
Abstract
Polycyclic aromatic hydrocarbons (PAHs) are common contaminants in groundwater. The remediation of PAH-contaminated groundwater often involves anaerobic biodegradation. The knowledge about the microorganisms responsible for PAH degradation in anaerobic subsurface environment is still lacking. DNA-based stable isotope probing (SIP) was applied to discover the microorganisms responsible for anaerobic anthracene degradation within microcosms inoculated with aquifer sediment from landfill leachate-contaminated site. Three phylotypes were identified as the degraders, all falling within the phylum Proteobacteria. Two anthracene degraders were classified within the genera Methylibium and Legionella, while another one was an unclassified Rhizobiales species. They all were first linked to PAH degradation. These findings also provide an illustration of the utility of SIP to discover the roles of uncultured microorganisms in PAH-degrading processes.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The existence of polycyclic aromatic hydrocarbons (PAHs) in the natural environments has caused great concerns, due to their potential threat to environmental ecosystem and human health. PAH compounds are usually present in landfill leachate-contaminated groundwater (Cornelissen et al. 2009; Jiries et al. 2005). Bioremediation is usually an economic and efficient way for PAH reduction at contaminated sites. Up to date, aerobic PAH-degrading bacteria have been well documented (Bamforth and Singleton 2005; Haritash and Kaushik 2009). Species from many genera such as Pseudomonas, Burkholderia, Comamonas, and Ralstonia are able to biodegrade an exceptionally wide variety of PAHs, using them as carbon or energy source (Chang et al. 2007). Moreover, numerous studies have investigated PAH biodegradation under different redox conditions, mostly with nitrate or sulfate as terminal electron acceptors (Chang et al. 2005b; Meckenstock et al. 2004). Pure cultures of several anaerobic nitrate- and sulfate-reducing bacteria, capable of degrading PAHs, have also been identified and most of the species are from the genus Pseudomonas (Haritash and Kaushik 2009). However, little information is available for anaerobic PAH biodegradation under methanogenic condition (Chang et al. 2005b, 2006).The anaerobic isolate able to degrade PAHs under methanogenic condition is still lacking.
Although many isolates have been obtained, microorganisms able to transform PAHs in a pure culture may not be able to perform the same function in the field. Different culture conditions or habitats may select for specific PAH degraders. Molecular approaches (e.g., denaturing gradient gel electrophoresis (DGGE), clone libraries, or terminal restriction fragment length polymorphism (TRFLP)) have enabled the identification of community members in a mixed culture sample. For example, clone library analysis showed the δ-proteobacteria was the major bacterial group in naphthalene-degrading methanogenic cultures initiated with Baltimore Harbor sediments, but the Firmicutes predominated in phenanthrene-degrading methanogenic culture (Chang et al. 2005b). However, these techniques typically do not directly link the ability to degrade a specific contaminant to individual species (Xie et al. 2010). Therefore, little is known about the microorganisms actually responsible for PAH transformation in mixed cultures or in situ at contaminated sites.
Stable isotope probing (SIP) has gained increasing attention as a novel tool for linking function with identity for environmental samples (Radajewski et al. 2000). Recently, SIP has been applied to identify naphthalene degraders in coal tar-contaminated sediment and soil (Jeon et al. 2003; Yu and Chu 2005), organisms capable of degrading naphthalene, phenanthrene, or pyrene in a bioreactor treating soil from a former manufactured-gas plant (Singleton et al. 2005, 2006), pyrene-degrading bacteria in creosote-contaminated soil (Jones et al. 2008). However, all these previous SIP works have been targeted on aerobic PAH-degrading microorganisms.
