
Abstract
Reduction of atmospheric sulfur (S) deposition and recovery of terrestrial ecosystems from acidification are matters of concerns in Asia. The Lake Ijira catchment (IJR) is located in the downwind region of the Chukyo Industrial Area in central Japan and has historically experienced large-scale deposition of S and nitrogen (N) from the atmosphere. Long-term monitoring data on the river water (RW) chemistry since 1988 were assessed with intensive-survey datasets on the input–output material budgets and S isotopic analysis (δ34S). Previous studies have suggested that IJR was acidified and N-saturated, which was triggered by climatic anomalies (cold summer and drought summer) in the mid-1990s. Analysis shows that with a decline in \( {\text{NO}}_{3}^{ - } \) concentrations, RW pH recovered to its original level in the early 2000s. Reductions in atmospheric deposition, diminished effects of climatic anomalies, and forest management practices, have all contributed to RW chemistry recovery. Although the \( {\text{SO}}_{4}^{2 - } \) concentration in the RW has declined continuously after the peak in 1994, the mean \( {\text{SO}}_{4}^{2 - } \) output significantly exceeded the input; the 5-year means from 2010 to 2014 are 2.5 kmolc ha−1 year−1 and 0.86 kmolc ha−1 year−1, respectively. The mean δ34S values of \( {\text{SO}}_{4}^{2 - } \) in rainwater and soil solution at 20 cm depth were 4.3 ‰ and 3.5 ‰, respectively, and that in the RW was −13.2‰. The contributions of groundwater S to the RW S were estimated to range from 75 to 91%. Geological S with significantly low δ34S values largely contributed to the discrepancy of the input–output budget, while atmospheric S appeared to accumulate in soil as organic S. The tree-ring δ34S profile recorded historical changes in the atmospheric inputs in the region. With recovery of the RW, the relative contribution of atmospheric S became smaller. However, most of the atmospheric S is still retained in soil. Over the last few decades, RW chemistry has sensitively responded to changes in the atmospheric environment, including the atmospheric deposition of S and N and climatic anomalies, and as future changes are likely, long-term monitoring is essential.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sulfur (S) deposited from the atmosphere is retained in the soil–plant system in various forms, such as organic S and adsorbed \( {\text{SO}}_{4}^{2 - } \) (e.g., Likens et al. 2002), and the S accumulating capacities vary depending on the soil type (Tanikawa and Takenaka 1999; Tanikawa et al. 2009). Retention and release processes of \( {\text{SO}}_{4}^{2 - } \) in terrestrial ecosystems are accompanied by H+ transfer (Van Breemen et al. 1983). Additionally, changes in S biogeochemistry affect the mobilization of cations, including H+, Al3+, Ca2+, Mg2+, and K+, and the acidification of watersheds (Mitchell and Likens 2011). Therefore, reduction of emissions and recovery of terrestrial ecosystems from acidification remain matters of concern, even in Europe and North America where atmospheric S deposition has already declined significantly over the last decades (e.g., Mitchell et al. 2011, 2013; Vuorenmaa et al. 2017). Meanwhile, the atmospheric environment in Asia has been changing dynamically. SO2 emissions in Japan have been declining continuously since the significant peak in the 1960s/1970s (Smith et al. 2011; Kurokawa et al. 2013). In China, SO2 emissions increased significantly from the 1980s until a peak in 2006 and then started declining gradually (Ohara et al. 2007; Lu et al. 2011; Kurokawa et al. 2013). Responses of terrestrial ecosystems to the changing atmospheric environment should be investigated carefully in the region.
The Chukyo Industrial Area near Nagoya in central Japan is the largest industrial area in Japan, and the surrounding area has historically experienced industrially-derived air pollution, in particular in the 1950s/1960s. Effects of the atmospheric deposition on ecosystems have been suggested, mainly in central Japan (Kurita and Ueda 2006; Yamada et al. 2007; Matsubara et al. 2009; Nakahara et al. 2010; Ishida et al. 2015a, b). Long-term declining trends of river water (RW) pH have been found in the neighboring three prefectures of central Japan, namely Nagano in the 1980s/1990s (Kurita and Ueda 2006) and Gifu and Niigata in the 1990s (Matsubara et al. 2009), especially in the rivers draining from acid-sensitive lithologies, such as granite, rhyolite, and chert. These acidified rivers in central Japan were included in the high-risk areas in the risk map for surface water acidification developed by Yamashita et al. (2016). Moreover, the S isotopic ratio (δ34S) in tree rings collected near the Chukyo Industrial Area indicates effects of the large atmospheric inputs in the 1960s/1970s (Ishida et al. 2015a, b).
The Lake Ijira catchment (IJR) in Gifu Prefecture, located approximately 45 km north–northwest from the central part of Nagoya City and in the downwind region of the Chukyo Industrial Area, has historically experienced large-scale deposition of S and nitrogen (N) from the atmosphere (Yamada et al. 2007; Nakahara et al. 2010). The Ministry of the Environment of Japan (MOEJ) has been monitoring the chemical properties of the lake, inflowing rivers, and soil in IJR since 1988. The MOEJ has been monitoring wet deposition using a wet-only sampler since 1999, and it has also monitored air concentrations by a filter-pack method and automatic monitors to estimate dry deposition flux (Nakahara et al. 2010). The monitoring data above in IJR have been submitted to the Acid Deposition Monitoring Network in East Asia (EANET) and disclosed annually since 2000 in the EANET data report (e.g., EANET 2015), as well as in the national report of MOEJ (e.g., Ministry of the Environment 2014). Based on the monitoring data, Yamada et al. (2007) found that the pH of the lake and RW had declined with an increase of \( {\text{NO}}_{3}^{ - } \) concentrations since the mid-1990s. Successively, Nakahara et al. (2010) identified a rapid decline of soil pH within the catchment over the 1990s and an increase of river \( {\text{NO}}_{3}^{ - } \) concentrations during the growing season since the mid-1990s. They suggested that climatic anomalies altered the biogeochemical processes, resulting in the acidification and N saturation of IJR in the mid-1990s. Moreover, \( {\text{SO}}_{4}^{2 - } \) concentrations in the RW increased greatly during the period of acidification (Nakahara et al. 2010), and the possibility of S accumulation in the ecosystems has also been discussed (EANET 2011).
Although the SO2 emissions in the region have been declining recently (Lu et al. 2011; Kurokawa et al. 2013), it has not yet been determined how forest ecosystems in Japan have responded to this decline in S deposition. Duan et al. (2016) showed that leaching of \( {\text{SO}}_{4}^{2 - } \) is relatively low in many stream waters in Japan and southwestern China, even in areas with high S deposition, which indicates that soil has a high \( {\text{SO}}_{4}^{2 - } \) adsorption capacity and that desorption of \( {\text{SO}}_{4}^{2 - } \) may delay soil recovery from acidification. The dynamics of S in ecosystems should be monitored carefully during the process of recovery, as was suggested in the cases in Europe (Vuorenmaa et al. 2017) and North America (Mitchell et al. 2011). Therefore, as IJR has experienced high S and N deposition, the S dynamics need to be carefully evaluated.
Isotopic analysis is a useful tool for evaluating such dynamics. The δ34S value of \( {\text{SO}}_{4}^{2 - } \) in rainwater (RF) is a source-specific value, which is useful for assessing the origin of the S, such as coal, oil, and their producing areas (e.g., Ohizumi et al. 2016). Isotopic values of samples from forests, such as from soil solution, tree rings, and RW, have also been utilized to discuss S dynamics in ecosystems (e.g., Ishida et al. 2015a, b; Novák et al. 2000). Water precipitated in mountainous forest area penetrates downward into unsaturated soil as soil solution, mixes with groundwater present in the lower soil and underlying rocks, and then flows out as RW. The \( {\text{SO}}_{4}^{2 - } \) concentration and the δ34S value in the RW can be determined using the mixing ratio of different-source waters that may have different \( {\text{SO}}_{4}^{2 - } \) concentrations and/or S isotopic values. A mixing model using the δ34S values can estimate contributions of the respective end members (such as RF, soil solution, and groundwater) to the RW (e.g. Hosono et al. 2007). Moreover, δ34S values in tree rings record historical changes in the S-cycle within soil–plant systems within forests, which also clearly reflects changes in the origin of S deposition (Ishida et al. 2015a, b).
In this paper, we synthetically assess monitoring data on atmospheric deposition and RW chemistry over the last few decades and recent integrated surveys providing input–output material budgets and S isotopic ratios. Based on these data, we clarify the response of RW in IJR to changes in the atmospheric environment, including the atmospheric deposition of S and N and climatic anomalies, and how atmospheric S has behaved in the ecosystem and contributed to the RW chemistry.
