
Abstract
Halyomorpha halys is an invasive stink bug pest originating from East Asia. In Europe, it was first detected in Switzerland in 2004. It is now present in thirteen countries, and seems to be spreading throughout the continent. In Italy, where it has been recorded since 2012, other than being an urban nuisance, it is causing severe damage in commercial fruit orchards. An integrated approach, using current and previous observational data in space and time and molecular information, was used to identify the genetic diversity of this pest in Europe, its invasion history, and the potential pathways of entry and diffusion. The analysis of 1175 bp of mitochondrial DNA cytochrome c oxidase I and II genes (cox1, cox2) led to the identification of twenty previously unknown haplotypes. The European distribution of H. halys is the result of multiple invasions that are still in progress, and, in some cases, it was possible to identify the specific Asian areas of origin. Moreover, secondary invasions could have occurred among European countries by a bridgehead effect. In Italy, the data were more clearly related to their temporal occurrence, allowing for a clearer reading of the patterns of invasion and dispersion. After having successfully established in localized areas, H. halys further expanded its range by an active dispersion process and/or by jump dispersal events due to passive transport. The multiple introductions from different areas of the native range together with the different patterns of diffusion of H. halys, may hamper the pest management strategies for its containment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Biological invasions result from accidental or intentional dispersal events, linked to human activities, of alien species. The potential for an alien population to become invasive is related to its ability to adapt to new or changing environmental conditions (Fisher 1930; Estoup and Guillemaud 2010). Inferring source populations, routes of invasions and patterns of dispersal of founding populations are important issues for defining and testing the evolutionary hypothesis involved in the success of invasive alien species (Estoup and Guillemaud 2010; Handley et al. 2011), projecting their spread (Kolar and Lodge 2001), and potentially directing the focus on conservation strategies and control management (Handley et al. 2011). The knowledge of life history and genetic traits that characterize an invasive species are therefore the starting point for a better understanding of the ecological and evolutionary dynamics underlying biological invasions. The genetic variability of invading populations, in terms of both molecular and quantitative traits, depends on the history of their populations of origin and the historical and demographic features of their introduction. For these reasons, it is important to estimate the number of specimens and the genetic diversity of the invasive population, the patterns of dispersal and diffusion into a new territory (Estoup and Guillemaud 2010), and the eventual presence of multiple introductions that usually increase the amounts of variation (see Dlugosch and Parker 2008).
A species that has been demonstrated to have an extremely high potential as global invasive organism is Halyomorpha halys (Stål, 1855) (Heteroptera, Pentatomidae), a phytophagous insect native to East Asia (Lee et al. 2013, 2017; Zhu et al. 2016), which is becoming an emerging pest of agricultural crops in Europe (Bariselli et al. 2016; Maistrello et al. unpublished). After the first report in Switzerland in 2007 (Wermelinger et al. 2008; Gariepy et al. 2014), very probably the initial entry point of this pest in Europe, H. halys was detected all over the continent: Liechtenstein (Arnold 2009), Greece (Milonas and Partsinevelos 2014), Germany (Heckmann 2012), Italy (Maistrello et al. 2014; Cesari et al. 2015); France (Callot and Brua 2013; Maurel et al. 2016), Hungary (Vétek et al. 2014), Austria (Rabitsch and Griebe 2015), Romania (Macavei et al. 2015), Serbia (Šeat 2015), Spain (Dioli et al. 2016), Russia (Mityushev 2016), and Bulgaria (Simov 2016). It has also been intercepted twice in Britain (Malumphy 2014). According to model predictions, under future climate scenarios, H. halys populations could also expand to previously unaffected regions of Europe, especially in the Mediterranean area (Haye et al. 2015; Kriticos et al. 2017) and in the Northern regions (Zhu et al. 2012). Trade increase with native and/or invaded areas could also lead to the expansion of H. halys populations to higher latitudes (i.e. Northern Europe). Moreover, in Italy, where H. halys is reported since 2012 (Cesari et al. 2015; Maistrello et al. 2016b), high damage to fruit orchards were reported during the summer of 2015 (Maistrello et al. 2016a, 2017), eliciting a high concern for Italian agricultural production (Fanfani and Pieri 2016). Other than Europe, H. halys has also invaded the U.S.A. (Zhu et al. 2016), where it was detected for the first time in 1996, although it was positively identified only in 2001 (Hoebeke and Carter 2003). From then on, H. halys has become a widespread agricultural pest occurring in all U.S. states east of the Mississippi River, as well as in several western coastal states and in Canada (Leskey et al. 2012; Rice et al. 2014; Haye et al. 2015). Interceptions of this species were reported in Australia and New Zealand (Walker 2009; Duthie et al. 2012; MacLellan 2013; Ward 2013) and reports of its presence have come from Guam (Mariana Islands, Pacific Ocean; Moore 2014) and India (Nikam and More 2016).