Due to both the low amount of dissolved oxygen and the slow diffusion of oxygen into subsurface, microorganisms can rapidly deplete oxygen, which suggests that anaerobic biodegradation might be more attractive in the subsurface environment (Chang et al. 2005a). SIP has recently been used to identify the active degraders of contaminants in various anaerobic environments, such as anaerobic benzene degraders in contaminated aquifer (Herrmann et al. 2010), or in freshwater sediment (Liou et al. 2008), sulfate-reducing toluene degrader in a BTEX contaminated aquifer (Bombach et al. 2010), benzoate-utilizing denitrifying bacteria in marine sediments (Gallagher et al. 2005), anaerobic phenol degraders in the treatment of synthetic coke-oven wastewater (Sueoka et al. 2009), and perchloroethene-respiring microorganisms in anoxic river sediment (Kittelmann and Friedrich 2008). These studies all illustrate SIP provides significant potential for understanding in situ anaerobic degradation.
Anthracene is selected as a PAHs model in this study. To obtain a more complete understanding of anthracene degradation in mixed culture, DNA-based SIP was applied here to investigate the microorganisms responsible for anaerobic anthracene degradation within microcosms inoculated with aquifer sediment from landfill leachate-contaminated sites. To the authors’ knowledge, this was the first report directly linking anaerobic PAH degradation to specific microorganisms in a mixed community sample using SIP. This work could provide additional insights on the microbial ecology of anaerobic PAH degradation in mixed culture samples.
Materials and methods
Soil incubation and chemical analyses
Subsurface aquifer sediment utilized in this study was collected from a landfill leachate-contaminated site. Following sample collection, aquifer sediment was homogenized and sieved through a 0.18-mm screen, and stored at 4°C until use. Sediment microcosms consisted of mineral salt medium (10 ml), as previously described (Yang and McCarty 1998), and sediment (3 g) in serum bottles (150 ml). Three treatments were prepared: sterile control, sample amended with unlabeled anthracene (120 μg, 99%, J&K China Chemical), and sample amended with labeled anthracene (120 μg, ring-13C6 anthracene, 99%, Cambridge Isotope Laboratories). Anthracene stock was first prepared in nonane. A volume of nonane containing the desired mass of anthracene was added to the serum bottle and the solvent was allowed to evaporate. Mineral salt medium and sediment was then added to the anthracene crystals in the bottles. No supplementary electron acceptor was added to the cultures in this study. To obtain anaerobic condition, the headspace in bottles was vacuumed for 10 s, and then refilled with purified N2 gas by puncturing the septa with a 1 ml syringe. This procedure was repeated three times to ensure that all of the oxygen inside the bottles was replaced by N2 gas (Li et al. 2010). The bottles were sealed with rubber stoppers and aluminum seals to maintain anaerobic conditions. The sterile controls were obtained by autoclaving repeatedly (three times). All samples exposed to anthracene (labeled or unlabeled) were prepared in duplicate, and thus the SIP investigation occurred in duplicate. Duplicate samples were not pooled, and the entire analysis for each was carried out separately.
Microcosms were incubated on a horizontal shaker (~100 rpm) at 20°C. A 120-day preliminary microcosm experiment (amended with unlabeled anthracene) indicated that percent anthracene remaining in solid phase on day 30, 60, 90, and 120 were 85–88, 42–45, 23–26, and 12–14%, respectively. The active degrader would be more enriched when the target compound was more significantly degraded (Luo et al. 2009). Therefore, on day 120, microcosms were sacrificed and sediment samples in duplicate were dried using a freeze drier (Alpha 1–2 LD plus, Martin Christ, German).Two grams of dried sediment samples were removed for DNA extraction (see below) and the remaining sediment was extracted with acetone for anthracene analysis, as previously described (Zhang et al. 2011). The anthracene analysis in solid phase was conducted using a HPLC apparatus (Shimadzu LC-10Avp, Agilent Technologies) equipped with a LC-10AT pump, a UV-detector, a Venusil PAH column (Agela Technologies). Methanol–water (90:10) was used as the mobile phase at a flow rate of 1 ml min−1. Anthracene was detected by absorbance at 251 nm with the mean recovery rate of 97%. Gas headspace samples were analyzed for methane with a gas chromatograph with a thermal conductivity detector. The operational temperature of the column was 40°C, and the carrier gas (N2) was at a flow rate of 30 ml min−1 (He et al. 2008).