Methods
Study site
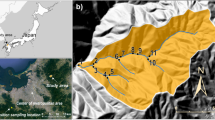
Lake Ijira (35:34:02.7 N 136:41:58.7 E) is a reservoir in Yamagata City, Gifu Prefecture, central Japan. The Kamagatani River, which flows into the lake, was mainly investigated in this study (Fig. 1). The bedrock of the catchment area (298 ha, within the bold line in Fig. 1) is mostly chert that was mainly formed in the Middle Triassic to Early Jurassic periods (Yoshida and Wakita 1999) but is also part of the melanges formed during the Late Jurassic to Early Cretaceous periods in the Mino Terrane (Wakita 1988). The Mino Terrane in Gifu and Shiga Prefectures is an accretionary wedge sedimentary complex that is connected to the Tanba Terrane in Kyoto and Hyogo Prefectures. Together, these terranes form a large sedimentary complex belt from central to western Japan called the Mino–Tanba Terrane. The dominant soil type is Cambisols. The dominant vegetation is Japanese cypress (Chamaecyparis obtusa Sieb. & Zucc., 49% of the catchment area), followed by broadleaf trees (a mixture of several species, 29%), Japanese red pine (Pinus densiflora Sieb. & Zucc., 13%), and Japanese cedar (Cryptomeria japonica D. Don, 8%). The forest floor is covered with various plant species: 22 species, such as Camellia sinensis (L.) Kuntze and Cleyera japonica Thunb, were recorded at the vegetation monitoring plot close to RW2 (EANET 2013a).

Sampling layout in IJR. RW1 and RW2, measurement points for river water chemistry and discharge rate; Station, Ijira deposition-monitoring site; WD, collection of “wet deposition” samples by a wet-only sampler; DD, measurement of air concentrations and meteorological data for estimation of the flux by “dry deposition”. Rain gauges were installed at three points on the mountain slope, namely, Akatani (260 m above sea level), Station (140 m a.s.l.), and Lakeside (110 m a.s.l.). The rainwater, soil solution, and tree rings for S isotopic analysis were collected at Station, near RW2, and along the Kobora River, respectively. The bold line indicates the catchment area for RW1 (298 ha). Nagoya and Yokkaichi are major cities that form the Chukyo Industrial Area. Yokkaichi and Inabu-cho are the sampling sites for tree ring analysis by Ishida et al. (2015b)
Sampling of river water and measurement of discharge
RW samples have been collected from the lake inflow quarterly since 1988 (shown as RW2 in Fig. 1). In addition to the historical sampling point RW2, samples have been collected at an upstream point (RW1 in Fig. 1) every 2 weeks or twice a month since 2005 to estimate material fluxes from the river more precisely. In this paper, the quarterly data collected at RW2 are used to assess the long-term trends of RW chemistry, and the biweekly/semimonthly data collected at RW1 are used to estimate material budgets in the catchment.
In addition to the regular surveys described above, intensive surveys were carried out during heavy-rain events to characterize typical high-flow events. To collect water samples during these events, an automatic liquid sampler (ISCO 6700, Teledyne ISCO, USA) was installed at RW1 and set to collect samples at intervals of 1 h. Intensive sampling for 48 h was carried out during each event. Finally, typical heavy-rain events were detected twice: once each in 2006 and 2007.
The discharge rate was estimated using the velocity area technique, whereby water depths and flow velocities are measured at a cross section of a river (Nakahara et al. 2010). Water depth was measured basically at 50-cm intervals across the RW flow, and flow velocity was measured at the middle points of each 50-cm split using a flow meter. Then, discharge was calculated based on the flow volume of each split. The measurement described above was conducted at RW1 and RW2. Water level was recorded continuously at 10-min intervals using a water level logger (U20, HOBO, USA) installed at RW1. Discharge was estimated based on the equation between water level and discharge. The discharge was expressed in mm based on the catchment area for RW1 (298 ha, bold lined area in Fig. 1). Because the discharge at RW2 seemed to be underestimated due to underground or hyporheic flow, especially for low-flow periods, the discharge at RW1 was used for evaluation of water and material budgets.
Measurement of wet deposition and air concentrations for dry deposition
Collection of RF using a wet-only sampler (US-420, Ogasawara, Japan) has been conducted weekly since 1999 at the Ijira deposition-monitoring site (Station in Fig. 1; 140 m above sea level), although biweekly sampling using a filtering-type bulk sampler (Tamaki et al. 2000) was conducted from 1988 to 1998 near RW2. The wet-only sampler was equipped with a refrigerator. The RF samples were transported biweekly to the laboratory. In this paper, the monthly data since 2000, which have been disclosed annually in the data reports of the EANET (e.g., EANET 2015), are used for assessment of the wet deposition.
Precipitation was measured using a standard rain gauge (Ogasawara, Japan) at Station. Additionally, two rain gauges were installed at Akatani (260 m a.s.l.) and Lakeside (110 m a.s.l.) in 2005 to check the precipitation distribution within the catchment. The rain gauge location of Lakeside was moved approximately 300 m from the point near RW2 to its current position in 2011.
Air concentrations of SO2 and NOX were measured using automatic monitors (APSA-365; APNA-365, Horiba, Japan). Concentrations of gaseous SO2, HNO3, and NH3 and particulate \( {\text{SO}}_{4}^{2 - } , \)\( {\text{NO}}_{3}^{ - } , \) and \( {\text{NH}}_{4}^{ + } \) in the atmosphere were determined biweekly using a four-stage filter-pack method (EANET 2013b). For SO2, the automatic data were used to estimate the flux. Meteorological factors (wind speed, temperature, relative humidity, solar radiation, and precipitation) were also measured at Station to calculate dry deposition velocities, Vd.
Chemical analysis
The pH, electrical conductivity (EC), and concentrations of major ions (Cl−, \( {\text{NO}}_{3}^{ - } , \)\( {\text{SO}}_{4}^{2 - } , \) Na+, K+, Ca2+, Mg2+, and \( {\text{NH}}_{4}^{ + } \)) in the water samples described above were determined according to the methods described in the EANET technical manual (2010a, b). The pH was measured using a glass electrode (HM-40V, HM-30R, HM-60G, DKK-TOA, Japan), and the EC was measured using a conductivity meter (AOL-40, CM-60G, DKK-TOA). RF samples, RW samples, and extract solutions from the filters for dry deposition were passed through a membrane filter (pore size, 0.45 μm) and stored in a refrigerator (4 °C) prior to chemical analysis. The concentrations of inorganic ions (Cl−, \( {\text{NO}}_{3}^{ - } , \)\( {\text{SO}}_{4}^{2 - } , \) Na+, K+, Ca2+, Mg2+, and \( {\text{NH}}_{4}^{ + } \)) were determined using ion chromatography (IC7000, Yokogawa, Japan; HIC-SP Super, HIC-10A Super, Shimadzu, Japan, DX-500, Dionex, USA, ICS-1100, ICS-2100, Thermo Fischer Scientific, USA), and concentrations of H+ ions were calculated from the pH values. The ion concentration was expressed in micromoles of charge per litre (μmolc L−1). The non-sea-salt (nss) fraction of \( {\text{SO}}_{4}^{2 - } \) was estimated from the concentrations of Na+ and the component ratio of seawater (EANET 2010a). In addition to the ion constituents above, since 1988 concentrations of dissolved organic carbon (DOC) in the RW (RW2) have been determined using an organic carbon analyzer (TOC-L, Shimadzu, Japan), although the data are missing for the period from 1999 to 2002. The DOC concentration is expressed in milligrams per litre (mg L−1).
Input and output budget
Atmospheric inputs (total deposition) of ion constituents to IJR were calculated as the sum of the fluxes by wet deposition and dry deposition. As described in “Water and material budgets” section, the annual precipitation amounts measured at Station, which is located at a middle altitude valley, were 10–20% lower than those at Akatani at higher altitude (see the locations in Fig. 1); therefore, the annual fluxes by wet deposition were estimated as the product of annual mean concentration of RF collected at Station and the mean precipitation amounts of three points including Lakeside. Fluxes by dry deposition were estimated by the inferential method (Endo et al. 2011; EANET 2013c). The land-use category was defined as “forest” to estimate the fluxes. The annual input was expressed in kilomoles of charge per hectare per year (kmolc ha−1 year−1).
The output of ion constituents from the catchment for RW1 was determined by multiplying the average measured concentrations of dissolved constituents in RW sampled at the beginning and end of an interval by the flow volume during a given period (Likens and Bormann 1995). The annual output was also expressed in kmolc ha−1 year−1 by taking the catchment area (298 ha) into account. In this paper, budgets of S, dissolved inorganic N (DIN) as the sum of \( {\text{NO}}_{3}^{ - } \) and \( {\text{NH}}_{4}^{ + } , \) and Cl− are shown.
