
Abstract
Tellurite, the most soluble tellurium oxyanion, is extremely harmful for most microorganisms. Part of this toxicity is due to the generation of reactive oxygen species that in turn cause oxidative stress. However, the way in which tellurite interferes with cellular processes is not well understood to date. Looking for new cellular tellurite targets, we decided to evaluate the functioning of the electron transport chain in tellurite-exposed cells. In this communication we show that the E. coli ndh gene, encoding NDH-II dehydrogenase, is significantly induced in toxicant-exposed cells and that the enzyme displays tellurite-reducing activity that results in increased superoxide levels in vitro.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Although scarce in the earth’s crust, tellurium is commonly found in alloys with other metals like calaverite (AuTe2) and silvanite (AgAuTe4) and nagyagita AuPb(Sb,Bi)Te2–3S6, among others (Cairnes 1911). Te belongs to the chalcogens (Group 16), along with other biologically important elements such as oxygen, sulfur and selenium (Fischer 2001) and is generally recovered as a waste of the mining activity. In its elemental form (Te0), tellurium does not display toxicity to living organisms, mostly because of poor solubility. Conversely, the tellurium oxyanions tellurite (TeO3 2−, very soluble) and tellurate (TeO4 2−, poorly soluble) are highly toxic to most microorganisms, especially Gram-negative bacteria (Taylor 1999).
Tellurite-mediated damage includes—among other effects—altered ΔpH, decreased ATP levels and increased generation of reactive oxygen species (ROS) in Escherichia coli (Tantaleán et al. 2003; Lohmeier-Vogel et al. 2004; Pérez et al. 2008), Pseudomonas pseudoalcaligenes KF707 (Borsetti et al. 2005) and Rhodobacter capsulatus (Tremaroli et al. 2007, 2009). Once inside the cell, tellurite is reduced in a chemical or enzymatic manner and superoxide (O .−2 ) is generated (Pérez et al. 2007, 2008; Tremaroli et al. 2007, 2009). In turn, this oxygen radical may cause membrane lipid peroxidation either by increasing the production of toxic aldehydes or changing the monounsaturated fatty acids ratio (Pérez et al. 2007; Pradenas et al. 2012, 2013). Tellurite also affects the activity of key metabolic enzymes such as phosphofructo kinase, pyruvate kinase, pyruvate dehydrogenase and α-ketoglutarate dehydrogenase while a number of metabolites including pyruvate, α-ketoglutarate and some phosphorylated sugars are accumulated in the presence of the toxicant (Castro et al. 2008; Reinoso et al. 2012; Valdivia-González et al. 2012). In addition, tellurite-mediated damage is reflected in protein carbonylation (Pérez et al. 2007, 2008; Contreras and Vásquez 2010), dismantling of [4Fe–4S] clusters from enzymes such as fumarase A and aconitase B (Imlay 2006; Calderón et al. 2009) and in a rapid decrease of the cell’s reduced thiol content, especially that of glutathione (Turner et al. 1999, 2001). Induction of oxidative stress response-regulator genes such as oxyR and soxRS along with those encoding catalase (katA, katG), superoxide dismutase (sodA, sodB, sodC), fumarase C (fumC), aconitase A (acnA), aldehyde reductase YqhD (yqhD) and glutathione peroxidase (btuE) has also been observed upon tellurite exposure (Imlay 2003; Pérez et al. 2007, 2008; Arenas et al. 2010, 2011).
Tellurite (Te4+) can be enzymatically reduced in vivo to elemental tellurium (Te0) in a NAD(P)H-dependent manner by branch activities exhibited by some enzymes that include catalase, dihydrolipoil dehydrogenase from the pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase complexes and isocitrate dehydrogenase (Calderón et al. 2006; Castro et al. 2008, 2009; Reinoso et al. 2012), among others. Interestingly, enzymes involved in electron transport, such as nitrate reductases of E. coli, Paracoccus denitrificans, P. pantotrophus and R. sphaeroides (Avazeri et al. 1997; Sabaty et al. 2001), as well as the E. coli terminal oxidases have been also involved in tellurite reduction (Trutko et al. 1998, 2000), which is concomitant with superoxide generation and with the accumulation of intracellular black deposits whose cell location would probably be associated with redox sites such as the cell membrane (Taylor et al. 1988; Lloyd-Jones et al. 1994; Calderón et al. 2006).
The electron transport chain (ETC) is responsible for generating a proton motive force that is used to transport molecules through the membrane, allowing processes such as flagellar rotation and ATP synthesis during oxidative phosphorylation. In aerobic conditions, the E. coli ETC is formed by the NADH dehydrogenase complex I (NDH-I), NADH dehydrogenase II (NDH-II), succinate quinone oxide reductase complex and the terminal oxidases bo, bd-I and bd-II (Matsushita et al. 1987; Hayashi et al. 1989; Puustinen et al. 1991; Calhoun et al. 1993; Nakamura et al. 1996).