DNA extraction and ultracentrifugation
Following the depletion of anthracene, DNA was extracted from sediment samples of replicate labeled and replicate unlabeled anthracene amended microcosms with the Powersoil kit (Mobio Laboratories) following the manufacturer’s instructions. Separation of the unlabeled and 13C-labeled DNA was accomplished by density gradient ultracentrifugation in cesium chloride according to the standard method (Xie et al. 2010, 2011), except that an Optima LE-80K Preparative Ultracentrifuge (Beckman Instruments) was used. Following ultracentrifugation, the tubes were placed into a fraction recovery system (Beckman Coulter) for fraction (200 μl) collection. The buoyant density (BD) of each fraction was measured (model AR200 digital refractometer, Leica Microsystems Inc.). CsCl was removed by glycogen-assisted ethanol precipitation and purified DNA fractions were stored at −20°C.
TRFLP and 16S rRNA gene sequencing
Each ultracentrifugation fraction for both labeled and unlabeled microcosms was subject to TRFLP analysis. Bacterial 16S rRNA genes were amplified using primers 27F-FAM (5′-GAGTTTGATCMTGGCTCAG-3′, 5′ end-labeled with carboxyfluorescine) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) (Tiangen BioTech, China) (Xie et al. 2010, 2011). The PCR program was run as follows: 94°C (5 min); 94°C (30 s); 55°C (30 s); 72°C (1.5 min) (30 cycles); 72°C (5 min). PCR products (300 ng) were purified with QIAquick PCR purification kit (Qiagen Inc.), following the manufacturer’s instructions. Purified PCR products were digested with HaeIII with a 6 h incubation period at 37°C. Digests HhaI and MspI were used to correlate the enriched TRFLP fragment lengths in the heavy fractions to the in silico cut sites of the cloned sequences (Xie et al. 2010). The in silico analysis was performed online (http://www.restrictionmapper.org/). The same purified PCR product was used for digestion with HaeIII, HhaI and MspI. The clone sequence was first identified, whose in silico cut site (HaeIII digest) matched the length of the enriched fragment (HaeIII digest) in the heavy fractions. If the clone restriction enzyme (HhaI and MspI) cut site predicted from sequence also matched the observed fragment length (HhaI and MspI digest) in the heavy fractions, the taxonomic identity of the enriched fragment (HaeIII digest) could be confirmed (Cupples and Sims 2007). The fragment pattern was detected by using an ABI 3730 DNA Analyzer (Applied Biosystems) with an internal lane standard (GeneScan-500 LIZ Size Standard, Applied Biosystems). The percent abundance of each fragment was determined, as previously described (Yu and Chu 2005).
For 16S rRNA gene sequencing, heavy fraction 13C-DNA was amplified as above, except the forward primer was unlabeled. PCR products were purified and cloned into pMD19-T vector (Takara Corp, Japan) following the manufacturer’s instruction. E. coli clones were grown on Luria–Bertani (LB) medium solidified with 15 g l−1 agar with 50 μg l−1 ampicillin for 16 h at 37°C. The white colonies were verified by PCR with primers M13 F (5′-TGTAAAACGACGGCCAGT-3′) and M13 R (5′-AACAGCTATGACCATG-3′). Clones were sequenced at SinoGenoMax Co., Ltd. (Beijing) and chimerism was checked using the Chimera Check program available at the MSU Center for Microbial Ecology. The closest GenBank sequences to microorganisms identified in this study were searched from the National Center for Biotechnology Information (NCBI) database (Altschul et al. 1990). The Ribosomal Database Project (MSU Center for Microbial Ecology) analysis tool “classifier” was utilized to determine taxonomic identity (Wang et al. 2007). The bacterial partial 16S rRNA gene sequences (800 bp) of microorganisms linked to anthracene degradation were deposited with GenBank under accession numbers HQ888823–HQ888825.
Results
Methane production in microcosms was observed during the whole biodegradation experiment, with an increase from 0 (day 0) to approximately 120 μmol l−1 (day 120), while the amount was negligible in the sterile controls, indicating the occurrence of methanogenesis in microcosms. The degradation pattern was similar for sediment amended with either the labeled or unlabeled anthracene. Approximately half of the anthracene remained after 60 days and a more significant depletion was observed after 120 days, compared with very limited decline in the autoclaved control, confirmed a biological removal mechanism (Table 1).