Sulfur isotopic analysis
The δ34S value of \( {\text{SO}}_{4}^{2 - } \) in RF, soil solution, and RW was determined monthly from April 2014 to January 2017. The RF samples were collected using the filtering-type bulk sampler at Station. The collection was carried out twice each month, but the composite sample for each month was determined to have obtained sufficient amounts of S for the isotopic analysis (> 5 mg as BaSO4) and was therefore used for analysis. The soil solution samples were collected once each month after precipitation using a porous-cap sampler (DIK-8390-11, DIK-8390-58, Daiki, Japan) from 20 cm depth at three positions, upper, middle and lower, on the slope close to RW2. Samples from 60 cm depth were collected only at the lower position. Four samplers were installed for each depth of each position. Composite samples of the four samplers were used for determination of the isotopic ratio. The RW samples were collected monthly at RW1 and RW2. The \( {\text{SO}}_{4}^{2 - } \) in the samples was concentrated by evaporating the water and was precipitated as BaSO4 for the isotopic analysis (Ohizumi et al. 2016). The BaSO4 precipitates were analyzed using a mass spectrometer equipped with an elemental analyzer, EA-IRMS (EA2500—Delta Plus via Conflo II, Thermo Fisher Scientific). The S isotopic ratio was expressed in conventional delta notation:
where x and CDT indicate a sample and Canyon Diablo Troilite (as the standard substance), respectively. The overall precision of the S isotopic analysis was controlled to be less than 0.2 ‰.
Tree disk samples were collected in July 2013 from three stumps of 110-year-old Japanese cedar trees along the Kobora River, near the point of inflow to the lake (see Fig. 1). The trees were cut in 2010. The S isotopic analysis of the disk samples was conducted according to the methods described in Ishida et al. (2015b). The disk samples were divided with a chisel into groups from the bark to the pith in 5-year tree ring intervals. After washing with deionized water and drying for 48 h at 80 °C, the samples were ground using an electric-powered mill. The ground samples were digested using 69% HNO3 and 30% H2O2. After evaporating the acids to dryness, the samples were dissolved again in 30% H2O2 and then the \( {\text{SO}}_{4}^{2 - } \) was precipitated as BaSO4 for the isotopic analysis. The δ34S of the BaSO4 precipitates was analyzed using an EA-IRMS (Flash HT—Delta V Plus via Conflo IV, Thermo Fisher Scientific). The precision was within 0.3 ‰.
Mixing model using the isotopic data
To estimate contributions of possible S sources to the RW, a mixing model was applied with three end members, namely, wet deposition (RF), soil solution, and groundwater. Referring to the mixing model by Hosono et al. (2007), the following mass balance equations were considered:
where S is \( {\text{SO}}_{4}^{2 - } \) concentration in mg L−1 and the subscripts rw, wd, ss, and gw represent RW, wet deposition, soil solution, and ground water, respectively. The fraction of each end member to form the target concentration of the RW is indicated by f. Contributions of the respective end members to the RW S in percentage can be calculated as follows:
Quality control and assurance
The quality of all the measurement processes was controlled according to the quality control and quality assurance procedures specified in the technical manuals of the EANET (2010a, b). Ion balances and theoretical values of EC were assessed for all measured data of the water samples. The RF and stream water used for this analysis generally had no significant error component based on the monitoring criteria. However, for the wet deposition data in 2006, the data completeness regarding the coverage of precipitation period and/or collected precipitation amounts was lower than 80% in 3 months (EANET 2007). Moreover, the monthly data in April 2006 is missing, which indicates an underestimation of the annual fluxes in the year. Therefore, these annual fluxes by wet deposition in 2006 are flagged with asterisks in Fig. 4. The laboratories have been participating in inter-laboratory comparison projects annually for RF samples since 1991, for the filter-pack method since 2005, and for inland water samples since 2000 to ensure and control the analytical quality of the data obtained (e.g., Network Center for EANET 2015).
Data analysis
In this paper, we define a water year (WY) as the time interval from November of one year to October of the next year, taking account of the hydrological pattern, and, for convenience, we identify each WY by the year in which it ended. For example, the WY from November 2013 to October 2014 is called ‘WY 2014’.
For the input–output budgets, the monthly data of wet deposition based on weekly sampling were used to calculate the annual inputs, and the actual collection dates at RW1 in the end of October or the beginning of November were applied to calculate the annual water balance and annual outputs from the RW. Although the total number of days for the respective WYs fluctuated by several days depending on the actual collection dates, the annual precipitation amounts based on the monthly wet deposition data and those based on the daily rain-gauge data on the collection dates agreed well within 3%.
Statistical analysis was conducted using R version 3.0.2 (R Core Team 2013) with the package “Rcmdr” version 2.0-0 (Fox 2005). The Pearson correlation coefficient (Pearson’s r) was used to assess correlations among the RW chemistry. The seasonal Mann–Kendall trend test with the package “Kendall” version 2.2 (Mcleod 2011) was used to test for trends of quarterly data of the RW or monthly data of the wet deposition during the observation period. As a dynamic change in RW chemistry has been evident in IJR over the past few decades, a trend analysis was applied to periods before and after the inflection points.
Results
River water chemistry
Long-term trends of the RW chemistry at RW2 are shown in Fig. 2. The pH increased until 1996 (p = 0.0044) and then declined until 2003 (p < 0.0001). Then, pH recovered suddenly to the original level of around 7.0 in 2004 and became stable. Concentrations of \( {\text{SO}}_{4}^{2 - } \) increased rapidly after a slight declining trend until 1996 (not significant, p = 0.072), peaked in summer 1994, and then declined thereafter (p < 0.0001). Concentrations of \( {\text{NO}}_{3}^{ - } \) increased until 2005/2006 (p < 0.0001) and then declined significantly (p < 0.0001). A significant high concentration of \( {\text{NO}}_{3}^{ - } \) was also observed in summer 1994. The ratio of basic cations to strong-acid anions (BCs/SAAs) generally increased until 1996 (p = 0.0054) and then declined until 2003 (p = 0.0001), although a sudden drop and peak were observed in 1994 and 2001, respectively. Then, the BC/SAA ratio increased gradually again from 2003 (p = 0.030). Concentrations of DOC increased until 1997/1998 (p < 0.0001). Although the data were missing for the period from 1999 to 2000, DOC declined thereafter (p = 0.0028).

Long-term trends of pH (a), concentrations of \( {\text{SO}}_{4}^{2 - } \) and \( {\text{NO}}_{3}^{ - } \) (b), ratio of basic cations to strong-acid anions (BCs/SAAs), (Ca2+ + Mg2+ + K+)/(\( {\text{SO}}_{4}^{2 - } \) + \( {\text{NO}}_{3}^{ - } \)) (c), and concentration of dissolved organic carbon, DOC (d) in the Kamagatani River. Quarterly data collected at RW2 from 1988 to 2014 are shown. The data until 2004 were already shown in a previous paper (Nakahara et al. 2010). The figures were updated with an additional 10-year increment of data, until 2014
Changes in concentrations of \( {\text{SO}}_{4}^{2 - } \) and \( {\text{NO}}_{3}^{ - } \) with the discharge rate of RW are shown in Fig. 3. Two typical heavy-rain events, which were detected in 18–20 July 2006 and 13–15 July 2007, are plotted with the regular biweekly/semimonthly sampling data since 2005. The \( {\text{SO}}_{4}^{2 - } \) concentration declined with increase of the discharge rate, and the lowest concentration, 74.7 μmolc L−1, was observed at the discharge rate of 0.000879 mm s−1. Then, the \( {\text{SO}}_{4}^{2 - } \) concentration rather increased with the discharge rate, although it tended to decrease in high discharge rates of more than 0.0025 mm s−1. The \( {\text{NO}}_{3}^{ - } \) concentration increased with the discharge rate in general, although it also tended to decrease in the high discharge rates. During the high-discharge period (> 0.000879 mm s−1) until 0.0025 mm s−1, concentrations of \( {\text{SO}}_{4}^{2 - } \) correlated positively with those of \( {\text{NO}}_{3}^{ - } \) (Pearson’s r 0.577, p = 0.0001) and the discharge rate (0.487, p = 0.0017), while negative correlations (− 0.310 and − 0.707, p < 0.0001 and < 0.0001, respectively) were observed during the low-discharge period (< 0.000879 mm s−1). However, no seasonality was found for the \( {\text{NO}}_{3}^{ - } \) concentration, and no correlation was observed between \( {\text{NO}}_{3}^{ - } \) concentrations and RW temperature (Pearson’s r 0.0639, p = 0.316), while the \( {\text{SO}}_{4}^{2 - } \) concentration was negatively correlated with temperature (−0.423, p < 0.0001, the detailed data are not shown here).