The accessibility and affordability of molecular technologies, coupled with the expansion of databases housing publicly available DNA sequence data, have helped in the use of molecular tools to evaluate the diversity and potential origin of source populations of H. halys. However, the records are still few, considering the spread and importance of this species as a pest (Gariepy et al. 2014, 2015; Xu et al. 2014; Cesari et al. 2015; Zhu et al. 2016; Lee et al. 2017). Some of these studies tried to identify the potential pathways of diffusion of H. halys in Europe (Gariepy et al. 2014, 2015; Cesari et al. 2015), but due to the early stage in the detection of this pest, the relatively low numbers of analysed specimens and molecular markers, and especially the very low data available from the area of origin, their conclusions were generally speculative and preliminary. However, patterns of haplotype diversity seemed to indicate the movement of successful invasive populations to generate secondary invasions within Europe, as well as the occurrence of multiple invasions from Asia (Gariepy et al. 2015), while in Italy, the invasion probably occurred from two different pathways, both from Switzerland and from Asia and/or North America (Cesari et al. 2015). Since biological invasion events often occur over large temporal and spatial scales, and can involve multiple introductions of a small number of individuals from one or more source populations, many of these events go undetected, unreported, and/or the data are not readily available to those outside of phytosanitary control organizations (Corin et al. 2007; Hulme et al. 2008). For this reason, to rapidly obtain data from a very large area, we collaborated with plant protection services at local, national, and international levels, and with the general public, applying a crowd-sourcing approach (Maistrello et al. 2016b).
The present study aims to identify the genetic diversity of this pest in Europe and the potential pathways of entry and diffusion of H. halys populations in Italy. Previous papers presenting molecular studies on H. halys showed a single picture in a defined specific time, not considering the timeframe in which the invasion occurred, thus reducing the interpretative potential of the data. In this study, we examined the genetic diversity of H. halys populations both in space and time to better understand the dispersal and dispersion patterns of this highly invasive species. We therefore integrated the two types of methods that have traditionally been used to make inferences concerning the routes of introduction of alien species: direct methods based on current and historical observations, and indirect methods based on patterns in population genetic data (Estoup and Guillemaud 2010). Newly collected specimens in Italy, Switzerland, Greece and Romania were analysed and the data were compared with others from all over Europe, North America and Asia. The integration of genetic, geographical and temporal data has proven to be useful for giving clearer indications on the pathways that this highly invasive pest is following throughout Europe and Italy.
Materials and methods
Halyomorpha halys specimens were collected in a single locality in Switzerland, Greece, Romania, and in ten Italian regions (Table 1, Online Resource 1) from agricultural crops, urban ornamental plants, and private residences. Moreover, the numbers and frequencies of H. halys in Italian provinces and regions in the years 2012–2016 were obtained during a survey (Maistrello et al., unpublished) that included both active sampling by entomologists, students and personnel of plant protection services as well as crowd sourcing data obtained with the involvement of volunteers via multimedia channels, using a procedure and database similar to that described in Maistrello et al. (2016b). Basically, in all cases, contributors were invited to collect or take high quality photographs of any brown-grey marmorated bugs and to send them to the project coordinators, together with details on the collection/sighting specifying when, where (location and context of occurrence) and how many bugs were detected (using an abundance index). Records, validated by the coordinators by examining the collected specimens and pictures, were included in a database.