Our work has focused on NDH-II, a 47 kDa enzyme encoded by the ndh gene. Similar to NDH-I, NDH-II catalyzes NADH oxidation (Young et al. 1978) but unlike it, NDH-II does not participate in translocating protons from the cytoplasm to the periplasmic space (Matsushita et al. 1987). Recently it was shown that E. coli NDH-II displays the ability to reduce Cu2+ to Cu1+ when incubated in the presence of FAD and NADH (Rapisarda et al. 1999, 2002; Volentini et al. 2011). Increased copper sensitivity has been observed in ndh mutants, suggesting that NDH-II can alleviate the oxidative damage generated by this metal (Rodríguez-Montelongo et al. 2006).
In this communication we show that the ndh gene is induced in tellurite-exposed E. coli and that NDH-II catalyzes the NADH-dependent tellurite reduction while generating superoxide in vitro.
Materials and methods
Bacterial strains and growth conditions
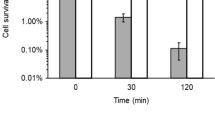
Bacterial strains used in this work are indicated in Table 1. Cells were grown at 37 °C with vigorous shaking in non-fermentative minimal medium (NFMM) that contained M9 1× salts (Na2HPO4 34 g, KH2PO4 15 g, NH4Cl 5 g and NaCl 2.5 g in 1 l of a 5× stock solution), casaminoacids (0.1 %) and glycerol (1 %) (Lin 1976). Solid media contained 2 % agar. When required, the medium was amended with the appropriate antibiotic. Growth was initiated with 1:10,000 dilutions of saturated cultures. For tellurite treatments, cultures in early exponential growth phase (OD600 ~0.3) were exposed for 15 or 30 min to sub-lethal (1.5 μM) tellurite concentrations (corresponding to one half of the minimal inhibitory concentration, MIC, as determined in NFMM medium).
Gene cloning
Using E. coli genome sequences deposited in databases, specific primers were designed to amplify the genes of interest (Table S1). PCR products were ligated to the pBAD/TOPO (Invitrogen) vector and transformed into competent E. coli JW2279 (nuoF) and JW1095 (ndh) strains. Plasmid purification was accomplished by using the Miniprep Kit QIAGEN (Darmstadt, Germany). Analysis of recombinant plasmids was carried out by PCR and NcoI restriction.
RNA purification
After exposing E. coli to tellurite for 15 or 30 min, cells were harvested at 8,000×g for 5 min and suspended in 20 mM sodium acetate pH 5.5 buffer, 1 mM EDTA, 5 % SDS (lysis buffer) and heated at 70 °C for 10 min. Then 500 μl of acid phenol (Trisure) were added and the mix was gently stirred once per min for 10 min at 70 °C. Chilling was at room temperature and after adding 200 μl of chloroform the solution was mixed as before. After centrifuging for 15 min at 15,000×g at 4 °C, the aqueous phase was rescued and nucleic acids were precipitated with two volumes of isopropanol. The tube was kept at −80 °C for 1 h and centrifuged as above. The pellet was air dried, suspended in 20 μl of a buffer (Qiagen) that contained RNase-free DNase I and incubated for 1 h at 37 °C. DNase was inactivated at 75 °C for 10 min and RNA was precipitated as described above. The pellet was suspended in nuclease-free water that contained diethylpirocarbonate (DEPC) and 0.01 % RNAsin (Invitrogen). RNA quantification was assessed using the Quanti-iT RNA kit (Invitrogen). Total RNA was finally diluted to a concentration of 250 ng/μl for qPCR reactions. Specific primers used are shown in Table S1.
Real-time PCR
Primers for qPCR assays (~300 bp) (Table S1) were designed utilizing the Vector NTI© 11 software (Invitrogen). Two hundred-fifty ng of RNA were used for qPCR with the LightCycler RNA Amplification SYBR Green I Kit (Roche Applied Science). The reaction mix contained 2 μl of each primer (10 pmol/ml), 0.4 μl of Light Cycler RT-PCR Enzyme Mix, 4 μl of resolution buffer, 2 μl of 10 mM nucleotide mix (dATP, dUTP, dCTP, dGTP), 4 μl SYBR Green I and 2.4 μl of 25 mM MgCl2. PCR-grade water was added to 20 μl final volume. The RT reaction was carried out at 30 °C using an amplification program that consisted of 45 cycles (30 s denaturation at 95 °C, 10 s annealing at 55 °C, 30 s at 72 °C); rpoD gene was used as the housekeeping gene. The Light Cycler 2.0 software was used to record the fluorescence increase in each cycle. Finally the results were analyzed as relative expression as described earlier (Pfaffl 2001).