Changes in concentrations of \( {\text{SO}}_{4}^{2 - } \) (a) and \( {\text{NO}}_{3}^{ - } \) (b) with the river water discharge rate during heavy rain events. The 1-h sampling data at RW2 during the heavy rain events of 18–20 July 2006 and 13–15 July 2007 are plotted together with the regular biweekly/semimonthly sampling data since 2005
Trends of wet deposition
Changes in annual fluxes of \( {\text{SO}}_{4}^{2 - } , \) DIN, and H+ by wet deposition are shown for the period from 2001 to 2014 in Fig. 4. The fluxes showed a declining trend for the observation period (by seasonal Mann–Kendall test for monthly data, p = 0.00068, 0.022, and 0.0080, respectively), while the precipitation amounts showed no trend (Fig. 4). During the period above, distinct peaks in \( {\text{SO}}_{4}^{2 - } \) and DIN were observed in 2006, even though the fluxes must be underestimated due to the missing data in April (see “Quality control and assurance” section). In particular, after the peaks in 2006, the fluxes showed a clear declining trend (p = 0.0010, 0.010, and 0.0061, respectively). For DIN, the fluxes of both \( {\text{NO}}_{3}^{ - } \) and \( {\text{NH}}_{4}^{ + } \) by wet deposition showed a clear declining trend since 2006 (p = 0.0096 and 0.0055, respectively), and that of nss \( {\text{SO}}_{4}^{2 - } \) also showed a declining trend simultaneously (p = 0.00051, detailed data are not shown here). The BC/SAA ratio of the fluxes declined significantly for the observation period (p = 0.00015), although the fluxes of base cations, such as Ca2+, Mg2+, and K−, also declined (p = 0.00052, 0.015, and < 0.0001, respectively, the detailed data are not shown here).

Changes in precipitation amount and annual fluxes of \( {\text{SO}}_{4}^{2 - } , \) dissolved inorganic nitrogen (DIN), and H+ by wet deposition. *, The data completeness regarding the coverage of precipitation period and/or collected precipitation amounts was lower than 80% in 3 months (EANET 2007). The water year (WY) is defined as the time interval from November of one year to October of the next year. The WY 2001 indicates the period from November 2000 to October 2001. DIN was calculated as the sum of \( {\text{NO}}_{3}^{ - } \) and \( {\text{NH}}_{4}^{ + } \)
Water and material budgets
The mean annual water budget for the five WYs from 2010 to 2014 is shown in Fig. 5a. Precipitation amounts were generally larger at Akatani than at Station and Lakeside throughout the survey period. The five-year mean precipitation amount at Akatani was approximately 20% larger than that at Station. The mean discharge amount (1964 mm year−1) corresponded to 58% of the mean precipitation amount (3345 mm year−1). The mean annual budgets of \( {\text{SO}}_{4}^{2 - } , \) DIN, and Cl− are shown in Fig. 5b. For the inputs, the contributions of dry deposition were very small, 4.3, 8.2, and 0.4% for \( {\text{SO}}_{4}^{2 - } , \) DIN, and Cl−, respectively (the detailed data are not shown here). The mean outputs of \( {\text{SO}}_{4}^{2 - } \) and Cl− significantly exceeded the inputs, while the DIN output was smaller than the input. In particular, the \( {\text{SO}}_{4}^{2 - } \) output was three times larger than the input: 2.5 ± 0.34 and 0.86 ± 0.038 kmolc ha−1 year−1, respectively.

Water budget (a) and the budgets of \( {\text{SO}}_{4}^{2 - } , \) dissolved inorganic nitrogen, and Cl− (b). The mean annual fluxes for the five water years from 2010 to 2014 are shown with standard errors in the respective items. Mean precipitation amount was calculated based on the precipitation amounts at Akatani, Station, and Lakeside in Fig. 1, whereas discharge amount was calculated from the discharge rate of river water at RW1. Mean inputs and mean outputs for \( {\text{SO}}_{4}^{2 - } , \) DIN, and Cl− were calculated from total fluxes and ion concentrations of wet deposition and dry deposition and from the discharge amount and ion concentrations of river water at RW1, respectively. DIN was calculated as the sum of \( {\text{NO}}_{3}^{ - } \) and \( {\text{NH}}_{4}^{ + } \)
Sulfur emission trends in Japan and the isotopic profile recorded in tree disks
The δ34S values of tree disks are plotted with the age of tree ring in Fig. 6b, together with the tree-ring data obtained in Yokkaichi City (YOK) and Inabu-cho in Toyota City (INA) within the Chukyo Industrial Area (Ishida et al. 2015b) and the S emission trends over the last century in Japan (Ohara et al. 2007; Smith et al. 2011; Kurokawa et al. 2013) in Fig. 6a for comparison. Anthropogenic S emissions to the atmosphere in Japan increased rapidly in the 1960s, peaked in 1969, and then declined steeply thereafter until the beginning of the 1980s (Fig. 6a). The δ34S profiles in tree disks were the converse of the emission trend profiles (Fig. 6b). The δ34S values of tree disks in YOK (which is famous for its petrochemical complexes) declined steeply in the 1960s and the lowest values (− 7.0 ± 0.2 ‰, the mean value of three stumps with its standard error) were observed in 1970, as shown in Fig. 6b. The values recovered promptly in the 1980s and then increased gradually. The tree disks in INA, which is far from the largest source, YOK (see the location in Fig. 1), showed a similar profile and the lowest value was − 0.3 ± 0.8 ‰. The δ34S values of tree disks in IJR also declined in the 1960s, but the timing was slightly later than those in YOK and INA. The lowest values in IJR (− 1.6 ± 0.5 ‰) were observed in 1983 (Fig. 6b), then the values increased gradually. In the early 2000s, the δ34S values of tree disks in the three points became approximately 2 ‰.

S emission trends in Japan (a) and δ34S profiles in Japanese cedar tree disks collected in the Chukyo Industrial Area and IJR (b). S emission trends in Japan are based on the emission inventories estimated by Smith et al. (2011) for the period from 1850 to 2005, Ohara et al. (2007) for the period from 1980 to 2003, and Kurokawa et al. (2013) for the period from 2000 to 2008. Tree disks YOK and INA were collected in Yokkaichi City and Inabu-cho in Toyota City, respectively (Ishida et al. 2015b). The δ34S was analyzed in 5-year tree ring intervals
Sulfur isotopic ratios of rainwater, soil solution, and river water
Seasonal changes in the δ34S values of RF, soil solutions, and RWs for the period from April 2014 to January 2017 are shown in Fig. 7. The δ34S values in RF showed a distinct seasonality, low in summer and high in winter. The lowest δ34S values, recorded in September 2014, August 2015, and August 2016, were 2.8, 1.5, and 1.4 ‰, and the highest values, recorded in December 2014, January 2016, and January 2017, were 8.0, 7.5, and 6.6‰, respectively. The flux-weighted mean δ34S value of RF was 4.3 ‰. The δ34S values of soil solutions in the shallow soil layer (20 cm depth) were relatively stable during the observation period: the mean values with standard errors were 3.8 ± 0.1 ‰, 3.7 ± 0.2 ‰, and 3.3 ± 0.1 ‰ in U20, M20, and L20, respectively. The δ34S values in the deeper soil layer (60 cm depth) were slightly lower than those in the shallow soil layer: the mean value in L60 was 2.5 ± 0.1 ‰. The δ34S values of RWs were also relatively stable during the observation period and were significantly lower than those of RF and soil solutions: the mean values were − 13.2 ± 0.1 ‰ and − 13.0 ± 0.1 ‰ at RW1 and RW2, respectively.

Seasonal changes in sulfur isotopic ratio (δ34S) in rainwater (RF), soil solution (SS), and river water (RW). U, M, and L in soil solution indicate sampling positions, upper, middle, and lower positions on the slope, respectively. The 20 and 60 indicate the sampling depth in cm. An outlier (− 6.9) at RW2 in April 2016 is not plotted here
The relationship between the δ34S values and the discharge rate at the sampling time is shown for RW1 in Fig. 8. The δ34S values were positively correlated with the discharge rate (Pearson’s r 0.473, p = 0.00474). More negative values were observed at lower discharge rates.

Relationships between δ34S value and discharge rate in river water. Data of RW1 are plotted here with the discharge rate at the sampling time
Mixing model to estimate the contribution of each end member
The relationships between the δ34S values and the reciprocal of the \( {\text{SO}}_{4}^{2 - } \) concentrations for the three waters are shown in Fig. 9. For the RW, only the data of RW1 are plotted, because the data of RW2 almost fully overlap those of RW1. The δ34S values of RF did not show a meaningful relationship with the \( {\text{SO}}_{4}^{2 - } \) concentration. No significant correlation was found between them (Pearson’s r 0.0432, p = 0.808). The δ34S value of soil solution tended to decrease with increase of \( {\text{SO}}_{4}^{2 - } \) concentration at all the three sites, although both values fluctuated largely at individual points. Consequently, the δ34S values showed a significant positive correlation with the reciprocal of \( {\text{SO}}_{4}^{2 - } \) concentration (r 0.439, p < 0.0001). The δ34S values of the RW showed stronger correlations with the reciprocal of \( {\text{SO}}_{4}^{2 - } \) concentration (r 0.781, p < 0.0001). The maximum and minimum δ34S values in RW1 were − 11.1 ‰ and − 14.3 ‰, respectively. The linear regression line (a) for RW1 passed close to the mean value of RF and its y-axis intercept was approximately − 17.7 ‰.