The collected samples arrived in laboratory either alive, dried or in ethanol and were stored at − 20 °C until used for molecular analysis. A single leg was removed from the specimens using sterilized tweezers, while the remaining stink bugs were retained as vouchers and stored at − 20 °C at the Department of Life Sciences of the University of Modena and Reggio Emilia (Italy). Genomic DNA was extracted following the method described in Cesari et al. (2015). Briefly, DNA was isolated from a single stink bug leg using the MasterPure™ Complete DNA and RNA Purification (Epicentre, Illumina, Madison, WI, U.S.A.) kit, following the manufacturer’s protocol. A negative extraction control was also carried out with all the kit solutions but without insect tissue in order to check for contaminations.
The genetic diversity of the H. halys population was detected by analysing the mitochondrial genes cytochrome c oxidase subunit I (cox1) and cytochrome c oxidase subunit II (cox2). These markers were chosen because of their fairly high variability (both markers, Xu et al. 2014; Cesari et al. 2015), and because of the high number of previously scored specimens (cox1; Gariepy et al. 2014, 2015; Xu et al. 2014; Cesari et al. 2015; Zhu et al. 2016; Lee et al. 2017). PCR amplification of a portion of the mtDNA cox1 gene was carried out using primers LCO-1490 (5′-GGT CAA CAA ATC ATA AAG ATA TTG G-3′; Folmer et al. 1994) and HCO-2198 (5′-TAA ACT TCA GGG TGA CCA AAA AAT CA-3′; Folmer et al. 1994), with the following protocol: 35 cycles with 30 s at 94 °C, 30 s at 48 °C, and 30 s at 72 °C, with a final elongation step at 72 °C for 7 min. In the case of cox2 gene, after several failed attempts on different specimens with the primers HhalysCO2F2 and HhalysCO2R2 (Xu et al. 2014; Cesari et al. 2015), a new set of primers specific to H. halys, were designed using the program Primer3 (Untergrasser et al. 2012): HhalysCO2altF (5′-TTT TAA CCC AAG ATG CAA ATT C-3′) and HhalysCO2altR (5′-TTG TTC TTT CAA TTA CGA TTG G-3′). The newly amplified products were 518 bp long and corresponded to positions 3042–3559 of the complete mitochondrial genome of H. halys (GenBank Acc. No. NC_013272). This allowed to obtain sequences that could be compared to ones present in literature (Xu et al. 2014). The PCR amplification protocol utilized for the cox2 gene was the same as the one utilized for the cox1 gene and described above. The amplified products of both genes were gel purified using the Wizard Gel and PCR cleaning (Promega, Madison, WI, U.S.A.) kit. Both strands were then subjected to sequencing reactions using the Big Dye Terminator 1.1 kit (Applied Biosystems, Foster City, CA, U.S.A.) and sequenced using an ABI Prism 3100 sequencer (Applied Biosystems). Nucleotide sequences of the newly analysed specimens were submitted to GenBank (accession numbers: KY710272-460 for cox1 and KY710461-681 for cox2). The chromatograms were checked for the presence of ambiguous bases as the sequences were translated to amino acids using the invertebrate mitochondrial code implemented in MEGA6 (Tamura et al. 2013) in order to check for the presence of stop codons and therefore of pseudogenes. Nucleotide sequences were aligned with the Clustal algorithm implemented in MEGA6 (pairwise and multiple alignment parameters: Gap opening penalty: 15, Gap extend penalty: 6.66) and checked by visual inspection. For appropriate molecular comparisons, we included in our analysis cox1 and cox2 sequences of H. halys from GenBank (see Online Resource 2) derived from the following studies: Lee et al. (2009, 2017), Xu et al. (2014), Gariepy et al. (2014, 2015), Zhu et al. (2016) and Dhami et al. (2016). Uncorrected p-distances between scored haplotypes were determined using MEGA6, after checking that scored cox1 and cox2 sequences were not subject to saturation using the program DAMBE (Xia and Xie 2001). The relationships among haplotypes were estimated using a parsimony network by applying the method described by Templeton et al. (1992), as implemented in TCS 1.21 (Clement et al. 2000) and visualized using tcsBU (Santos et al. 2015). Both newly analysed and reference sequences were included in the analysis, in order to provide better qualitative information for potential pathways of invasion. Number of haplotypes (H), haplotype (h) and nucleotide (π) diversities were estimated using the program DnaSP v5 (Librado and Rozas 2009), whereas Tajima’s D and Fu’s Fs values were estimated by using Arlequin 3.5 (Excoffier and Lischer 2010), with P values generated using 1000 simulations under a model of selective neutrality. Differentiation between populations and the number of migrants following Wright (1969) were computed with conventional FST statistics from haplotypes with 1000 permutations with Arlequin 3.5. The population genetic analysis was carried out for all countries for the cox1 gene, while, due to the lack of sequences, it was restricted to Italian, Swiss, Greek, Romanian, U.S., Korean, Japanese and Chinese specimens for the cox2 gene, and to the latter countries, excluding Japan, for the combined analyses. The procedures and the experiments in this study were carried out with respect to the ethical standards of the Helsinki declaration of 1975, as revised in 2000 and 2008, as well as Italian law.
