
Abstract
Pregnant rats were treated with 0.4% lead acetate through drinking water from 6th day of gestation and this treatment was continued till 21 post natal days (PND). Four regions of the brain namely hippocampus, cerebellum, frontal cortex and brain stem were dissected at 10, 20, 30 and 40 PND for estimation of lipid peroxidation products (LPP), catalase (CAT) and superoxide dismutase (SOD). The results indicate that there was a significant (P < 0.05) increase of LPP in exposed rats than their corresponding control at 10, 20 and 30 PND both in hippocampus and cerebellum. At PND 40, the LPP of control and exposed were found to be almost same in both the tissues indicating recovery from lead toxicity. CAT activity was significantly (P < 0.05) high in hippocampus of exposed rats up to PND 30 but up to PND 20 in cerebellum and frontal cortex. However, in brain stem, a significant (P < 0.05) increase in CAT activity was observed only at PND 10. A significant (P < 0.05) increase in SOD activity was observed up to PND 30 both in hippocampus and cerebellum on lead exposure. Frontal cortex exhibited a similar significant (P < 0.05) increase of SOD activity up to PND 20 and for brain stem up to PND 10. There was no significant change in the activity of antioxidant enzymes (CAT and SOD) and LPP in all the four brain tissues of control and exposed rats at PND 40 indicating recovery from lead-induced oxidative stress.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lead is a ubiquitous environmental and industrial pollutant that has been detected in every facet of environmental and biological systems. Lead can be found in water pipes, insecticides, lining of equipment where corrosion resistance and pliability are required, in petroleum refining, in construction, bullets of gun, x-ray and atomic radiation protection and is a major industrial byproduct. The manipulation of lead for these uses has caused lead contamination of air, dust, and soil. Lead has been known to cause mental retardation and learning disorders (Burdette and Goldstein 1986; Toscano and Guilarte 2005; Garza et al. 2006). Lead antagonizes biological systems by attracting strong oxidizers (O2) and active metals (sodium, potassium) and inevitably disrupting normal cellular metabolism. This interference leads to the generation of highly reactive oxygen species (ROS), namely superoxides (O2), hydrogen peroxide (H2O2), hydroxyl radicals (OH−) and lipid peroxides causing damage to cellular components including proteins, membrane lipids, and nucleic acids (Halliwell and Gutteridge 1989). One of the proposed mechanisms for lead toxicity is that lead-induced oxidative stress contributes to the deleterious effects by disrupting the delicate prooxidant/antioxidant balance that exists within mammalian cells (Lima-Hermes et al. 1991; Monteiro et al. 1995). Lead has been found to produce a wide range of toxic biochemical effects, besides behavioral dysfunction in man and in experimental animals (Klassen 1990). The antioxidant enzymes released into the body give protection against the detrimental effects of the reactive oxygen species (ROS). The neurodegenerative diseases like Parkinson’s (Zhang et al. 2000) and Alzheimer’s (Selkoe 2001) are also reported to be caused by excessive generation of ROS and also developmental exposure to xenobitoic metals like lead (Zawia and Basha 2005; Bolin et al. 2006). In a normal cell there is a delicate prooxidant/antioxidant balance that prevails and if this balance is shifted towards prooxidants, this state can be termed as “oxidative stress”. This can result in serious cell damage if the stress is massive or prolonged. Antioxidant enzymes namely superoxide dismutase and catalase are important in the preservation of homeostasis for normal cell function.
Burdette and Goldstein (1986) observed that lead exerts its most severe consequences in the developing brain and this fact can be attributed to the intense cellular proliferation and synaptogenesis which takes place in the developing brain. Lockitch (1993) reported that the developing organism presents a five fold greater absorption of lead than the adult organism. According to Goyer (1993), this is due to lack of a functional blood brain barrier in the developing organism. Moreira et al. (2001) found evidence that prenatal exposure to low levels of lead has been involved in behavioral and neurochemical alterations detected in both suckling and adult rats. Low and high levels of lead exposure lactactionally resulted in alterations in cholinergic system (Reddy et al. 2006) and catecholamines (Devi et al. 2005) in different regions of developing rat brain. There has been much research on developing organisms and lead has its own importance because of its effects seen in children. The available research (Villeda-Hernandez et al. 2001) also shows that oxidative stress can cause damage to biomolecules by free radical attachment to polyunsaturated fatty acid (PUFA) side chains in cells and this leads to lipid peroxidation. The lipid peroxidation of cells produces malondialdehyde, which is toxic in nature.