Relationships between the δ34S values and the reciprocal of the \( {\text{SO}}_{4}^{2 - } \) concentrations. Refer to the caption of Fig. 7 for the abbreviations. [\( {\text{SO}}_{4}^{2 - } \)], \( {\text{SO}}_{4}^{2 - } \) concentrations in mg L−1. The lines (a) and (b) indicate the linear regressions for the samples of RW1 (y = 29.1910x − 17.7247, R2 0.598, p < 0.0001, n = 34) and the samples for soil solutions (y = 2.3634x + 2.1511, R2 0.184, p < 0.0001, n = 97), respectively. The points A, B, and C indicate the mean RF value, the highest concentration \( {\text{SO}}_{4}^{2 - } \) sample of soil solution, and the expected value of groundwater, which are defined in Table 1
A linear relationship between the δ34S values and the reciprocal of the \( {\text{SO}}_{4}^{2 - } \) concentrations suggests that mixing of two end members regulates the δ34S values and that the y-intercept indicates the δ34S value of the source responsible for elevated \( {\text{SO}}_{4}^{2 - } \) concentrations (Krouse 1980; Novák et al. 2001; Hosono et al. 2007; Mayer et al. 2010). Points A, B, and C indicate the selected end members to represent wet deposition, soil solution, and groundwater (Table 1). With this assumption, the groundwater δ34S value was estimated as the mean value of the lowest δ34S value (− 14.3 ‰) and the intercept (− 17.7 ‰) of the regression line a) in RW1. Fractions of the respective end members and contributions to the RW S calculated by the mixing model are summarized for the respective cases in Table 2. When the mean values in RW1 (Case 1) were applied to Srw and δ34Srw of Eqs. (2) and (3), respectively, the calculated fractions of the wet deposition, soil solution, and groundwater to form the target concentration (6.49 mg L−1) were 0.64, 0.03, and 0.33, respectively. The contribution of S derived from the groundwater to the RW S was significantly high, 86%, due to its high \( {\text{SO}}_{4}^{2 - } \) concentration, while the contribution from the wet deposition was small, 11.4%, due to its low \( {\text{SO}}_{4}^{2 - } \) concentration.
Discussion
Recovery from acidification and nitrogen saturation
The declining trends of the fluxes by wet deposition seem to reflect the national emission trends. According to the latest emission inventories of the pollutants, emissions of SO2 and NH3 in Japan decreased from 2000 to 2008 by 12% and 10%, respectively, and NOX emissions also began decreasing in 2005 after the constant period until 2004 (Kurokawa et al. 2013). As for the peaks in 2006, the high precipitation amount in 2006 compared to those in the year before and the years after may have contributed to them (see Fig. 4), although the peak of SO2 emissions in China that year (Lu et al. 2011) may also have partly contributed to the \( {\text{SO}}_{4}^{2 - } \) flux. In the case of IJR, the contribution of wet deposition was significantly larger than that of dry deposition (Endo et al. 2011), as discussed later; therefore it is suggested that the trends of the fluxes by wet deposition reflected the trends of atmospheric inputs to the ecosystem. The trend in \( {\text{SO}}_{4}^{2 - } \) deposition appears also to be clearly recorded in the profile of tree ring S isotopic values, as discussed in the “Historic record in tree rings and origins of S in river water” section.
The RW in IJR in the mid-1990s was considered as acidified and N saturated due to the climatic anomalies at this time (Nakahara et al. 2010). Since this period, the RW has shown a stepwise recovery from the acidification and N saturation. High concentrations of \( {\text{SO}}_{4}^{2 - } \) and DOC were observed for the period from the mid-1990s to the early 2000s after the climatic anomalies, although the timings of the peaks were slightly different (Fig. 2b, d). Slight increases in \( {\text{SO}}_{4}^{2 - } \) concentration observed during the high-flow events (see Fig. 3a) suggest that S that had accumulated in the shallow soil layer was leached out by the events. Contamination with sea-salt S was not the cause of the increase in \( {\text{SO}}_{4}^{2 - } \) concentrations, as the Cl− concentration declined during the high-flow events (detailed data are not shown here). The speciation of S compounds in the soils collected within IJR indicated that total S contents in shallow soil layers (A horizons) were quite large (> 1000 mg S kg−1) and that more than half of the total S was stored as S forms non-extractable by phosphate solvent, of which the majority might be organic S (unpublished data). Nakahara et al. (2010) suggested that enhanced mineralization of organic matter contributed to high concentrations of \( {\text{SO}}_{4}^{2 - } , \) particularly after the dry summer in 1994 when the annual precipitation was 1604 mm. Thereafter, as of 2014, precipitation amounts lower than 2000 mm were not recorded (EANET 2015; see also Fig. 4). The decline of DOC concentration indicated that effects of the enhanced mineralization became diminished in the early 2000s. Accordingly, leaching of \( {\text{SO}}_{4}^{2 - } , \) which produces H+ (van Breemen et al. 1983), declined. Finally, the RW pH recovered to the original level of around 7.0 in 2004, just after the BC/SAA ratios started increasing in 2003 (see Fig. 2). According to the tree-ring analysis of thinned trees within the catchment, tree growth rates declined in the mid-1990s, just after the climatic anomalies, but increased again in 2002/2003 (Nakahara et al. 2010), which suggested recovery of N uptake by the trees. Moreover, the thinning of trees as a forest management practice was implemented within the catchment area in 2002–2004 and 2006–2007 (Ministry of the Environment 2012). This may also have enhanced N uptake by trees and understory vegetation. Then, \( {\text{NO}}_{3}^{ - } \) concentration started declining in 2005/2006. The enhanced biological N uptake appears to be related to a reduction in the atmospheric \( {\text{NO}}_{3}^{ - } \) contribution to the RW chemistry, as suggested by Tsunogai et al. (2014). Based on an analysis of 17O excess of \( {\text{NO}}_{3}^{ - } , \) Nakagawa et al. (2018) reported that the export flux of unprocessed atmospheric \( {\text{NO}}_{3}^{ - } \) relative to the deposition flux could be used as the new index of N saturation, since the flux ratio showed a clear normal correlation with the mean \( {\text{NO}}_{3}^{ - } \) concentration in streams of several forested catchments, including IJR. In their evaluation, both the flux ratio of unprocessed atmospheric \( {\text{NO}}_{3}^{ - } \) (6.5 ± 1.8%) and the mean \( {\text{NO}}_{3}^{ - } \) concentration (24.4 μmolc L−1) in the period from 2012 to 2014 in IJR were relatively low compared with those in other catchments. The \( {\text{NO}}_{3}^{ - } \) concentration was much higher in 2005/2006 (> 40 μmolc L−1, see Fig. 2), and it appears that the flux ratio of unprocessed atmospheric \( {\text{NO}}_{3}^{ - } \) was also higher at that time. The recent recovery of biological N uptake may have lowered the flux ratio of unprocessed atmospheric \( {\text{NO}}_{3}^{ - } \) as well as the \( {\text{NO}}_{3}^{ - } \) concentration in RW. Therefore, in addition to the decline of atmospheric inputs, it is considered that the diminished effects of climatic anomalies and recent forest management have contributed to the recovery of RW from acidification and N saturation.
Changes in RW chemistry with atmospheric acid inputs have been reported recently in China. Increase of \( {\text{SO}}_{4}^{2 - } \) concentrations and decrease of pH, which were observed in the 1990s in the tributary rivers of the Upper Yangtze River, had stopped in the 2000s due to stabilized SO2 emissions (Duan et al. 2011). Moreover, Qiao et al. (2016) found that increasing trends of RW pH occurred in the area with reductions of SO2 and NOX emissions for the period from 2004 to 2014, while decreasing trends continued to occur in the area with increased NOX emissions. These studies suggest that the recovery of RW from acidification began in the 2000s in some areas of China. As described above, declining trends of RW pH were observed in the 1980s/1990s in central Japan, especially in the rivers draining from acid-sensitive geology (Kurita and Ueda 2006; Matsubara et al. 2009). Moreover, according to the water quality data at 638 observation points for monitoring of public water bodies in Japan, a declining trend of pH was detected at 65, 61, and 31 points while an increasing trend was detected at 91, 92, and 134 observation points in 1981–1990, 1991–2000, and 2001–2009, respectively (Yamashita et al. 2016). Also, the number of points with increasing pH in Japan increased in the 2000s. Therefore, recovery of RW from acidification due to declining atmospheric deposition seems to have been the trend at the regional scale in China and Japan in the 2000s. For IJR, the decline in atmospheric deposition and the diminished effects relating to climatic anomalies appear to have contributed to the recovery from acidification and N saturation.