Isolation of E. coli membrane fractions
NFMM-grown E. coli cultures were harvested at 8,000×g for 5 min at 4 °C and washed with 50 mM Tris–HCl pH 7.4 buffer (buffer A) or, when indicated with 50 mM MES-NaOH pH 7.4 buffer (buffer B). Cell extracts were prepared according to the method previously described (Messner and Imlay 1999) with minor modifications. Membrane protein concentration was assessed as described (Bradford 1976) using 1:1 membrane fraction:guanidine hydrochloride (GndCl, 6 M) to improve both membrane solubility and sensitivity. BSA was used as protein standard.
NADH dehydrogenase activity in E. coli membrane fractions
NADH dehydrogenase activity was assessed at 25 °C in membrane fractions by monitoring NADH oxidation at 340 nm. The reaction mix (500 μl) contained buffer A and 60 μM NADH. Reactions were initiated by adding 25 μg of membrane proteins as previously described (Matsushita et al. 1987). A unit was defined as the amount of enzyme that oxidized 1 μmol of NADH per min at 25 °C.
Tellurite reductase (TR) activity
Escherichia coli membrane fractions were incubated for 10 min at 37 °C in a mix that contained 50 mM Tris–HCl pH 7.4 buffer, 1 mM K2TeO3, 1 mM 2-mercaptoethanol and 0.5 mM NADH (TR) buffer as previously described (Calderón et al. 2006). A unit was defined as the amount of enzyme that increased the OD500 by 0.001 units per min at 37 °C.
Protein carbonylation in E. coli membrane proteins
Carbonyl group content was assessed in membrane fractions as described earlier (Semchyshyn et al. 2005) with minor modifications (Contreras and Vásquez 2010).
Protein purification
NDH-II, FumA and dihydrolipoil dehydrogenase were purified after inducing ndh, fumA and lpd genes with 1 mM IPTG for 4 h in appropriate E. coli strains (Table 1). Briefly, cell extracts were prepared by sonication and after eliminating the cell debris by low speed centrifugation, cleared supernatants were loaded onto HisTrap affinity columns under the vendor’s instructions (GE Healthcare Life Sciences).
In vitro superoxide generation
Affinity-purified NDH-II, FumA or Lpd (E3) were mixed with TR buffer (TRB) that contained the water soluble probe 2-(4-Iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt (WST-1, Sigma) and incubated for 5 min at 25 °C. Superoxide formation was monitored at 439 nm as described earlier (Calderón et al. 2006).
Te quantification by inductively coupled plasma-optical emission spectrometry (ICP-OES)
Membrane fractions from cells previously exposed to tellurite were suspended in 1 ml of buffer A and gently sonicated. Samples were diluted tenfold with 10 % HNO3 and the whole volume was used for Te determination using a Spectro CIROS Vision ICP-OES instrument using the 214 nm Te line. A calibration curves were constructed with commercially-available tellurium standards (Sigma-Aldrich) also containing 10 % HNO3.
Scanning electron microscopy (SEM)
Purified membrane fractions were incubated with sodium desoxycholate (2 %) for 2 h to form membrane vesicles. After centrifuging at 120,000×g for 1 h, the sediment—consisting mainly of membrane vesicles—was washed with buffer A and fixed with glutaraldehyde (1.25 %) for later viewing in a SEM EVO MA 10 (Karl Zeiss) microscope.
Results and discussion
Tellurite reductase activity in E. coli membranes
It is well known that the Te oxyanion tellurite is toxic for most living organisms. Once inside the cell this toxicant causes a number of alterations including [FeS] cluster dismantling, decreased glutathione pool, increased ROS levels and accumulation of key metabolites, among other effects (Tremaroli et al. 2007; Chasteen et al. 2009). Nevertheless, the complete description of tellurite-mediated damage is currently unknown.
Our laboratory has been interested for a long time in unveiling tellurite-mediated damage to the cell’s general metabolism. In these investigations it has been observed that important metabolic pathways are also affected by TeO3 2− including glycolysis (Valdivia-González et al. 2012), Krebs cycle (Calderón et al. 2009; Reinoso et al. 2012) and the pentose phosphate shunt (Sandoval et al. 2011). Given that these paths provide the real ETC substrates, namely NADH and FADH2, it was very interesting to focus on tellurite effects on the E. coli ETC. This goal finds relevance in the fact that tellurite damage occurs mainly in the presence of oxygen (Tantaleán et al. 2003) and in recent results showing that the bacterial membrane is likewise affected by the toxicant (Pérez et al. 2007; Pradenas et al. 2012, 2013).
On the other hand, it has been shown that some NAD(P)H-dependent dehydrogenases like dihydrolipoil dehydrogenase (E3) (Castro et al. 2008, 2009) and isocitrate dehydrogenase display tellurite-reducing ability (Reinoso et al. 2013). In this context, preliminary results from our laboratory indicate that while NADH dehydrogenase activity from NDH-I is completely abolished by tellurite in toxicant-exposed cells, that of NDH-II remains almost unaltered (to be published elsewhere). The obvious question to answer therefore was if NDH-II exhibits or not TR activity.