Though there are some conflicting results in the literature, most of the investigations reported that lead induces oxidative stress and oxidative stress in turn causes the pathogenicity (Villeda-Hernandez et al. 2006). Ercal et al. (1996) and Gurer et al. (1998) through their investigations suggest oxidative stress as one of the important mechanisms of the toxic effects of lead. Halliwell (1994) and Adonaylo and Oteiza (1999) have also indicated that oxidative stress contributes to Pb-associated tissue injury in the brain and other organs. Moreover, it is widely accepted that oxidative stress causes the lipid peroxidation of PUFA side chains, which in turn produces the toxic substance malonaldehyde that interferes with normal cell function. It is clear from the literature that both low (0.2%) and high (1%) levels of lead have been reported to cause oxidative stress and other neurochemical perturbations in developing rat brain. However, in the present investigation, an attempt has been made to study the effect of moderate levels (0.4%) of lead on lipid peroxidation products and antioxidant enzymes in different regions of developing rat brain.
Materials and methods
Chemicals
All biochemicals and lead acetate were obtained from Sigma Chemical Co. (St. Louis, MO).
Treatment
Wistar female and male rats weighing approximately 100–120 g were obtained from Ghosh Enterprises, Calcutta (India). They were maintained in the animal facility for about four days before use. They were given a commercial feed (Hindustan Lever Ltd., Mumbai, India) and water ad libitum. Then, they were allowed to mate in the ratio of two females to one male and the pregnancy was confirmed by looking the swollen vaginal swabs and observing the presence of viable sperms in the vaginal smear. After pregnancy confirmation, the mother rats were given 0.4% of lead acetate dissolved in double distilled water after sixth day of pregnancy. The control group received an equal amount of sodium acetate through drinking water. Sodium acetate was also dissolved in distilled water. Both control and exposed groups were given free access to food. The pups were dissected at the intervals of 10, 20, 30 and 40 post natal days (PND) and the whole brains were extracted immediately. The brains were washed separately with ice-cold normal saline. Different regions of brain namely the frontal cortex, the cerebellum, the hippocampus and the brain stems were isolated on ice individually. Due to small amount of tissue, the hippocampus was pooled separately both in exposed and control animals. The tissues were immediately used for the estimation of lipid peroxidation products, catalase and superoxide dismutase as described below:
Lipid peroxidation products
These products were estimated by the method of Hiroshi et al. (1979). Briefly, the homogenate (10%) of the different isolated tissues was prepared in 1.5% potassium chloride solution. One milliliter of the homogenate was added to 2.5 ml of 20% trichloroacetic acid (TCA). The mixture was centrifuged at 3,500 rpm for 10 min at 4°C. The pellet was then dissolved in 0.05 M sulphuric acid and 3 ml of 2 M thiobarbituric acid was added to it. The test tubes were incubated in boiling water bath for 30 min. The contents were cooled and the color was extracted into 4 ml of n-butanol. The colour was read at 530 nm using a spectrophotometer against the blank. The results were expressed as micromoles of MDA formed/gm weight of tissue.
Catalase
The enzyme activity was determined by the method of Aebi and Packer (1984). Different regions of brain were homogenized separately in 0.05 M phosphate buffer (pH 7) containing 0.1 mM EDTA. The homogenate (10%) was centrifuged at 4,000 rpm for 15 min at 4°C. The supernatant was decanted and centrifuged at 16,000 rpm for 60 min at 4°C. The supernatant was used for the enzyme assay. The reaction mixture consisted of 100 μl of supernatant and 10 μl of alcoholic ethanol was added to it. This was vortexed well and kept on ice water bath for 10 min. Then, the tubes were brought to room temperature and 10 μl of Triton X-100 was added and vortexed well till the whole tissue extract was completely dissolved. Afterwards, 0.66 M hydrogen peroxide (H2O2) in phosphate buffer was prepared afresh at the time of reaction and 100 μl of this buffer containing H2O2 was added to the above reaction mixture and decrease in the absorbance was read in a spectrophotometer at 240 nm against a blank for 60 sec. Care was taken that the reactions were carried out in dark. The data were expressed as micromoles of H2O2 metabolized/mg protein/min.