Results
Our analysis was carried out on 1271 sequences obtained from H. halys specimens (234 individuals were analysed for the first time in this study; Online Resource 1) on a total of 1175 base pairs (657 bp for cox1 and 518 bp for cox2). Haplotype designation followed that of Xu et al. (2014), Gariepy et al. (2014, 2015), Cesari et al. (2015), Zhu et al. (2016), and Lee et al. (2017). The obtained cox1 tract was shorter (657 vs 1349 bp) than the one used by Zhu et al. (2016), resulting in the coalescing of some haplotypes (H1 = Hap6 = Hap14 = Hap45 = Hap54 = Hap83 = Hap96 = Hap112 = Hap119 = Hap123; H2 = Hap92 = Hap93; H3 = Hap10 = Hap16 = Hap24 = Hap32 = Hap34 = Hap46 = Hap58; H14 = Hap21 = Hap30 = Hap121 = Hap126; H22 = Hap4 = Hap5 = Hap33 = Hap47 = Hap49 = Hap52 = Hap56 = Hap59 = Hap61 = Hap70 = Hap73 = Hap75 = Hap84 = Hap87 = Hap88 = Hap89 = Hap91 = Hap109 = Hap110 = Hap117 = Hap133; H23 = Hap62 = Hap63 = Hap65; H33 = Hap76 = Hap86 = Hap90; H37 = Hap53 = Hap 68 = Hap124; Hap17 = Hap29; Hap38 = Hap40; Hap39 = Hap 43; Hap44 = Hap 120; Hap57 = Hap115; Hap71 = Hap74; Hap79 = Hap82; Hap94 = Hap95; Hap107 = Hap129; Hap113 = Hap130).
cox1
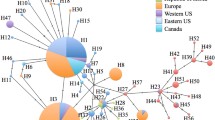
For the cox1 marker, among the 111 haplotypes scored, seven of them (H150–156), all originating from Italian specimens, were detected for the first time (Online Resource 1; Fig. 1). Haplotype diversity for all populations was 0.757, while nucleotide diversity was 0.00398 (Table 2). The haplotypes differed between 1 and 12 substitutions, with p-distances ranging from 0.002 to 0.019. Two haplotypes (H1 and H3) were the most abundant and widespread (Fig. 1): H1 (n = 440) was present in all analysed countries, excluding Japan and South Korea, while H3 (n = 393) was found in China, France, Switzerland, Hungary, Greece, and Italy. All other haplotypes were retrieved in a maximum of two countries, with the exception of H8 (found in Switzerland, France, and Italy) and H22 (in China, Korea, and Greece). Most of the scored Greek haplotypes pointed to Asian origins: H22 (see above) and H33 (which was also found in Chinese specimens of the provinces of Shanxi and Shaanxi). Other than the new haplotypes found for the first time in Italy, the haplotypes H8 (see above), H30, H31, H32 (Greece) were exclusively detected in Europe.