Input–output budget and its uncertainties
The mean discharge rate in IJR was 58% compared to the mean precipitation amounts of three different slope positions. The data from the Japan Forestry Agency showed that the runoff rates in forest watersheds in Japan were in the range of 60 to 80% when the precipitation amount was more than 2500 mm (Ogawa 2005). Although the discharge rate in IJR is at the lower end of this range (or below it), the same discharge rate level (57%) was also observed in a forested catchment covered by similar tree species, such as C. japonica and C. obtusa, in Kyushu Prefecture, Japan (Shimizu et al. 2003). It is considered that the water balance in IJR could be used to discuss the material budget.
The wet fluxes of S and DIN at Station had the highest levels among 80 monitoring sites in Japan (Matsubara et al. 2009), even though the precipitation amounts at Station were relatively small compared with those at Akatani. The ionic composition of the RF at Station was not largely different from those at other monitoring sites within Gifu Prefecture (Ministry of the Environment 2014). The current estimates may be practical levels for the fluxes by wet deposition in Japan. The contribution of dry deposition to the total inputs in IJR is relatively small. Endo et al. (2011) previously suggested this when they estimated fluxes by dry deposition at 10 EANET sites in Japan. They pointed out that atmospheric concentrations at Station were similar to those at other EANET sites in western Japan but that Vd at Station was the lowest among the sites because of the lowest wind speed there, that is, because of the stationary air in the catchment. Ban et al. (2016) assumed the total uncertainty for the dry deposition estimated by the inferential method to be 10–50%. Even if the uncertainty was 50%, there was not much influence of dry deposition to the total inputs (2, 4, and 0.2% for \( {\text{SO}}_{4}^{2 - } , \) DIN, and Cl−, respectively).
Even though these uncertainties were taken into account, the \( {\text{SO}}_{4}^{2 - } \) output may have exceeded the input, as shown in Fig. 5b. In the current estimates, the Cl− output also exceeded the input. Cl− is considered as a conservative ion in ecosystems with high Cl− deposition (> 6 kg ha−1 year−1) (Svensson et al. 2012). The Cl− input is relatively large in IJR. If we assume that the Cl− input is equivalent to the output, the input should be 77% larger than the current estimate. Accordingly, the \( {\text{SO}}_{4}^{2 - } \) input should be 1.5 kmolc ha−1 year−1, which is still significantly smaller than the output, 2.5 kmolc ha−1 year−1. Thus, it is suggested that the \( {\text{SO}}_{4}^{2 - } \) output certainly exceeded the input in IJR. A S-budget discrepancy has been suggested in Europe (e.g., Novák et al. 2000; Vuorenmaa et al. 2017), in the US (e.g., Mitchell et al. 2011, 2013; Mitchell and Likens 2011), and even in Japan (Kobayashi et al. 2013). However, the causes of the discrepancy have depended on the site conditions. Possible origins of the excess S and its dynamics in the case of IJR are discussed in the next section.
Historic record in tree rings and origins of S in river water
It has been suggested that the profile of δ34S values in tree disks of C. japonica reflects historical changes in emission sources of S deposited in Japanese forests (Kawamura et al. 2006; Ishida et al. 2015a, b), although it has been suggested that isotopic fractionations of S occur before it is fixed in tree tissue (Novák et al. 2000, 2001; Wynn et al. 2014; Ishida et al. 2015b). The tree-ring δ34S profiles in YOK and INA collected near the Chukyo Industrial Area (Ishida et al. 2015b) were similar but opposite to that of the SO2 emission trend in Japan (Smith et al. 2011), as shown in Fig. 6. The value in IJR also started declining in the 1960s, but the lowest values were found in the early 1980s, which was slightly later than those in the cases of YOK and INA. It has been suggested that the significant decline of the tree-ring δ34S in the 1970s was the result of large inputs of S with lower δ34S values derived from imported oils (Kawamura et al. 2006; Ishida et al. 2015a, b), because the oils imported from countries in the Middle East generally had negative δ34S values, ranging from − 10.3 to − 0.7 ‰ (Maruyama et al. 2000). In fact, Nakai et al. (1991) reported that the mean δ34S value of anthropogenic SO2 collected in industrial areas, including Nagoya, in the period from 1971 to 1977 was − 4.0 ‰. This suggests that the decline of the δ34S values of tree rings in IJR in the 1960s/1970s also reflected the large S inputs derived from anthropogenic SO2 in Japan. As described above, a large amount of S (> 1000 mg kg−1) is accumulated in soil in IJR. The large S storage in soil–plant systems in IJR may have buffered the effects of atmospheric S inputs and delayed the timing of decline of δ34S values. The recent tree-ring values in IJR (around 2 ‰) were slightly lower than those of RF (4.3 ‰) in IJR. This is consistent with the investigation by Ishida et al. (2015b) in the Chukyo Industrial Area. Novák et al. (2001) pointed out that the δ34S values of forest floor were lower than those of local throughfall deposition and suggested that plant tissues assimilate systematically lighter isotopes (on average, by 2 ‰). Thus, it is suggested that anthropogenic SO2 emitted in the Chukyo Industrial Area was deposited in IJR through several decades and that its historical changes were recorded in the tree rings of C. japonica.
The \( {\text{SO}}_{4}^{2 - } \) output exceeded the input in IJR, as discussed above. The S isotopic analysis and the mixing model provided information on origins of the excess S. The δ34S values of RW in IJR (− 13.2 ‰ at RW1) were significantly lower than those of RF and soil solutions. The mean δ34S values of soil solution collected from shallow soil layers at the three points were within the range of 3.3–3.8 ‰. These values appear to reflect both the current atmospheric S (4.3 ‰ in RF) and the large S storage in soil. The lowest δ34S values in the IJR tree rings, which occurred in the early 1980s, were around − 1.6 ‰, and the recent values were around 2 ‰, as discussed above. Therefore, it is suggested that S derived from atmospheric deposition is not a major source of \( {\text{SO}}_{4}^{2 - } \) in the RW.
The y-intercept in the linear relationship between the δ34S values and the reciprocal of \( {\text{SO}}_{4}^{2 - } \) concentrations in the RW (see Fig. 9) indicates the δ34S value of the source responsible for elevated \( {\text{SO}}_{4}^{2 - } \) concentrations (Krouse 1980; Novák et al. 2001; Mayer et al. 2010). The y-intercept was approximately − 17.5 ‰, which is certainly lower than the possible range of δ34S values in the imported oils reported by Maruyama et al. (2000). As another possible source, geology can be considered. The bedrock of IJR is mainly chert, which is part of the melanges in the large sedimentary complex belt, the Mino–Tanba Terrane (Wakita 1988; Yoshida and Wakita 1999). Kajiwara et al. (1994) investigated S contents and δ34S values in the vertical profile of the Mino–Tanba Terrane and reported that the δ34S values of shale, mudstone, and chert in the terrane range from − 40 to − 10 ‰. The δ34S value of the possible S source in IJR, − 17.7 ‰, is within this range. Moreover, as shown in Fig. 8, δ34S values that are even more negative were observed in lower discharge rates during base flow conditions, which also supports the notion of a contribution from bedrock. Thus, the sedimentary rocks of the Mino–Tanba Terrane appear to be the most possible major source of \( {\text{SO}}_{4}^{2 - } \) in the RW. It can be considered that the groundwater selected as one of the end members (point C) in the mixing model is strongly affected by the rocks mentioned above.
The regression line (b) for the soil solution samples passes through the points A and B and its intercept could be approximately 2 ‰ (see Fig. 9). The \( {\text{SO}}_{4}^{2 - } \) concentrations and their δ34S values in the soil solution seem to be regulated by mixing of the atmospheric S and a S source with a δ34S value of approximately 2 ‰, which is similar to the δ34S values of the recent tree-ring S. Thus, it is suggested that the point B, with the highest \( {\text{SO}}_{4}^{2 - } \) concentration among the soil solutions, is strongly affected by the retained/cycled S in the soil–plant system. In particular, as described above, more than half of S is likely to be accumulated as organic S in the soil, and this appears to largely contribute to the soil solution.
