in breast cancer patients receiving neoadjuvant paclitaxel. Breast Cancer Res Treat 134, 401–410 (2012). https://doi.org/10.1007/s10549-012-2054-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-012-2054-0