Haplotype network of the cox1 gene from 1271 Halyomorpha halys specimens. Circles represent haplotypes, circle size denotes the total haplotype frequency, while each slice represents the haplotype frequency in different countries. Small white squares indicate missing/ideal haplotypes. Numbers point out the haplotypes found in Europe. Haplotypes detected for the first time (150–156) are in italic and bold. Data from: present study; Gariepy et al. 2014, 2015; Xu et al. 2014; Cesari et al. 2015; Zhu et al. 2016; Lee et al. 2017
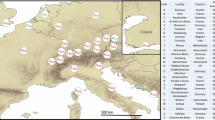
The highest haplotype diversity was found in China (h = 0.804), with the presence of 75 haplotypes. High haplotype diversities were also recorded in Korea and Japan (h = 0.669 and 0.533, respectively). Outside of the native area of H. halys, the highest diversities were found in Greece (π = 0.00366; h = 0.707; Table 2, Figs. 1, 2), confirming the data of Gariepy et al. (2015), and in Italy (see below). On the other hand, a high haplotype uniformity was found in Romania, as only one haplotype was found (H1; Table 2, Figs. 1, 2). In Italy, 13 haplotypes were scored (Figs. 1, 2), with high haplotypic (h = 0.702; Table 2) and nucleotide (π = 0.00545; Table 2) diversities; these values were the highest (π) or the second highest (h) found all over the world. Seven Italian cox1 haplotypes (H1; H3; H8; Hap78, Hap80; Hap81) were also detected in other countries, in some cases indicating their possible origins. In fact, other than in Italy, haplotype H8 was found in Switzerland and France, while haplotypes Hap78, Hap80, and Hap 81 were scored in the Shandong province of China.

Geographic distribution of cox1 haplotypes detected in 731 European Halyomorpha halys specimens. Circle size denotes number of analyzed specimens, while the size of each slice denotes the haplotype frequency. Haplotypes detected for the first time (H150–H156) are in bold. Data from: present study; Gariepy et al. 2014, 2015; Xu et al. 2014; Cesari et al. 2015; Zhu et al. 2016; Lee et al. 2017
Demographic expansion was detected using both Fu’s Fs and Tajima’s D values (negative values, Table 2) both in the native continent and in Europe and North America. Almost all invaded countries showed negative values, indicating a probable demographic expansion after a bottleneck, even though no significance was detected for one or both selective neutrality tests for most invaded countries. The analysis of the pairwise φ ST and number of migrants (Table 3) showed a clear subdivision among populations from all countries, with the exception of Hungary and Romania (probably due to their low diversity) and between specimens from China and those intercepted in New Zealand.
cox2
For the cox2 gene, the analysis was carried out on fewer individuals (367). Of these, 219 were analysed for the first time in this study. A total of 21 haplotypes were scored (h1–h21, Fig. 3); of these, two (h20, h21) were new to science, both retrieved from Italian specimens. The haplotypes differed between 1 and 9 substitutions, with p-distances ranging from 0.002 to 0.017. The haplotype diversity for all populations was 0.553, while the nucleotide diversity was 0.00225 (Online Resource 3). Two haplotypes (h1 and h3) were the most abundant and widespread: h1 (n = 186) was present in all analysed countries (Switzerland and Japan excluded); h3 (n = 107) was found in China, South Korea, Switzerland, Greece, and Italy (Fig. 3). All other haplotypes were detected in only one country, with the exception of haplotypes h11 (in Korea, Japan and Italy) and h15 (in Japan and Italy). In Asia, 19 cox2 haplotypes were scored (h = 0.893), a value much higher than those found in the U.S.A. (1 haplotype) and in Europe (6 haplotypes; h = 0.510). One haplotype was detected in Switzerland (h3) and in Romania (h1) while two were found in Greece (h1 and h3). Italian H. halys individuals included all of the haplotypes found in Europe (h1, h3, h11, h15, h20, h21; Fig. 3).