The regression lines for the RW and the soil solution pass close to the point A, which is the mean value of RF. The point A seems to be reasonable as one of the end members. On the other hand, the soil solution samples from the shallow soil layer (20 cm depth) plotted close to point A and occasionally had higher δ34S values than some RF samples. Although the point A was the mean RF value representing atmospheric S, it appears to also reflect the contribution of the soil solution in shallow soil layers. The contributions of the atmospheric S and the retained/cycled S seemed not to be clearly distinguished in the mixing model. However, in any case, the geological S mainly contributed to the RW S. As estimated above, the mean S input and the mean S output were 0.86 kmolc ha−1 year−1 and 2.5 kmolc ha−1 year−1, respectively. If we apply the mean contributions (Case 1, See Table 2) to the S output, the portions of the output derived from the wet deposition, the soil solution, and the groundwater are 0.29 kmolc ha−1 year−1, 0.06 kmolc ha−1 year−1, and 2.15 kmolc ha−1 year−1, respectively. The large S output is attributable to the groundwater, which is strongly affected by the geological S. This clearly explains the discrepancy of the S budget. Given that the wet deposition (point A) is certainly regulated by atmospheric S, 33% of the atmospheric S input is discharged into the river, while 67% is retained/cycled within the soil–plant system. The high S content in soil also supports the model result.
The tree-ring δ34S profile recorded clearly the historical changes in the atmospheric S in Japan, which suggested the active S cycle in the soil–plant system. Therefore, the atmospheric S appears to have been accumulated as organic S in soil. This is also consistent with the slight increases in \( {\text{SO}}_{4}^{2 - } \) concentration observed during the high-flow events (see Fig. 3a). As shown in Fig. 2b, the \( {\text{SO}}_{4}^{2 - } \) concentrations in the RW were significantly high (over 200 μmolc L−1) in the mid-1990s compared to the current level (annual mean, 138 μmolc L−1: EANET 2015). Nakahara et al. (2010) suggested that enhanced mineralization of the organic S in soil caused the \( {\text{SO}}_{4}^{2 - } \) peak in the mid-1990s, which is supported by the high concentrations of DOC in the corresponding period. Therefore, it is possible that the accumulated organic S derived from the atmospheric S contributed to the high \( {\text{SO}}_{4}^{2 - } \) concentrations in the RW during the acidifying period in the mid-1990s. After this period, the relative contribution of the atmospheric S appeared to become smaller with recovery of the river from acidification.
Conclusion
The IJR in central Japan has historically suffered from atmospheric deposition derived from both domestic and trans-boundary air pollution. Although the catchment was once acidified and N-saturated due to climatic anomalies in the mid-1990s, recovery has been observed over the last decade. In addition to the recent decline in atmospheric deposition, diminished effects of the climatic anomalies and enhanced N uptake due to forest management appear to have contributed to the recovery. The S output exceeded the input in IJR, and the S isotopic analysis suggested that geological S largely contributed to the discrepancy of the budget, while atmospheric S continued to be accumulated in the ecosystem. In the case of IJR, the RW seems to be recovering smoothly. However, as shown in the history of the catchment, the climate may affect the acidification of its terrestrial ecosystems and its recovery. As suggested by previous studies, climate will also play an important role in regulating S dynamics when the atmospheric S input declines. In this study, we clarified the dynamic changes in RW chemistry of IJR due to changes in the atmospheric deposition and climatic conditions over the last few decades. As most of the atmospheric S input appears to be currently accumulated within the catchment of IJR, it is possible that climate change or possible climatic anomalies, such as those occurring in the mid-1990s, will alter the RW chemistry again. Therefore, atmospheric deposition and RW chemistry should be monitored carefully in the future, and particularly in areas that are sensitive to changing atmospheric environments, such as IJR.
References
Ban S, Matsuda K, Sato K, Ohizumi T (2016) Long-term assessment of nitrogen deposition at remote EANET sites in Japan. Atmos Environ 146:70–78. https://doi.org/10.1016/j.atmosenv.2016.04.015
Duan L, Ma X, Larssen T, Mulder J, Hao J (2011) Response of surface water acidification in Upper Yangtze River to SO2 emissions abatement in China. Environ Sci Technol 45:3275–3281
Duan L, Yu Q, Zhang Q, Wang Z, Pan Y, Larssen T, Tang J, Mulder J (2016) Acid deposition in Asia: emissions, deposition, and ecosystem effects. Atmos Environ 146:55–69
EANET (2007) Data report 2006. Network Center for EANET, Acid Deposition and Oxidant Research Center, Niigata
EANET (2010a) Technical manual for inland aquatic environment monitoring in East Asia-2010. Network Center for the EANET, Asia Center for Air Pollution Research, Niigata
EANET (2010b) Technical manual for wet deposition monitoring in East Asia-2010. Network Center for the EANET, Asia Center for Air Pollution Research, Niigata
EANET (2011) Part II national assessments, second periodic report on the state of acid deposition in East Asia. Acid Deposition Monitoring Network in East Asia (EANET), Secretariat for the EANET, United Nations Environment Programme Regional Resource Centre for Asia and Pacific (UNEP/RRC.AP), Pathumthani, Network Center for the EANET, Asia Center for Air Pollution Research (ACAP), Niigata
EANET (2013a) Data report 2011. Network Center for EANET, Acid Deposition and Oxidant Research Center, Niigata
EANET (2013b) Technical manual for air concentration monitoring in East Asia. Network Center for the EANET, Asia Center for Air Pollution Research, Niigata
EANET (2013c) Technical manual on dry deposition flux estimation in East Asia. Network Center for the EANET, Asia Center for Air Pollution Research, Niigata
EANET (2015) Data report 2014. Network Center for EANET, Asia Center for Air Pollution Research, Niigata
Endo T, Yagoh H, Sato K, Matsuda K, Hayashi K, Noguchi I, Sawada K (2011) Regional characteristics of dry deposition of sulfur and nitrogen compounds at EANET sites in Japan from 2003 to 2008. Atmos Environ 45:1259–1267. https://doi.org/10.1016/j.atmosenv.2010.12.003
Fox J (2005) The R Commander: a basic statistics graphical user interface to R. J Stat Softw 14(9):1–42
Hosono T, Nakano T, Igeta A, Tayasu I, Tanaka T, Yachi S (2007) Impact of fertilizer on a small watershed of Lake Biwa: use of sulfur and strontium isotopes in environmental diagnosis. Sci Total Environ 384:342–354
Ishida T, Tayasu I, Takenaka C (2015a) Characterization of sulfur deposition over the period of industrialization in Japan using sulfur isotope ratio in Japanese cedar tree rings taken from stumps. Environ Monit Assess 187:459
Ishida T, Tayasu I, Takenaka C (2015b) Quantitative reconstruction of sulfur deposition using a mixing model based on sulfur isotope ratios in tree rings. J Environ Qual 44:1800–1808
Kajiwara Y, Yamakita S, Ishida K, Ishiga H, Imai A (1994) Development of a largely anoxic stratified ocean and its temporary massive mixing at the Permian/Triassic boundary supported by the sulfur isotopic record. Palaeogeogr Palaeoclimatol Palaeoecol 111:367–379
Kawamura H, Matsuoka N, Momoshima N, Koike M, Takashima Y (2006) Isotopic evidence in tree rings for historical changes in atmospheric sulfur sources. Environ Sci Technol 40:5750–5754
Kobayashi R, Sumarriani Y, Yamashita N, Ohta T, Matsubara H, Yago H, Nakata M, Sase H (2013) Seasonal variation of water chemistry and sulfur budget in an acid-sensitive river along the Sea of Japan. Limnology 14:195–209
Krouse HR (1980) Sulphur isotopes in our environment. In: Fritz P, Fontes J-C (eds) Handbook of environmental isotope geochemistry. Elsevier, Amsterdam, pp 435–471
Kurita H, Ueda H (2006) Long term decrease of pH of river and lake water in the upper-most stream part of the mountainous region in central Japan—decrease of pH in past 30 years in relation with acid rain. J Jpn Soc Atmos Environ 41(2):45–64 (in Japanese with English summary)
Kurokawa J, Ohara T, Morikawa T, Hanayama S, Janssens-Maenhout G, Fukui T, Kawashima K, Akimoto H (2013) Emissions of air pollutants and greenhouse gases over Asian regions during 2000–2008: Regional Emission Inventory in Asia (REAS) version 2. Atmos Chem Phys 13:11019–11058