Purified membrane fractions from tellurite-exposed E. coli displayed a characteristic black color, indicative of metallic tellurium deposits (Fig. S1A). Similar results using whole cells were communicated earlier (Taylor et al. 1988; Trutko et al. 2000). Membrane vesicles were clearly identifiable in SEM images (Fig. S1B). It is not clear if elemental Te has an impact or not on membrane structure or fluidity. If so, transport and movement of lipid-soluble molecules could be altered, thus affecting the whole functioning of the membrane. For instance, altered quinone diffusion could be reflected in decreased electron flux (and hence proton motive force), terminal oxidase activity and/or ATP synthesis.
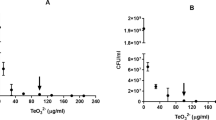
To determine if tellurite reduction occurs at the level of the ETC’s dehydrogenases in vitro, membranes from the parental strain were incubated with TR buffer that contained cyanide to eliminate the potential contribution of terminal oxidases. This experiment was undertaken because it had been previously communicated that ETC terminal oxidases can use tellurite as the final electron acceptor (Trutko et al. 1998, 2000; Baesman et al. 2007). Opposite to the findings of Trutko et al. (2000), who observed that tellurite reduction as well as the accumulation of elemental Te crystals decreased in cyanide-exposed cells, our results clearly show that tellurite reduction actually increases in the presence of KCN (Fig. 1a). This occurs probably because instead of being conducted from the dehydrogenases to the terminal oxidases, electrons are being directly transferred to tellurite. These data allow us to infer that the cyanide target in the terminal oxidases is not related to that where tellurite reduction is taking place. Alternatively, this could also mean that the terminal oxidases would not be the only ETC macromolecules involved in tellurite reduction.

Tellurite reductase activity in E. coli membrane fractions. TR activity was assayed in membrane fractions from the indicated bacteria unexposed (a) or exposed (b). KCN was used to inhibit cytochrome oxidase activity. SA specific activity. Bars represent the average of three independent trials ± SE. P < 0.001 (***), NS not significant
With this in mind, tellurite reduction was assessed in membrane fractions from wild type and mutant E. coli strains, exposed or not to the toxicant. After the treatment, no change was observed in tellurite reduction by wild type and NDH-II− strains. However, TeO3 2−-exposed membranes from NDH-I− cells showed an important augment of tellurite reduction (Fig. 1b). These results suggest that exposing cells to the toxicant causes important changes in membrane composition and/or structure that could promote Te4+→Te0 reduction. In this context, NDH-II could represent a NADH-dependent tellurite-reducing enzyme whose expression could be increased in the tellurite-provoked absence of the NDH-I complex. Further evidence stating that NDH-II but not NDH-I is able of tellurite reduction came from experiments in which dNADH, an exclusive substrate for NDH-I, was used instead of NADH. No tellurite reduction was observed at all under these conditions (not shown). On the other hand and with the exception of lawsome, quinone participation in tellurite reduction has been recently ruled out (Wang et al. 2011).
NDH-II displays TR activity
Given that NDH-II is the ETC’s most important dehydrogenase during the E. coli aerobic growth (Unden and Bongaerts 1997), it would not be unexpected that its structural gene, ndh, would be induced in the presence of oxygen (see below). This could also represent a cell response against tellurite damage to the ETC, which is in agreement with the observation that, as mentioned before, NDH-I and not NDH-II is affected by tellurite.
To determine if effectively NDH-II is the membrane component that is responsible for TR activity, the enzyme was purified and used for assessing tellurite-reducing activity. The E3 component of the E. coli pyruvate dehydrogenase complex was also purified and used as positive control for NADH-dependent TR activity (Castro et al. 2008, 2009)(Fig. S2a). NAD-II’s TR activity was determined in the presence of NADH-containing TR buffer (Fig. 2a) and also in situ after being fractioned by native PAGE (Fig. 2b).

TR activity of purified E. coli NDH-II. TR activity was assayed visually (a) and in situ after fractionation by native PAGE (b), as described in “Materials and methods” section. c TR activity of NDH-II previously incubated with 20 μM FAD. Purified E. coli dihydrolipoil dehydrogenase (E3) was used as positive control for TR activity. Bars represent the average of three independent trials ± SE. P < 0.05 (*)
Since some flavoproteins lose activity upon FAD withdrawing (Hefti et al. 2003), it was of interest to determine if this situation affects DH and TR activities of the NDH-II flavoprotein. Thus, purified NDH-II was preincubated with FAD and assayed for DH and TR activity. The results showed that NDH-II’s DH and TR activities increased 56 and 62 %, respectively, in regard to the control without FAD (Fig. S2b and 2c).