Haplotype network of the cox2 gene from 367 Halyomorpha halys specimens. Circles represent haplotypes, circle size denotes the total haplotype frequency, while each slice represents the haplotype frequency in different countries. Small white squares indicate missing/ideal haplotypes. Haplotype denotation follows that proposed by Cesari et al. (2015). Haplotypes detected for the first time (h20–h21) are shown in bold. Data from: present study; Xu et al. 2014; Cesari et al. 2015
Combined analysis (cox1 + cox2)
In the combined analysis, given that only 10 cox1 + cox2 sequences were available in GenBank, we focused on the newly produced data (n = 228), and mainly on Italian specimens (n = 206). Twenty-six haplotypes were found (TH1–TH26, the haplotypes found in Cesari et al. (2015) have this correspondence: Ha = TH1; Hb = TH2; Hc = TH3; Hd = TH4; He = TH11; Hf = TH12), of which 20 were new to science (TH5–TH10; TH13–TH26). These were all found in Italian and Greek specimens. Haplotypes differed between 1 and 11 substitutions, with p-distances ranging from 0.001 to 0.010. The total haplotype diversity was 0.00361, while the total nucleotide diversity was 0.728 (Table 4). Haplotype TH1 was the most abundant and widespread (Fig. 4), being present in all analysed countries, Switzerland and Korea excluded. All other haplotypes were each found in only one country, with the exception of TH4, which was scored in both Swiss and Italian H. halys specimens. Twenty Italian haplotypes (TH5–TH24) were also unique and therefore not found anywhere else (Fig. 4). Italy had also the highest nucleotide (π = 0.00376) and haplotype (h = 0.734) diversities (Table 4). The most diverse Italian regions were in the North: Piedmont (10 haplotypes, h = 0.758), Lombardy (10 haplotypes, h = 0.776) and Veneto (8 haplotypes, h = 0.792).

Haplotype network of the cox1 + cox2 genes from 238 Halyomorpha halys specimens. Circles represent haplotypes, circle size denotes the total haplotype frequency, while each slice represents the haplotype frequency in different countries/Italian regions. Small white squares indicate missing/ideal haplotypes
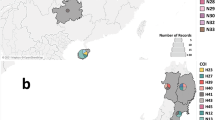
The analysis of the pairwise φ ST and number of migrants in the different Italian regions (Table 4) showed a marked subdivision between different regions, excluding those colonized very recently (Tuscany, Lazio, Calabria, and in most comparisons, Trentino-South Tyrol). Figures 5 and 6 show the distribution of combined haplotypes in the different provinces and regions of Italy in a temporal scale (2013–2014 in Fig. 5, and 2015–2016 in Fig. 6). Both figures show how the number of haplotypes and their distribution have increased over time, leading to a marked differentiation between the regions of North-West (Piedmont and Lombardy) and those of North–East (Emilia-Romagna, Friuli-Venezia Giulia and Trentino-South Tyrol) Italy. Halyomorpha halys has been recently found (2016) in Central and South Italian regions, which have likely been colonized by individuals coming from Lombardy/Piedmont (TH12 in Lazio and Calabria), and Emilia-Romagna (TH1 in Tuscany). Moreover, some haplotypes typically found in Lombardy and Piedmont thus far (TH4 and TH7) have finally been detected both in the Western (TH4) and Eastern provinces of Emilia-Romagna (TH6, Fig. 6).