DNA extracts (day 120) from labeled and unlabeled anthracene amended sediment samples were subject to ultracentrifugation, fractionation, followed by TRFLP on each fraction. Analyses of TRFLP profiles illustrated the enrichment of three different fragments in the heavy fractions obtained from the 13C-anthracene microcosms but not in the unlabeled microcosms (Fig. 1). Relative abundances of the three fragments in TRFLP profiles over a range of BD values are illustrated in Fig. 2. Notably, three TRFLP fragments (193, 198 and 206 bp) were enriched in the heavy fractions. This suggests three different microorganisms were responsible for anthracene biodegradation. Peak relative abundance values in 13C-anthracene amended samples were 6.3 or 7.5% (at 1.734 g ml−1), 7 or 8% (at 1.715 g ml−1), and 8.1 or 9.3% (at 1.737 g ml−1) for TRFLP fragments 193, 198 and 206 bp, respectively (Fig. 2).

Comparison of heavy fraction TRFLP profiles from unlabeled (12C) and labeled (13C) anthracene amended sediments to illustrate the enriched fragments in labeled fractions compared to the unlabeled fractions control. The duplicate illustrated the same trend

Relative abundance of the three enriched TRFLP fragments (a–c) in fractions of three 13C-anthracene amended sediment compared to unlabeled anthracene amended sediment (day 120)
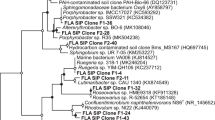
To determine the identity of these enriched fragments approximately 60 clones were partially sequenced. In addition, abundant TRFLP fragments (relative abundance above 1%) obtained with selected 13C fractions with HhaI or MspI were used to provide a confirmative identification of each HaeIII TRFLP enriched fragment (Fig. 3). The abundant fragments obtained from all digests were compared to those obtained from in silico digests to determine the sequence of each enriched fragment. Comparison of TRFLP and in silico cut sites for each identified fragment is presented (Table 2). A slight difference (2–3 bases) between the observed fragment lengths and those predicted using sequence data has also been noted elsewhere (Luo et al. 2009; Xie et al. 2010, 2011). The taxonomic identity of each enriched fragment (cloned sequence) is presented (Table 3).The enriched HaeIII TRFLP fragments 193, 198 and 206 bp in 13C heavy DNA fraction were identified as unclassified Rhizobiales, genera Methylibium and Legionella, respectively.

TRFLP profiles from fractions (1.715 and 1.737 g ml−1) of 13C labeled anthracene amended sediment with two different restriction enzymes
Discussion
A field-based SIP work has shown that a β-proteobacterium strain was responsible for naphthalene degradation in coal tar-contaminated sediment (Jeon et al. 2003). β- and γ-Proteobacteria were responsible for naphthalene degradation in coal tar-contaminated soil (Yu and Chu 2005). The primary pyrene degraders identified in a bioreactor treating soil from a former manufactured-gas plant site were uncultivated β- and γ-proteobacteria (Singleton et al. 2006). The γ-proteobacteria responsible for pyrene degradation was the most prominent sequences recovered in 13C heavy DNA fraction from creosote-contaminated soil treated either by the addition of inorganic nutrients or by slurrying (Jones et al. 2008). These previous reports provide strong evidence that phylum Proteobacteria is widely involved in PAH degradation. The current study was to investigate the active anaerobic PAH degraders in microcosm inoculated with subsurface sediment a landfill leachate-contaminated site. DGGE analysis showed the dominant bacterial species in groundwater contaminated with landfill leachate were also α-, β- or γ-proteobacteria (Röling et al. 2001). Sequence analysis of 16S rDNA clone library indicated the presence of β-, γ-, δ-, ε-proteobacteria, Bacteroidetes, Firmicutes, Actinobacteria, and Cyanobacteria in leachate-polluted aquifer (Tian et al. 2005). In this study, members of α-, β- and γ-proteobacteria were directly linked to anthracene degradation under methanogenic condition. This finding sustained the role of phylum Proteobacteria in PAH degradation under methanogenic condition. However, given the slow anthracene degradation rates observed, the possibility of label cross-feeding can not entirely be ruled out.