NDH-II-mediated tellurite reduction results in superoxide generation
Since (i) enzymes exhibiting TR activity such as catalase generate superoxide (Calderón et al. 2006), (ii) NDH-II generates high ROS amounts under normal conditions (Messner and Imlay 1999) and (iii) that preliminary experiments from our laboratory show that the E. coli ETC’s terminal oxidases would not participate in ROS generation, these facts support the idea that the tellurite-mediated membrane oxidative unbalance would be significantly related to the ETC’s dehydrogenases.
Next, we determined if the in vitro, NDH-II-mediated tellurite reduction results in superoxide generation which in turn, and among other effects, could provoke the loss of activity of some ETC components. The results showed that NDH-II displays higher superoxide levels than catalase (Calderón et al. 2006) (Fig. 3a). Thus, NDH-II can generate ROS at the membrane and as a consequence, an oxidative unbalance that would result in membrane lipid peroxidation (Pradenas et al. 2012). The carbonyl group content was found to increase in membrane proteins from tellurite-exposed cells as compared with the respective controls. This observation was particularly evident for the NDH-I− strain, suggesting the generation of an oxidative unbalance provoked either by the absence of a functional NDH-I complex and/or by an increased amount of NDH-II in the membrane (Fig. 3b). However, regarding the control situation, the NDH-I− strain showed a lower basal carbonylation than that observed for the NDH-II− mutant.

In vitro superoxide generation by NDH-II and oxidative damage to E. coli membrane proteins. a superoxide generation by purified NDH-II was determined using the probe WST-1 as described in “Materials and methods” section. Catalase (bovine liver) was used as positive control for O2 − production. b carbonyl group content was assessed in membranes of the indicated tellurite-exposed E. coli strains. Hydrogen peroxide (2.5 mM) was used as positive control for oxidative damage. Bars represent the average of three independent trials ± SE. P < 0.05 (*), P < 0.01(**)
Tellurium content in membranes from tellurite-exposed E. coli
To assess if nuoF or ndh over expression influences the observed increase of in vitro tellurite reduction (Fig. 1b), membranes from E. coli BW25113, pBnuoF and pBndh previously grown in the presence of arabinose and exposed to tellurite were analyzed by ICP-OES as described in “Materials and methods” section. While the parental strain showed no difference in the intracelular Te, genetically-complemented mutant strains pBnuoF and pBndh showed increased tellurium content. Interestingly pBndh cells showed higher amounts of Te in the presence of arabinose (Fig. 4a). Similar results were obtained with nuo, ndh and the double mutant nuo/ndh strains, where nuo cells showed the highest level of elemental tellurium (Fig. 4b). This result rules out the participation of NDH-I-mediated tellurite reduction in vivo and defines the E. coli ETC’s NDH-II dehydrogenase as a new membrane enzyme exhibiting TR activity.

Intracellular Te content in E. coli as determined by ICP-OES. Te content in membranes from genetically-complemented strains previously grown to early exponential phase, induced with 0.02 % arabinose and then exposed to tellurite (a) and in the indicated mutant strains previously exposed to tellurite (b). Bars represent the average of 3 independent trials ± SE. P < 0.05 (*), P < 0.001(***), NS not significant
Relative gene expression studies in tellurite-exposed E. coli
To determine if the enhanced level of tellurite reduction by NDH-II is the result of increased ndh expression, assays for relative gene expression were carried out by qPCR after 15 or 30 min of toxicant exposure. At 15 min, while nuoF and ndh expression was repressed and slightly induced, respectively, that of sdhB (which encodes the succinate dehydrogenase B subunit) showed no apparent changes (Table 2, Fig. S3a). In addition, while fnr and arcA genes (related to aerobic/anaerobic respiration regulation) showed a faint repression, napA (encodes the major subunit of cytoplasmic nitrate reductase) and narZ (encoding the α-subunit of nitrate reductase Z) showed no expression changes. The only exception was narG, encoding a nitrate reductase involved in tellurite reduction at the membrane (Avazeri et al. 1997), whose expression was enhanced over sixfold. These results could be explained since, in addition to the important role of NDH-II in aerobic respiration, the enzyme continues functioning during nitrate respiration (Jackson et al. 2004). The fact that NDH-II and NarG are capable of tellurite reduction is indicative that the toxicant is being continuously reduced at the membrane during aerobic respiration. On the other hand and as expected, soxS expression was also found enhanced (fivefold), which most probably reflects the activation of the antioxidant machinery as consequence of tellurite exposure.