Geographic distribution of the cox1 + cox2 haplotypes detected in 2013 (above) and in 2014 (below) in Italy. On the left, maps with different colours show the distribution and abundance of Halyomorpha halys individuals. On the right, the maps with different diagrams represent the haplotype diversities in the different provinces (close provinces with the same haplotypes were grouped). The grey lines delimit the provinces, the black lines the regions

Geographic distribution of the cox1 + cox2 haplotypes detected in 2015 (above) and in 2016 (below) in Italy. On the left, maps with different colours show the distribution and abundance of Halyomorpha halys individuals. On the right, the maps with different diagrams represent the haplotype diversities in the different provinces (close provinces with the same haplotypes were grouped). The grey lines delimit the provinces, the black lines the regions
Discussion
The present study allowed us to gain new insights on the genetic diversity of H. halys in Europe, and the correlation of genetic data with temporal and spatial information has produced new information that will be useful for the control and management of this alien pest. The inference of H. halys invasion routes, geographical origin of introduced specimens and their genetic diversity should facilitate the design of effective measures for both preventing further invasions (by targeting control of goods from targeted source areas) and controlling invasive populations (by choosing potential auxiliary agents from the same geographical areas from which invasive H. halys come from). The number of detected haplotypes in Europe represent only the 31% of those found in Asia (China, Korea, and Japan), but the European populations of H. halys do not seem to have been affected by a high reduction in genetic diversity during invasion processes as one might expect from a bottleneck effect. This high diversity is related to a pattern of multiple invasions in Europe by H. halys, as pointed out by the different haplotypes and their different distributions among the European countries (Fig. 2). The higher haplotype diversity recorded in southern Europe (Italy and Greece) can be explained by a variety of reasons which are not mutually exclusive: a sampling bias, multiple invasions, and more favourable environmental conditions for reproduction and growth. In fact, modelling approaches have shown that southern Europe has a more favourable habitat for H. halys populations than higher latitudes (Haye et al. 2015), especially in prime horticultural production areas (Kriticos et al. 2017).
Most haplotypes detected in Europe were also found in China. Therefore, it can be hypothesized that most specimens arrived from this country, especially given the large amount of commercial trading that occurs between Europe and Asia. However, it is important to note that some secondary invasions from North America cannot be excluded. Some European haplotypes were also detected in specific areas of China, such as haplotype H33 which was found in Greece and in the Chinese provinces of Shanxi and Shaanxi, or haplotypes Hap78, Hap80, Hap81 which were found in Italy and in the province of Shandong. These data can therefore identify the possible sources for the introduction. Moreover, some Italian specimens share haplotypes (h11, h15) with Korean and Japanese organisms (Fig. 3).
On the other hand, other haplotypes were found only in Europe (H8, H30–32, H150–156), clearly indicating how the biodiversity of the native range is still understudied. The temporal distribution of some of these European haplotypes clearly shows how a secondary invasion has occurred among European countries, e.g. haplotype H8 was detected in 2012 in Switzerland (Gariepy et al. 2014) and then was found in 2013 both in France (Gariepy et al. 2015) and in Northern Italy, close to the Swiss border (Cesari et al. 2015).
Multiple introductions seem to be common in invasions (Ellstrand and Schierenbeck 2000; Bossdorf et al. 2005; Novak and Mack 2005), and they can bring together large amounts of variation, as we detected for H. halys. This leads to the hypothesis that the successful European invasion by H. halys is a direct result of an influx of genetic variation from multiple introductions. The inclusion of the data from Lee et al. (2017), mainly coming from the U.S. East coast (West Virginia, New Jersey), show that the cox1 haplotype diversities in North America are higher than that reported so far (Gariepy et al. 2014; Xu et al. 2014), pointing to the possibility that different scenarios could also be taken into account also for that continent, even though so far all molecular data were sampled in the East coast.
The most common haplotypes (TH1, TH4, H1, H3) present in Europe comprise also that one more commonly found in North America (H1). It is not clear if these haplotypes are more common because they are also more common in Asia (such as H1 and H3) and/or more commonly transported passively, and/or because they are associated with the genomes of specimens that more easily adapt to newly colonized territories. Further analyses involving more variable markers (i.e. microsatellites, SNPs) will help in clarifying this scenario in the future.
In Italy, more data are available with respect to other invaded countries, and these data are more clearly related to their temporal occurrence, allowing for a clearer reading of the patterns of invasion and dispersion of H. halys. It is evident that, as the areas colonized by H. halys increased during time, so did the number and distribution of the haplotypes. In just a few years, the number of haplotypes increased from two (detected in 2013 in only few provinces) to 22 (distributed almost all over Italy), providing evidence for a progressive, continuous and frequent invasion due to the introduction of new haplotypes. These multiple invasions, very probably, are due to stink bugs arriving from different countries, both from other continents (e.g. Asia) and from Europe (e.g. Switzerland).