Superoxide dismutase
The enzyme assay (total activity) was performed by using the method described by Beauchamp and Fridovich (1971). A 10% homogenate was prepared using a homogenizing buffer (100 mM phosphate buffer, pH 7.5) and then centrifuged at 4,000 rpm for 10 min at 4°C. The supernatant was decanted and then centrifuged at 16,000 rpm for 60 min at 4°C and the supernatant obtained during this spin was used for the assay. The reaction mixture contained 1.5 ml of 100 mM phosphate buffer (pH 7.5), 0.3 ml of 130 mM methionine, 0.3 ml of 750 mM nitroblue tetrazonium (NBT), 0.3 ml of 10 mM EDTA and 0.1 ml of enzyme source (supernatant). The reaction was started by addition of 0.1 ml riboflavin (60 mM) which was prepared afresh. After addition of the riboflavin, the tubes were placed under light for 30 min. A similar set of controls was maintained under dark condition. The optical density was read at 560 nm against the controls kept in dark condition. The results were reported as units/mg protein. One unit of enzyme activity was defined as the amount of enzyme that decreases the initial rate to 50% of its maximal value for the particular tissue being assayed.
Statistics
At least five samples were used for the assay and all the assays were done in triplicate. The mean and standard deviations were calculated using standard statistical procedures. Comparison between control and exposed was made by following Student’s ‘t’ test Snedecor and Cochran (1967) and significant differences were calculated at P < 0.05
Results
Different regions of brain
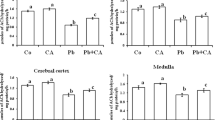
The results on lipid peroxidation products, catalase and superoxide dismutase for hippocampus, cerebellum, frontal cortex, brain stem were presented in Figs. 1–3 respectively.

Lipid peroxidation products in different regions of developing rat brain on exposure to lead. Each value represents the mean ± standard deviation. *Significantly different from its respective control at P < 0.05

Catalase activity in different regions of developing of rat brain on exposure to lead. Each value represents the mean ± standard deviation. *Significantly different from its respective control at P < 0.05

Superoxide dismutase activity in different regions of developing of rat brain on exposure to lead. Each value represents the mean ± standard deviation. *Significantly different from its respective control at P < 0.05
Hippocampus
The data on lipid peroxidation products (LPP) were presented in Fig. 1 and there was a significant (P < 0.05) increase in LPP up to 30 PND in the exposed rats when compared to their respective controls. These levels almost resumed to normal level of the control at 40 PND (Fig. 1). These results indicate that Pb induces lipid peroxidation up to 30 PND. The catalase activity also showed a gradual and significant (P < 0.05) increase in the exposed rats up to 30 PND with respect to their controls (Fig. 2). However, the activity remained almost the same as that of control at 40 PND. There was a significant (P < 0.05) increase in superoxide dismutase activity of exposed rats at 10, 20 and 30 PND (Fig. 3). The enzyme activities for both control and exposed were found to be almost the same at 40 PND (Fig. 3).
Cerebellum
The LPP showed a gradual and significant (P < 0.05) increase in their levels up to 30 PND and resumed to the levels of control at 40 PND (Fig. 1). There was an increase in catalase activity at 10 and 20 PND with respect to their controls and this increase was also significant (P < 0.05) (Fig. 2). Later at 30 and 40 PND, the enzyme levels also showed a marginal increase but not significant resuming to their normal levels. A significant (P < 0.05) enhancement in superoxide dismutase activity was observed at 10, 20 and 30 PND for exposed rats compared to their respective controls (Fig. 3). However, there was not much difference in the enzyme activities between control and exposed rats at 40 PND.