After 30 min of toxicant exposure, ndh showed an important over expression which may explain the observed increase of tellurite reduction (Figs. 1b, 4b). While the relative expression of fnr, arcA, narG and narZ did not show almost any change that of napA was highly induced (6.4-fold) (Table 2, Fig. S3b). To date, it is not known if the periplasmic nitrate reductase NapABC is able of tellurite reduction. Most probably pyruvate accumulation occurring upon tellurite exposure (Valdivia-González et al. 2012), makes the PDH repressor no longer blocking ndh and pyruvate dehydrogenase complex genes (aceE, aceF and lpd) transcription (Ogasawara et al. 2007), thus allowing to maintain the required oxidized NAD+ pool for pyruvate oxidative metabolism. It is worth to notice that the Lpd dihydrolipoamide dehydrogenase is also a tellurite reductase (Castro et al. 2008). In turn, soxS showed a lower induction than that observed at 15 min, suggesting that genes related to the oxidative stress response reach a maximal expression at short times of toxicant exposure.
Finally, in this communication we showed that NADH dehydrogenase and tellurite reductase activities of E. coli NDH-II are not affected by tellurite. However, the enzyme can still be harmful because of its ability to generate superoxide while reducing the toxicant. Additional experiments to address this issue are being carried out in our laboratory.
References
Arenas FA, Díaz WA, Leal CA, Pérez-Donoso JM, Imlay JA et al (2010) The Escherichia coli btuE gene, encodes a glutathione peroxidase that is induced under oxidative stress conditions. Biochem Biophys Res Commun 398:690–694
Arenas FA, Covarrubias PC, Sandoval JM, Pérez-Donoso JM, Imlay JA et al (2011) The Escherichia coli BtuE protein functions as a resistance determinant against reactive oxygen species. PLoS ONE 6:e15979
Avazeri C, Turner RJ, Pommier J, Weiner J, Giordano G et al (1997) Tellurite reductase activity of nitrate reductase is responsible for the basal resistance of Escherichia coli to tellurite. Microbiology 143:1181–1189
Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y et al (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008
Baesman SM, Bullen TD, Dewald J, Zhang D, Curran S et al (2007) Formation of tellurium nanocrystals during anaerobic growth of bacteria that use Te oxyanions as respiratory electron acceptors. Appl Environ Microbiol 73:2135–2143
Borsetti F, Tremaroli V, Michelacci F, Winterstein C, Daldal F et al (2005) Tellurite effects on Rhodobacter capsulatus cell viability and superoxide dismutase activity under oxidative stress conditions. Res Microbiol 156:807–813
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Cairnes DD (1911) Canadian-containing ores. J Can Min Inst 14:185–202
Calderón IL, Arenas FA, Pérez JM, Fuentes DE, Araya MA et al (2006) Catalases are NAD(P)H-dependent tellurite reductases. PLoS ONE 1:e70
Calderón IL, Elías AO, Fuentes EL, Pradenas GA, Castro ME et al (2009) Tellurite-mediated disabling of [4Fe–4S] clusters of Escherichia coli dehydratases. Microbiology 155:1840–1846
Calhoun MW, Oden KL, Gennis RB, de Mattos MJ, Neijssel OM (1993) Energetic efficiency of Escherichia coli: effects of mutations in components of the aerobic respiratory chain. J Bacteriol 175:3020–3025
Castro ME, Molina R, Díaz W, Pichuantes S, Vásquez CC (2008) The dihydrolipoamide dehydrogenase of Aeromonas caviae ST exhibits NADH-dependent tellurite reductase activity. Biochem Biophys Res Commun 75:91–94
Castro ME, Molina R, Díaz W, Pradenas G, Vásquez CC (2009) Expression of Aeromonas caviae ST pyruvate dehydrogenase complex components mediate tellurite resistance in Escherichia coli. Biochem Biophys Res Commun 380:148–152
Chasteen TG, Fuentes DE, Tantaleán JC, Vásquez CC (2009) Tellurite: history, oxidative stress, and molecular mechanisms of resistance. FEMS Microbiol Rev 33:820–832
Contreras NP, Vásquez CC (2010) Tellurite-induced carbonylation of the Escherichia coli pyruvate dehydrogenase multienzyme complex. Arch Microbiol 192:969–973
Fischer W (2001) Second note on the term “chalcogen”. J Chem Ed 78:1333
Hayashi M, Miyoshi T, Takashina S, Unemoto T (1989) Purification of NADH-ferricyanide dehydrogenase and NADH-quinone reductase from Escherichia coli membranes and their roles in the respiratory chain. Biochim Biophys Acta 977:62–69
Hefti MH, Vervoort J, van Berkel WJ (2003) Deflavination and reconstitution of flavoproteins. Eur J Biochem 270:4227–4242