Clone libraries of 13C labeled heavy DNA from the aerobic phenanthrene and naphthalene incubations were composed primarily of genus Acidovorax, and genera Pseudomonas and Ralstonia, respectively (Singleton et al. 2005). Another two previous SIP works showed genus Polaromonas or Intrasporangium was responsible for aerobic naphthalene degradation in coal tar-contaminated sites (Jeon et al. 2003; Yu and Chu 2005). In this study, genera Methylibium and Legionella, and unclassified Rhizobiales were responsible for anaerobic anthracene degradation.
Many different bacteria able to degrade anthracene aerobically, previously isolated from PAH-contaminated site, belong to genera Mycobacterium (Pizzul et al. 2007), Pseudomonas (Al-Thani et al. 2009), Achromobacter (Al-Thani et al. 2009), Janibacter (Yamazoe et al. 2004), and Rhodococcus (Tongpim and Pickard 1996). It is of interest to be noted that any previously reported anthracene degraders were not identified here. The active anthracene degraders might be site-dependent. In this study, the microcosms were inoculated with aquifer sediment in landfill leachate-contaminated site. To the authors’ knowledge, no isolate of PAH degrader has been obtained from the kind of habitat.
The enriched HaeIII TRFLP fragment 193 bp in 13C heavy DNA fraction was identified here as an unclassified Rhizobiales species within the α-proteobacteria. The 16S rRNA gene sequence of the Rhizobiales (HQ888823) illustrated high similarity to reported sequences within Genbank. The three most similar partial 16S rRNA gene sequences (99% similarity) (CU918797.1, CU925735.1 and CU927124.1) originated from anaerobic sludge digesters (Rivière et al. 2009). The closest cultivated match was a Rhizobiales species (AJ810382.1, 96% identity) from legume (Lafay and Burdon 2006). Some members of the order Rhizobiales could degrade benzene, toluene, ethylbenzene and xylene (Cavalca et al. 2004), hydroxy- and dihydroxy-benzoates, halogenated aromatic compounds (Vela et al. 2002), and phenol (Baek et al. 2003). However, to the authors’ knowledge, this was also the first study to directly link the order Rhizobiales to the degradation of any PAH compounds. Moreover, although some α-proteobacteria species are well known for their ability to transform PAHs (Lafortune et al. 2009), the uniqueness of the 16S rRNA gene sequence identified here appears to represent a truly novel PAH degrader within the class.
The microorganism (with representative HaeIII fragment 198 bp) was classified within the β-proteobacteria as a Methylibium species. The three closest matches of the sequence (HQ888824) of Methylibium strain identified here were obtained from a restored grassland (EU300364.1, 100%, unpublished GenBank data), trichloroethene-contaminated groundwater with propane-stimulated bioremediation (AY435511.1, 98%) (Connon et al. 2005), and tallgrass prairie soil (FJ479220.1, 97%). Moreover, the 16S rRNA gene sequence of the Methylibium obtained in this study was closely related to a Methylibium strain (DQ664244.1, 97%) and an Azonexus strain (DQ664241.1, 97%) isolated from a eutrophic freshwater pond (Chou et al. 2008).Some species of Methylibium have usually been linked to aerobic methyl tert-butyl ether (MTBE) degradation (Kane et al. 2007; Nakatsu et al. 2006). However, a Methylibium species could also degrade aromatic (benzene, toluene, and xylene) and straight-chain hydrocarbons present in petroleum products (Kane et al. 2007). To the authors’ knowledge, this was also the first study to directly link a microorganism within this genus to the degradation of any PAH compounds.