Frontal cortex
There was a little and significant (P < 0.05) increase in LPP levels at 10 PND of exposed rats in comparison with controls (Fig. 1). At 20, 30, 40 PND, these levels were almost found to be the same with out much difference both in control and exposed rats. The increase in catalase activity was significant (P < 0.05) at 10 and 20 PND of exposed rats with respect to their controls (Fig. 2). Later, the enzyme activity resumed to their control value at 30 and 40 PND. There was an initial significant (P < 0.05) increase in superoxide dismutase activity at 10 and 20 PND of exposed rats in comparison to their controls (Fig. 3). However, their activities resumed gradually to their control levels at 30 and 40 PND.
Brain stem
There was a significant (P < 0.05) increase in LPP levels at 10 PND (Fig. 1) of the exposed rats with respect to control but the levels resumed to normal from 20 PND showing not much difference between control and exposed rats. The catalase activity showed an initial and significant (P < 0.05) increase at 10 PND (Fig. 2) but however their levels are not significant from 20 PND onwards both in the control and exposed rats. The superoxide dismutase activity had an initial and significant (P < 0.05) increase in their levels at 10 PND compared with their respective control (Fig. 3) but the levels have not showed any significant increase from 20 PND onwards with respective to their controls.
Discussion
Lead exposure resulted in oxidative stress and this was well extrapolated from the increase in lipid peroxidation products (LPP). An increase in lipid peroxides damages various cellular components of brain. Lipid peroxidation products increased in all four regions of the brain from PND 10–PND 40 with significant values (P < 0.05) at 10, 20, and 30 PND in the cerebellum and hippocampus compared to their respective controls. The hippocampus was of primary concern due to its role in learning and the formation of memory and lead is thought to inhibit learning and memory (Altmann et al. 1993). The values in these regions incriminate oxidative stress as a mechanism of lead toxicity. However, further tests are needed to confirm. There were no significant values recorded for the brain stem except at 10 PND. Significant values (P < 0.05) were recorded in the frontal cortex at 10 and 20 PND. The values of LPP in control and exposed rats were almost the same at 40 PND indicating recovery from Pb toxicity. Similar Pb-induced disruption through free radical involvement was reported by Jiun and Hsien (1994) and West et al. (1994). Our results also corroborate well with that of Bachara et al. (1993), Oteiza et al. (1995) and Adonaylo and Oteiza (1999) who demonstrated that lead increases the rate of lipid peroxidation in brain. An enhancement of lipid peroxidation was reported in rat brain homogenates by Gerber and Maes (1978). Shafiqu-ur-Rehman (1984) and Rehman et al. (1995) also demonstrated lipid peroxidative damage with increase in lead concentration in different regions of brain. According to Kodavanti (1999) the central nervous system is vulnerable to reactive oxygen species (ROS)-mediated injury because of a high rate of oxidative metabolic activity, high concentration of readily oxidizable substances (membrane polyunsaturated fatty acids), and endogenous generation of ROS by specific neurochemical reactions and high ratio of membrane surface area to cytoplasmic volume.
Catalase activity was significantly (P < 0.05) high in hippocampus of exposed rats as compared with their respective controls up to PND 30. A similar increase in catalase was observed in cerebellum and frontal cortex but only up to 20 PND. However, brain stem showed a significant (P < 0.05) increase only at 10 PND. The SOD activity was significantly high in hippocampus and cerebellum of exposed rats as compared to their respective controls up to 30 PND. Then recovery from lead toxicity was observed at PND 40 with almost similar values for control and exposed. In the frontal cortex, lead treatment resulted in significant (P < 0.05) increase of SOD up to 20 PND whereas brain stem showed a significantly (P < 0.05) high value only at 10 PND. Sandhir et al. (1994) also demonstrated different responses in different regions of brain exposed to lead. According to Patra (2001) and Gurer et al. (1998) the pro-oxidant role of lead is not known but the inhibition of delta amino levulinic acid dehydratase causes accumulation of delta amino levulinic acid which on auto-oxidation results in the formation of superoxide and hydrogen peroxide. While studying the protective role of alpha lipoic acid, Gurer et al. (1999) reported a similar increase in catalase and SOD activities in rats under lead-induced oxidative stress.