Imlay JA (2003) Pathways of oxidative damage. Annu Rev Microbiol 57:395–418
Imlay JA (2006) Iron–sulphur clusters and the problem with oxygen. Mol Microbiol 59:1073–1082
Jackson L, Blake T, Green J (2004) Regulation of ndh expression in Escherichia coli by Fis. Microbiology 150:407–413
Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E et al (2005) Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res 12:291–299
Lin ECC (1976) Glycerol dissimilation and its regulation in bacteria. Annu Rev Microbiol 30:535–578
Lloyd-Jones G, Osborn AM, Ritchie DA, Strike P, Hobman JL et al (1994) Accumulation and intracellular fate of tellurite in tellurite-resistant Escherichia coli: a model for the mechanism of resistance. FEMS Microbiol Lett 118:113–119
Lohmeier-Vogel EM, Ung S, Turner RJ (2004) In vivo 31P nuclear magnetic resonance investigation of tellurite toxicity in Escherichia coli. Appl Environ Microbiol 70:7342–7347
Matsushita K, Ohnishi T, Kaback HR (1987) NADH-ubiquinone oxidoreductases of the Escherichia coli aerobic respiratory chain. Biochemistry 26:7732–7737
Messner KR, Imlay JA (1999) The identification of primary sites of superoxide and hydrogen peroxide formation in the aerobic respiratory chain and sulfite reductase complex of Escherichia coli. J Biol Chem 274:10119–10128
Nakamura K, Yamaki M, Sarada M, Nakayama S, Vibat C et al (1996) Two hydrophobic subunits are essential for the Heme b ligation and functional assembly of complex II (succinate-ubiquinone oxidoreductase) from Escherichia coli. J Biol Chem 271:521–527
Ogasawara H, Ishida Y, Yamada K, Yamamoto K, Ishihama A (2007) PdhR (pyruvate dehydrogenase complex regulator) controls the respiratory electron transport system in Escherichia coli. J Bacteriol 189:5534–5541
Pérez JM, Calderón IL, Arenas FA, Fuentes DE, Pradenas GA et al (2007) Bacterial toxicity of potassium tellurite: unveiling an ancient enigma. PLoS ONE 2:e211
Pérez JM, Arenas FA, Pradenas GA, Sandoval JM, Vásquez CC (2008) Escherichia coli YqhD exhibits aldehyde reductase activity and protects from the harmful effect of lipid peroxidation-derived aldehydes. J Biol Chem 283:7346–7353
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Pradenas GA, Paillavil BA, Reyes-Cerpa S, Pérez-Donoso JM, Vásquez CC (2012) Reduction of the monounsaturated fatty acid content of Escherichia coli results in increased resistance to oxidative damage. Microbiology 158:1279–1283
Pradenas GA, Díaz-Vásquez WA, Pérez-Donoso JM, Vásquez CC (2013) Monounsaturated fatty acids are substrates for aldehyde generation in tellurite-exposed Escherichia coli. Biomed Res Int. doi:10.1155/2013/563756
Puustinen A, Finel M, Haltia T, Gennis R, Wikström M (1991) Properties of the two terminal oxidases of Escherichia coli. Biochemistry 30:3936–3942
Rapisarda VA, Montelongo LR, Farías RN, Massa EM (1999) Characterization of an NADH-linked cupric reductase activity from the Escherichia coli respiratory chain. Arch Biochem Biophys 370:143–150
Rapisarda VA, Chehín RN, De Las Rivas J, Rodríguez-Montelongo L (2002) Evidence for Cu(I)-thiolate ligation and prediction of a putative copper-binding site in the Escherichia coli NADH dehydrogenase-2. Arch Biochem Biophys 405:87–94
Reinoso CA, Auger C, Appanna VD, Vásquez CC (2012) Tellurite-exposed Escherichia coli exhibits increased intracellular alpha-ketoglutarate. Biochem Biophys Res Commun 421:721–726
Reinoso CA, Appanna VD, Vásquez CC (2013) α-Ketoglutarate accumulation is not dependent on isocitrate dehydrogenase activity during tellurite detoxification in Escherichia coli. Biomed Res Int. doi:10.1155/2013/784190
Rodríguez-Montelongo L, Volentini SI, Farías RN, Massa EM, Rapisarda VA (2006) The Cu(II)-reductase NADH dehydrogenase-2 of Escherichia coli improves the bacterial growth in extreme copper concentrations and increases the resistance to the damage caused by copper and hydroperoxide. Arch Biochem Biophys 451:1–7
Sabaty M, Avazeri C, Pignol D, Vermeglio A (2001) Characterization of the reduction of selenate and tellurite by nitrate reductases. Appl Environ Microbiol 67:5122–5126
Sandoval JM, Arenas FA, Vásquez CC (2011) Glucose-6-phosphate dehydrogenase protects Escherichia coli from tellurite-mediated oxidative stress. PLoS ONE 6:e25573
Semchyshyn H, Bagnyukova T, Storey K, Lushchak V (2005) Hydrogen peroxide increases the activities of soxRS regulon enzymes and the levels of oxidized proteins and lipids in Escherichia coli. Cell Biol Int 29:898–902