The third microorganism (with representative HaeIII fragment 206 bp) responsible for anthracene degradation under methanogenic condition was classified within the γ-proteobacteria as a Legionella species. The 16S rRNA gene sequence (HQ888825) of the Legionella strain identified in this study had a low similarity (95%) to the sequences in GenBank, representing a novel PAH degrader within the class γ-proteobacteria. Surprisingly, little information exists on microorganisms within the genus Legionella. Two members of genus Legionella have been linked to be opportunistic pathogen or pathogen, respectively (Jjemba et al. 2010; Solomon et al. 2000). However, to the authors’ knowledge, there has been no report concerning the role of genus Legionella in degradation of environmental contaminants. Therefore, the result obtained in the current study could contribute to the limited pool of knowledge on the function of this novel organism within mixed microbial communities.
Conclusions
SIP was used to identify the microorganisms responsible for anaerobic anthracene degradation within microcosms inoculated with aquifer sediment from landfill leachate-contaminated sites. Three phylotypes within the phylum Proteobacteria were identified as the degraders. Two were classified within the genera Methylibium and Legionella respectively, and another one was an unclassified Rhizobiales. They all were first linked to PAH degradation. These findings have the potential to add important new knowledge to PAH degradation under methanogenic conditions.
References
Al-Thani RF, Abd-El-Haleem DAM, Al-Shammri M (2009) Isolation and characterization of polyaromatic hydrocarbons-degrading bacteria from different Qatari soils. Afr J Microbiol Res 3:761–766
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Baek SH, Kim KH, Yin CR, Jeon CO, Im WT, Kim KK, Lee ST (2003) Isolation and characterization of bacteria capable of degrading phenol and reducing nitrate under low-oxygen conditions. Curr Microbiol 47:462–466
Bamforth SM, Singleton L (2005) Bioremediation of polycyclic aromatic hydrocarbons, current knowledge and future directions. J Chem Technol Biotechnol 80:723–736
Bombach P, Chatzinotas A, Neu TR, Kästner M, Lueders T, Vogt C (2010) Enrichment and characterization of a sulfate-reducing toluene degrading microbial consortium by combining in situ microcosms and stable isotope probing techniques. FEMS Microbiol Ecol 71:237–246
Cavalca L, Dell’Amico E, Andreoni V (2004) Intrinsic bioremediability of an aromatic hydrocarbon-polluted groundwater: diversity of bacterial population and toluene monooxygenase genes. Appl Microbiol Biotechnol 64:576–587
Chang W, Um YS, Holoman TRP (2005a) Molecular characterization of anaerobic microbial communities from benzene-degrading sediments under methanogenic conditions. Biotechnol Prog 21:1789–1794
Chang W, Um YS, Holoman TRP (2005b) Molecular characterization of polycyclic aromatic hydrocarbon (PAH)-degrading methanogenic communities. Biotechnol Prog 21:682–688
Chang W, Um YS, Holoman TRP (2006) Polycyclic aromatic hydrocarbon, PAH degradation coupled to methanogenesis. Biotechnol Lett 28:425–430
Chang YT, Lee JF, Chao HP (2007) Variability of communities and physiological characteristics between free-living bacteria and attached bacteria during the PAH biodegradation in a soil/water system. Eur J Soil Biol 43:283–296
Chou JH, Jiang SR, Cho JC, Song J, Lin MC, Chen WM (2008) Azonexus hydrophilus sp. nov., a nifH gene-harbouring bacterium isolated from freshwater. Int J Syst Evol Microbiol 58:946–951
Connon SA, Tovanabootr A, Dolan M, Vergin K, Giovannoni SJ, Semprini L (2005) Bacterial community composition determined by culture-independent and -dependent methods during propane-stimulated bioremediation in trichloroethene-contaminated groundwater. Environ Microbiol 7:165–178