There was no significant change in LPP or antioxidant enzymes (SOD, CAT) in all regions of brain at PND 40 indicating a recovery from lead-induced oxidative stress.
In conclusion, the investigation suggests that the increase in LPP and antioxidant enzymes such as superoxide dismutase and catalase were more in hippocampus and cerebellum compared to frontal cortex and brain stem. Overall, lead disturbs pro-and anti-oxidative balance in the brain causing oxidative stress which might explain the neurotoxic nature of lead.
References
Adonaylo VN, Oteiza PI (1999) Lead promotes lipid oxidation and alterations in membrane physical properties. Toxicology 132:19–32
Aebi H, Packer L (1984) Catalase in vitro. Methods Enzymol 105:121–126
Altmann L, Weinsberg F, Seinsson L, Llienthal H, Wiegand H, Winneke G (1993) Impairment of long term potentiation and learning following chronic lead. Toxicol Lett 66:105–112
Bachara EJH, Mediros MHG, Monteiro HP, Hermes-Lima M, Pereira B, Demasi M, Costa CA, Abdullah DSP, Onuki J, Wendel CMA, Di Mascio P (1993) A free radical hypothesis of lead poisoning and inborn porphyries associated with 5-aminolevulenic acid overload. Quirn Nova 16:385–392
Beauchamp PC, Fridovich I (1971) Superoxide dismutase: improved assay and an assay applicable to PAGE. Analyt Biochem 44:276–287
Bolin CM, Basha R, Cox D, Zawia NH, Maloney B, Lahiri DK, Cardozo-Palaez F (2006) Exposure to lead and the developmental origin of oxidative DNA damage in the aging brain. FASEB J 20:788–790
Burdette LJ, Goldstein R (1986) Long term behavioural and electrophysiological changes associated with lead exposure at different stages of brain development in the rat. Dev Brain Res 29:101–110
Devi CB, Reddy GH, Prasanthi RP, Chetty CS, Reddy GR (2005) Developmental lead exposure alters mitochondrial monoamine oxidase and synaptosomal catecholamine levels in rat brain. Int J Dev Neurosci 23:375–381
Ercal N, Teratphan RH, Grannermann NH, Spitz DR (1996) In vivo indices of oxidative stress in lead induced C57BL/6 mice are reduced by the treatment with meso-2 3-dimercatosuccinic acid or N-acetyl cysteine. Free Rad Biol Med 21:157–161
Garza A, Vega R, Soto E (2006) Cellular mechanisms of lead neurotoxicity. Med Sci Monit 12:57–65
Gerber GB, Maes J (1978) Increased ALA dehydratase activity and spleen weight in lead intoxicated rats. A consequence of increased blood cell destruction. J Experimentia 34:381–382
Goyer RA (1993) Lead toxicity: current concerns. Environ Health Perspect 86:177–181
Gurer H, Neal R, Yang P, Oztezcan S, Ercal N (1999) Capotril as an antioxidant in lead—fisher 344 rats. Hum Exp Toxicol 18:27–32
Gurer H, Ozgunes H, Neal R, Spitzand DR, Ercal N (1998) Antioxidant effects of N-acetyl cysteine and succimer in red blood cells from lead exposed rats. Toxicology 128:181–189
Halliwell B (1994) Free radicals, antioxidants, and human disease: curiosity, cause and consequences. Lancet 344:721–724
Halliwell B, Gutteridge JMC (1989) Free radicals in biology and medicine. Clarendon press, Oxford
Hiroshi O, Ohisi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by TBA reaction. Anal Biochem 95:351–358
Jiun YS, Hsien LT (1994) Lipid peroxidation in workers exposed to lead. Arch Environ Health 49:256–259
Klassen CD (1990) Heavy metals and heavy metal antagonist. In: Gilman TW, Nies A, Taylor P (eds) Goodman and Gilman’s the pharmacological basis of therapeutics. Pergamon Press, New York, pp 1592–1614
Kodavanti PRS (1999) Reactive oxygen species and antioxidant homeostasis in neurotoxicology. In: Tilson HA, Harry GJ (eds) Neurotoxicology. Taylor & Francis Press, USA