Tantaleán JC, Araya MA, Saavedra CP, Fuentes DE, Pérez JM et al (2003) The Geobacillus stearothermophilus V iscS gene, encoding cysteine desulfurase, confers resistance to potassium tellurite in Escherichia coli K-12. J Bacteriol 185:5831–5837
Taylor DE (1999) Bacterial tellurite resistance. Trends Microbiol 7:111–115
Taylor DE, Walter EG, Sherburne R, Bazett-Jones DP (1988) Structure and location of tellurium deposited in Escherichia coli cells harbouring tellurite resistance plasmids. J Ultrastruct Mol Struct Res 99:18–26
Tremaroli V, Fedi S, Zannoni D (2007) Evidence for a tellurite-dependent generation of reactive oxygen species and absence of a tellurite-mediated adaptive response to oxidative stress in cells of Pseudomonas pseudoalcaligenes KF707. Arch Microbiol 187:127–135
Tremaroli V, Workentine ML, Weljie AM, Vogel HJ, Ceri H et al (2009) Metabolomic investigation of the bacterial response to a metal challenge. Appl Environ Microbiol 75:719–728
Trutko SM, Suzina NE, Duda VI, Akimenko VK, Boronin AM (1998) Participation of the bacterial respiratory chain in reduction of potassium tellurite. Dokl Akad Nauk 358:836–838
Trutko SM, Akimenko VK, Suzina NE, Anisimova LA, Shlyapnikov MG et al (2000) Involvement of the respiratory chain of Gram-negative bacteria in the reduction of tellurite. Arch Microbiol 173:178–186
Turner RJ, Weiner JH, Taylor DE (1999) Tellurite-mediated thiol oxidation in Escherichia coli. Microbiology 145:2549–2557
Turner RJ, Aharonowitz Y, Weiner JH, Taylor DE (2001) Glutathione is a target in tellurite toxicity and is protected by tellurite resistance determinants in Escherichia coli. Can J Microbiol 47:33–40
Unden G, Bongaerts J (1997) Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim Biophys Acta 1320:217–234
Valdivia-González M, Pérez-Donoso JM, Vásquez CC (2012) Effect of tellurite-mediated oxidative stress on the Escherichia coli glycolytic pathway. Biometals 25:451–458
Volentini S, Farías R, Rodríguez-Montelongo L, Rapisarda VA (2011) Cu(II)-reduction by Escherichia coli cells is dependent on respiratory chain components. Biometals 24:827–835
Wang X, Guangfei L, Zhou J, Wang J, Roufei J et al (2011) Quinone-mediated reduction of selenite and tellurite by Escherichia coli. Biores Technol 102:3268–3271
Young IG, Jaworowski A, Poulis MI (1978) Amplification of the respiratory NADH dehydrogenase of Escherichia coli by gene cloning. Gene 4:25–36
Acknowledgments
The authors thank Dr. J.A. Imlay for providing some of the microorganisms detailed in Table 1. This work received financial support from FONDECYT (Fondo Nacional de Ciencia y Tecnología) 1090097 and DICYT (Dirección de Investigación en Ciencia y Tecnología, Universidad de Santiago de Chile) (CCV). A doctoral fellowship and Grant No. 24121087 from CONICYT (Comisión Nacional de Ciencia y Tecnología) to WAD-V is also acknowledged. Support from the Robert A. Welch Foundation (X-011) is also gratefully acknowledged for the work at Sam Houston State University.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10534_2013_9701_MOESM1_ESM.doc
Black deposits in membrane fractions from tellurite-exposed E. coli. a membrane fraction sediments from cells grown in the absence (control) or presence of the toxicant (tellurite). b SEM image of membrane vesicles of E. coli BW25113. A representative vesicle is indicated (arrow)
10534_2013_9701_MOESM2_ESM.doc
Purification of E. coli NDH-II. NDH-II and E3 (positive control for TR activity) were purified by affinity chromatography as described in “Materials and methods” section. a SDS-PAGE of the purified enzymes. b NADH dehydrogenase activity of NDH-II previously incubated or not with 20 μM FAD. Bars represent the average of three independent trials ± SE. P < 0.05 (*)
10534_2013_9701_MOESM3_ESM.doc
Relative expression of genes involved in aerobic/anaerobic respiration, respiration regulation and oxidative stress response in E. coli exposed to sub-lethal tellurite concentrations. a, b gene expression after 15 or 30 min of tellurite exposure, respectively. The rpoD gene was used as the housekeeping control (baseline). The data represent the average of three independent trials SE
Rights and permissions
About this article
Cite this article
Díaz-Vásquez, W.A., Abarca-Lagunas, M.J., Arenas, F.A. et al. Tellurite reduction by Escherichia coli NDH-II dehydrogenase results in superoxide production in membranes of toxicant-exposed cells. Biometals 27, 237–246 (2014). https://doi.org/10.1007/s10534-013-9701-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-013-9701-8