Cornelissen G, Okkenhaug G, Breedveld GD, Sorlie JE (2009) Transport of polycyclic aromatic hydrocarbons and polychlorinated biphenyls in a landfill: a novel equilibrium passive sampler to determine free and total dissolved concentrations in leachate water. J Hydrol 369:253–259
Cupples AM, Sims GK (2007) Identification of in situ 24-dichlorophenoxyacetic acid-degrading soil microorganisms using DNA-stable isotope probing. Soil Biol Biochem 39:232–238
Gallagher E, McGuinness L, Phelps C, Young LY, Kerkhof LJ (2005) 13C-carrier DNA shortens the incubation time needed to detect benzoate-utilizing denitrifying bacteria by stable-isotope probing. Appl Environ Microbiol 71:5192–5196
Haritash AK, Kaushik CP (2009) Biodegradation aspects of polycyclic aromatic hydrocarbons (PAHs): a review. J Hazard Mater 169:1–15
He R, Ruan AD, Jiang CJ, Shen DS (2008) Responses of oxidation rate and microbial communities to methane in simulated landfill cover soil microcosms. Bioresour Technol 99:7192–7199
Herrmann S, Kleinsteuber S, Chatzinotas A, Kuppardt S, Lueders T, Richnow HH, Vogt C (2010) Functional characterization of an anaerobic benzene-degrading enrichment culture by DNA stable isotope probing. Environ Microbiol 12:401–411
Jeon CO, Park W, Padmanabhan P, DeRito C, Snape JR, Madsen EL (2003) Discovery of a bacterium, with distinctive dioxygenase, that is responsible for in situ biodegradation in contaminated sediment. Proc Natl Acad Sci USA 100:13591–13596
Jiries A, Rimawi O, Lintelmann J, Batarseh M (2005) Polycyclic aromatic hydrocarbons (PAH) in top soil, leachate and groundwater from Ruseifa solid waste landfill, Jordan. Int J Environ Pollut 23:179–188
Jjemba PK, Weinrich LA, Cheng W, Giraldo E, LeChevallier MW (2010) Regrowth of potential opportunistic pathogens and algae in reclaimed-water distribution systems. Appl Environ Microbiol 76:4169–4178
Jones MD, Singleton DR, Carstensen DP, Powell SN, Swanson JS, Pfaender FK, Aitken MD (2008) Effect of incubation conditions on the enrichment of pyrene-degrading bacteria identified by stable-isotope probing in an aged, PAH-contaminated soil. Microb Ecol 56:341–349
Kane SR, Chakicherla AY, Chain PSG, Schmidt R, Shin MW, Legler TC, Scow KM, Larimer FW, Lucas SM, Richardson PM, Hristova KR (2007) Whole-genome analysis of the methyl tert-butyl ether-degrading beta-proteobacterium Methylibium petroleiphilum PM1. J Bacteriol 189:1931–1945
Kittelmann S, Friedrich MW (2008) Identification of novel perchloroethene-respiring microorganisms in anoxic river sediment by RNA-based stable isotope probing. Environ Microbiol 10:31–46
Lafay B, Burdon JJ (2006) Molecular diversity of rhizobia nodulating the invasive legume Cytisus scoparius in Australia. J Appl Microbiol 100:1228–1238
Lafortune I, Juteau P, Deziel E, Lepine F, Beaudet R, Villemur R (2009) Bacterial diversity of a consortium degrading high-molecular-weight polycyclic aromatic hydrocarbons in a two-liquid phase biosystem. Microb Ecol 57:455–468
Li CH, Wong YS, Tama NFY (2010) Anaerobic biodegradation of polycyclic aromatic hydrocarbons with amendment of iron III in mangrove sediment slurry. Bioresour Technol 101:8083–8092
Liou JSC, DeRito CM, Madsen EL (2008) Field-based and laboratory stable isotope probing surveys of the identities of both aerobic and anaerobic benzene-metabolizing microorganisms in freshwater sediment. Environ Microbiol 10:1964–1977
Luo CL, Xie SG, Sun WM, Li XD, Cupples AM (2009) Identification of a novel toluene-degrading bacterium from the candidate phylum TM7, as determined by DNA stable isotope probing. Appl Environ Microbiol 75:4644–4654
Meckenstock RU, Safinowski M, Griebler C (2004) Anaerobic degradation of polycyclic aromatic hydrocarbons. FEMS Microbiol Ecol 49:27–36