Likens GE, Bormann FH (1995) Chemistry. In: Likens GE, Bormann FH (eds) Biogeochemistry of a forested ecosystem. Springer, New York, pp 31–72
Likens GE, Driscoll CT, Buso DC, Mitchell MJ, Lovett GM, Bailey SW, Siccama TG, Reiners WA, Alewell C (2002) The biogeochemistry of sulfur at Hubbard Brook. Biogeochemistry 60:235–316
Lu Z, Zhang Q, Streets DG (2011) Sulfur dioxide and primary carbonaceous aerosol emission in China and India, 1996–2010. Atmos Chem Phys 11:9839–9864
Maruyama T, Ohizumi T, Taneoka Y, Minami N, Fukuzaki N, Mukai H, Murano K, Kusakabe M (2000) Sulfur isotope ratios of coals and oils used in China and Japan. Nippon Kagaku Kaishi 1:45–51. https://doi.org/10.1246/nikkashi.2000.45 (in Japanese with English summary)
Matsubara H, Morimoto S, Sase H, Ohizumi T, Sumida H, Nakata M, Ueda H (2009) Long-term declining trends in river water pH in central Japan. Water Air Soil Pollut 200:253–265. https://doi.org/10.1007/s11270-008-9909-3
Mayer B, Shanley JB, Bailey SW, Mitchell MJ (2010) Identifying sources of stream water sulfate after a summer drought in the Sleepers River watershed (Vermont, USA) using hydrological, chemical, and isotopic techniques. Appl Geochem 25:747–754
McLeod AI (2011) Kendall: Kendall rank correlation and Mann–Kendall trend test. R package version 2.2. https://CRAN.R-project.org/package=Kendall
Ministry of the Environment (2012) Mid-term report (JFY 2008–2010), long-term monitoring on transboundary air pollution and acid deposition. Ministry of the Environment, Japan (in Japanese)
Ministry of the Environment (2014) Report of the long-term monitoring on transboundary air pollution and acid deposition (JFY 2008–2012). Ministry of the Environment, Japan (in Japanese)
Mitchell MJ, Likens GE (2011) Watershed sulfur biogeochemistry: shift from atmospheric deposition dominance to climatic regulation. Environ Sci Technol 45:5267–5271
Mitchell MJ, Lovett G, Bailey S, Beall F, Burns D, Buso D, Clair TA, Courchesne F, Duchesne L, Eimers C, Fernandez I, Houle D, Jeffries DS, Likens GE, Moran MD, Rogers C, Schwede D, Shanley J, Weathers KC, Vet R (2011) Comparisons of watershed sulfur budgets in southeast Canada and northeast US: new approaches and implications. Biogeochemistry 103:181–207
Mitchell MJ, Driscoll CT, McHale PJ, Roy KM, Dong Z (2013) Lake/watershed sulfur budgets and their response to decreases in atmospheric sulfur deposition: watershed and climate controls. Hydrol Process 27:710–720
Nakagawa F, Tsunogai U, Obata Y, Ando K, Yamashita N, Saito T, Uchiyama S, Morohashi M, Sase H (2018) Export flux of unprocessed atmospheric nitrate from temperate forested catchments: a possible new index for nitrogen saturation. Biogeosciences 15:7025–7042. https://doi.org/10.5194/bg-15-7025-2018
Nakahara O, Takahashi M, Sase H, Yamada T, Matsuda K, Ohizumi T, Fukuhara H, Inoue T, Takahashi A, Kobayashi H, Hatano R, Hakamata T (2010) Soil and stream water acidification in a forested catchment in central Japan. Biogeochemistry 97:141–158
Nakai N, Tsuji Y, Takeuchi U (1991) Sources of atmospheric sulphur compounds based on the sulphur isotopic composition of SO4 2− in precipitation in Japan, 1960–79. In: Krouse HR, Grinenko VA (eds) SCOPE 43 Stable isotopes: natural and anthropogenic sulphur in the environment. Wiley, Chichester, pp 352–358
Network Center for EANET (2015) Report of the inter-laboratory comparison project 2014. Asia Center for Air Pollution Research, Niigata
Novák M, Kirchner JW, Groscheová H, Havel M, Cerný J, Krejčí R, Buzek F (2000) Sulfur isotope dynamics in two Central European watersheds affected by high atmospheric deposition of SOx. Geochim Cosmochim Acta 64:367–383
Novák M, Bottrell SH, Přechová E (2001) Sulfur isotope inventories of atmospheric deposition, spruce forest floor and living Sphagnum along a NW–SE transect across Europe. Biogeochemistry 53:23–50
Ogawa S (2005) Runoff of rainwater, sediment, and nutrient from forest areas with reference to forest management. Ecol Civ Eng 8:51–59 (in Japanese with English summary)
Ohara T, Akimoto H, Kurokawa J, Horii N, Yamaji K, Yan X, Hayasaka T (2007) An Asian emission inventory of anthropogenic emission sources for the period 1980–2020. Atmos Chem Phys 7:4419–4444
Ohizumi T, Take N, Inomata Y, Yagoh H, Endo T, Takahashi M, Yanahara K, Kusakabe M (2016) Long-term variation of the source of sulfate deposition in a leeward area of Asian continent in view of sulfur isotopic composition. Atmos Environ 140:42–51
Qiao Y, Feng J, Liu X, Wang W, Zhang P, Zhu L (2016) Surface water pH variations and trends in China from 2004 to 2014. Environ Monit Assess 188:443
R Core Team (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org/
Shimizu A, Shimizu T, Miyabuchi Y, Ogawa Y (2003) Evapotranspiration and runoff in a forest watershed in western Japan. Hydrol Process 17:3125–3139
Smith SJ, van Aardenne J, Klimont Z, Andres RJ, Volke A, Delgado Arias S (2011) Anthropogenic sulfur dioxide emissions: 1850–2005. Atmos Chem Phys 11:1101–1116
Svensson T, Lovett G, Likens GE (2012) Is chloride a conservative ion in forest ecosystems? Biogeochemistry 107:125–134
Tamaki M, Hiraki T, Aikawa M (2000) Progress in acid deposition monitoring technology in Japan. Glob Environ Res 4:25–38
Tanikawa T, Takenaka C (1999) Relating sulfate adsorption to soil properties in Japanese forest soils. J For Res 4:217–222
Tanikawa T, Takahashi M, Imaya A, Ishizuka K (2009) Highly accumulated sulfur constituents and their mineralogical relationships in Andisols from central Japan. Geoderma 151:42–49
Tsunogai U, Komatsu DD, Ohyama T, Suzuki A, Nakagawa F, Noguchi I, Takagi K, Nomura M, Fukuzawa K, Shibata H (2014) Quantifying the effects of clear-cutting and strip-cutting on nitrate dynamics in a forested watershed using triple oxygen isotopes as tracers. Biogeosciences 11:5411–5424. https://doi.org/10.5194/bg-11-5411-2014
Van Breemen N, Mulder J, Driscoll CT (1983) Acidification and alkalinization of soils. Plant Soil 75:283–308
Vuorenmaa J, Augustaitis A, Beudert B, Clarke N, de Wit HA, Dirnböck T, Frey J, Forsius M, Indriksone I, Kleemolaa S, Kobler J, Krám P, Lindroos A-J, Lundin L, Ruoho-Airola T, Ukonmaanaho L, Váňa M (2017) Long-term sulphate and inorganic nitrogen mass balance budgets in European ICP Integrated Monitoring catchments (1990–2012). Ecol Indic 76:15–29
Wakita K (1988) Origin of chaotically mixed rock bodies in the Early Jurassic to Early Cretaceous sedimentary complex of the Mino Terrane, central Japan. Bull Geol Surv Jpn 39(11):675–757
Wynn PM, Loader NJ, Fairchild IJ (2014) Interrogating trees for isotopic archives of atmospheric sulphur deposition and comparison to speleothem records. Environ Pollut 187:98–105. https://doi.org/10.1016/j.envpol.2013.12.017
Yamada T, Inoue T, Fukuhara H, Nakahara O, Izuta T, Suda R, Takahashi M, Sase H, Takahashi A, Kobayashi H, Ohizumi T, Hakamata T (2007) Long-term trend in surface water quality of five lakes in Japan. Water Air Soil Pollut Focus 7:259–266
Yamashita N, Sase H, Ohizumi T, Kurokawa J, Ohara T, Morino Y, Kuribayashi M, Ohta S, Kaneko S, Hayashi K, Fukuhara H, Hakamata T (2016) Mapping of the relative risk of surface water acidification based on cumulative acid deposition over the past 25 years in Japan. J For Res 21:115–124
Yoshida F, Wakita K (1999) Geology of the Gifu District. With geological sheet map at 1:50,000. Geological Survey of Japan (in Japanese with English summary)
Acknowledgements
This study was conducted using monitoring data obtained from the MOEJ and the related research outputs. Sulfur isotopic analysis of tree disks was conducted with the support of the Joint Research Grant for the Environmental Isotope Study of Research Institute for Humanity and Nature. The authors thank Emeritus Prof. M. Kusakabe and Prof. H. Chiba, Okayama University, for their advice on sulfur isotopic analysis of water samples. The authors also would like to thank the officers, experts and scientists from all the relevant organizations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Chris D. Evans.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sase, H., Takahashi, M., Matsuda, K. et al. Response of river water chemistry to changing atmospheric environment and sulfur dynamics in a forested catchment in central Japan. Biogeochemistry 142, 357–374 (2019). https://doi.org/10.1007/s10533-019-00540-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-019-00540-1