Lima-Hermes M, Pereira B, Bechara EJH (1991) Are free radicals involved in lead poisoning? Xenobiotica 21:1085–1090
Lockitch G (1993) Perspectives on lead toxicity. Clin Biochem 26:371–381
Monteiro H, Abdullah D, Arcuri A, Bechara E (1995) Oxygen toxicity related to exposure to lead. Clin Chem 31:1673–1676
Moreira EG, Vassilieff I, Vassilieff VS (2001) Developmental lead exposure: behaviour alterations in the short and long term. Neurotoxicol Teratol 23:489–495
Oteiza PI, Verstraeten SV, Adonaylo VN (1995) Oxidative damage induced by metals without redox capacity in biological systems. Ci Cult 47:330–335
Patra RC, Swarup D, Dwivedi SK (2001) Antioxidant effects of alpha tocopherol, ascorbic acid and l-methionine on lead induced oxidative stress to the liver, kidney and brain in rats. Toxicology 162:81–88
Reddy GR, Devi BC, Chetty CS (2006) Developmental lead neurotoxicity: alterations in brain cholinergic system. Neurotoxicol (In Press)
Rehman S, Shafiq-ur-Rehman O, Chandre O, Abdullah M (1995) Evaluation of malondialdehyde as an index of lead damage in rat brain homogenates. Biometals 8:275–279
Sandhir R, Julka D, Gill KD (1994) Lipoperoxidative damage on lead treatment in rat brain and its implications on membrane bound enzymes. Pharmacol Toxicol 74:66–71
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins and therapy. Physiol Rev 81:741–766
Snedecor GW, Cochran WG, (1967) Statistical methods. The Iowa State University Press, Iowa
Toscano CD, Guilarte TR (2005) Lead neurotoxicity: from exposure to molecular effects. Brain Res Rev 49:529–554
Villeda-Hernandez J, Barroso-Moguel R, Mendez-Armenta M, Nava-Ruiz C, Huerta-Romero R, Rios C (2001) Enhanced brain regional lipid peroxidation in developing rats exposed to low level lead acetate. Brain Res Bull 55:247–251
Villeda-Hernandez J, Mendez Armenta M, Barroso-Moguel R, Trejo-Solis MC, Guevara J, Rios C (2006) Morphometric analysis of brain lesions in rat fetuses prenatally exposed to low-level lead acetate: correlation with lipid peroxidation. Histol Histopathol 21:609–617
West WL, Knight EM, Edwards CH, Menning M, Spurlock B, James H, Johnson AA, Oyemade UJ, Jackson C, Westney LS (1994) Maternal low level lead and pregnancy outcomes. J Nutri 124:981–996
Shafiqu-ur-Rehman S (1984) Lead induced regional lipid peroxidation in brain. Toxicol Lett 21:333–337
Zawia NH, Basha MR (2005) Environmental risk factors and the developmental basis for Alzheimer’s disease. Rev Neurosci 16:325–337
Zhang Y, Dawson VL, Dawson TM, 2000 Oxidative stress and genetics in the pathogenesis of Parkinson’s disease. Neurobiol Dis 7:240–250
Acknowledgements
This research work was supported by NIH/FIC/MIRT #00132, NIH/NIGMS/MBRS-SCORE #GM55356 awarded to Bettaiya Rajanna at Alcorn State University, Mississippi, USA. Erika Brown and Rashidi McCormick are undergraduate student participants in MIRT Program. The authors thank the authorities of Andhra University for providing research facilities for this international collaboration of undergraduate student research training program.
Author information
Authors and Affiliations
Corresponding author
Additional information
This research work was presented as a poster in Annual Biomedical Research Conference for Minority Students (ABRCMS) at Dallas, Texas, USA, during November 10–13, 2004 and the abstract was printed on page 231 of the Conference Proceedings
Rights and permissions
About this article
Cite this article
Bokara, K.K., Brown, E., McCormick, R. et al. Lead-induced increase in antioxidant enzymes and lipid peroxidation products in developing rat brain. Biometals 21, 9–16 (2008). https://doi.org/10.1007/s10534-007-9088-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-007-9088-5