Nakatsu CH, Hristova K, Hanada S, Meng XY, Hanson JR, Scow KM, Kamagata Y (2006) Methylibium petrolelphilum gen. nov., sp nov., a novel methyl tert-butyl ether-degrading methylotroph of the Betaproteobacteria. Int J Syst Evol Microbiol 56:983–989
Pizzul L, Sjogren A, Castillo MD, Stenstroem J (2007) Degradation of polycyclic aromatic hydrocarbons in soil by a two-step sequential treatment. Biodegradation 18:607–616
Radajewski S, Ineson P, Parekh NR, Murrell JC (2000) Stable-isotope probing as a tool in microbial ecology. Nature 403:646–649
Rivière D, Desvignes V, Pelletier E, Chaussonnerie S, Guermazi S, Weissenbach J, Li T, Camacho P, Sghir A (2009) Towards the definition of a core of microorganisms involved in anaerobic digestion of sludge. ISME J 3:700–714
Röling WFM, Van Breukelen BM, Braster M, Lin B, Van Verseveld HW (2001) Relationships between microbial community structure and hydrochemistry in a landfill leachate-polluted aquifer. Appl Environ Microbiol 67:4619–4629
Singleton DR, Powell SN, Sangaiah R, Gold A, Ball LM, Aitken MD (2005) Stable-isotope probing of bacteria capable of degrading salicylate, naphthalene, or phenanthrene in a bioreactor treating contaminated soil. Appl Environ Microbiol 71:1202–1209
Singleton DR, Sangaiah R, Gold A, Ball LM, Aitken MD (2006) Identification and quantification of uncultivated proteobacteria associated with pyrene degradation in a bioreactor treating PAH contaminated soil. Environ Microbiol 8:1736–1745
Solomon JM, Rupper A, Cardelli JA, Isberg RR (2000) Intracellular growth of Legionella pneumophila in Dictyostelium discoideum, a system for genetic analysis of host–pathogen interactions. Infect Immun 68:2939–2947
Sueoka K, Satoh H, Onuki M, Mino T (2009) Microorganisms involved in anaerobic phenol degradation in the treatment of synthetic coke-oven wastewater detected by RNA stable-isotope probing. FEMS Microbiol Lett 291:169–174
Tian YJ, Yang H, Wu XJ, Li DT (2005) Molecular analysis of microbial community in a groundwater sample polluted by landfill leachate and seawater. J Zhejiang Univ Sci B 23:165–170
Tongpim S, Pickard MA (1996) Growth of Rhodococcus S1 on anthracene. Can J Microbiol 42:289–294
Vela S, Haggblom MM, Young LY (2002) Biodegradation of aromatic and aliphatic compounds by rhizobial species. Soil Sci 167:802–810
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Xie SG, Sun WM, Luo CL, Cupples AM (2010) Stable isotope probing identifies novel m-xylene degraders in soil microcosms from contaminated and uncontaminated sites. Water Air Soil Pollut 212:113–122
Xie SG, Sun WM, Luo CL, Cupples AM (2011) Novel aerobic benzene degrading microorganisms identified in three soils by stable isotope probing. Biodegradation 22:71–81
Yamazoe A, Yagi O, Oyaizu H (2004) Degradation of polycyclic aromatic hydrocarbons by a newly isolated dibenzofuran-utilizing Janibacter sp strain YY-1. Appl Microbiol Biotechnol 65:211–218
Yang Y, McCarty PL (1998) Competition for hydrogen within a chlorinated solvent dehalogenating anaerobic mixed culture. Environ Sci Technol 32:3591–3597
Yu CP, Chu KH (2005) A quantitative assay for linking microbial community function and structure of a naphthalene-degrading microbial consortium. Environ Sci Technol 39:9611–9619
Zhang SY, Wang QF, Xie SG (2011) Microbial community changes in contaminated soils in response to phenanthrene amendment. Int J Environ Sci Technol 8:321–330
Acknowledgments
This work was financially supported by National Natural Science Foundation of China (No. 50979002).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, S., Wang, Q. & Xie, S. Stable isotope probing identifies anthracene degraders under methanogenic conditions. Biodegradation 23, 221–230 (2012). https://doi.org/10.1007/s10532-011-9501-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10532-011-9501-1