Based on the different haplotype distributions, the diffusion of H. halys in Italy followed different invasion patterns. The two initial haplotypes found in Italy (TH1, TH4) remain the more abundant and widespread, and seem to be most highly invasive. The combined haplotype TH4, similar to the cox1 haplotype H8, is a haplotype found only in Switzerland and Italy (H8 is present also in France; Gariepy et al. 2015). Because Switzerland was the European country in which H. halys was first detected in 2004 (Wermelinger et al. 2008; Haye et al. 2015), it is very probable that Swiss specimens arrived in Northern Italy (Lombardy) from the Ticino valley (Maistrello et al. 2016b) and then spread to nearby areas (Fig. 5). In the same year of the first record in Italy (2012), some specimens were also found in the attic of the Natural History Museum of Milan (Dioli, pers. comm.), which could represent a possible earlier settlement connected with the commercial route Milan-Zurich. From this first inoculation, the population colonized new areas by being passively transported via jump dispersal events to other distant areas in northern (e.g. eastern Emilia-Romagna and Trentino-South Tyrol), central (Lazio) and southern Italy (Calabria) (Fig. 6). Therefore, specimens with this haplotype could have colonized new territories by a “bridgehead effect” (Lombaert et al. 2010) in which widespread secondary invasions stem from a particularly successful invasive population. A similar pattern, but with fewer occurrences, is shown by the haplotypes TH7, TH12, TH13, and TH22. Previous studies on the movement of H. halys in U.S. invaded territories pointed out its strong association with urban and commercial developments, with railroads and was helped by the ability of the stink bugs to overwinter in multiple habitats and to feed on different plants (Wallner et al. 2014). All these features are also found in Italy, especially in the Po valley (northern Italy), thus facilitating the ability of H. halys to colonize new areas.
On the other hand, the haplotype TH1 seems to have arrived in Italy and expanded its distribution range in a different way with respect to TH4. The first specimens of H. halys found in Italy had the TH1 haplotype (Cesari et al. 2015). This haplotype was initially confined to the central part of Emilia-Romagna (Fig. 5). It then expanded its range progressively colonizing nearby areas by diffusion without wide jump dispersal events, spreading all over Northern Italy. It is now moving south (Fig. 6). TH1 is probably the same haplotype of the specimens that were the focus of movement capacity investigations in the U.S.A. These studies found that adult H. halys can fly an average of 2 km/day and up to 117 km in the same time period (Lee and Leskey 2015), with individuals of the summer generation flying the longest distances (Wiman et al. 2015). Moreover, nymphs exhibited a strong walking capacity on both horizontal and vertical surfaces, and they can be even faster than adults in certain environments (Lee et al. 2014). On the other hand, other less-widespread haplotypes (e.g. TH9, TH10, TH16, TH19, TH20) show a local distribution, apparently they did not extend their range during the sampling interval. This difference of dispersal abilities could also be linked to different landscape structure and host plant diversity. In fact, it has been shown that H. halys can feed on both non reproductive parts and fruits of multiple host plants (Acebes-Doria et al. 2016), but some more suitable host plants can be less represented in different areas, thus negatively influencing the dispersal. Moreover, Rice et al. (2016) demonstrated that most damage in agricultural fields (i.e. more individuals) is often linked to forests bordering crops, a feature not so common in the Italian countryside.
Northeast and Northwest Italy show different haplotypes, providing evidence for different origins. For example, in the Veneto region (in Northeast Italy), clear events of multiple introductions are detectable. For instance, a new haplotype (TH20) was suddenly found in 2014. Then, in just 2 years the number of new haplotypes increased to seven, most of which being exclusively found in this area (Figs. 5, 6). In Piedmont (in Northwest Italy) the number of haplotypes increased rapidly in 3 years, leading to a total of nine haplotypes. Of these haplotypes, only two (TH1, TH4, the most frequent in Europe) are shared with Veneto.
Conclusion
The use of multiple molecular markers in this population genetic analysis has improved our ability to identify the different invasion patterns of H. halys. The current European distribution of H. halys is the result of multiple invasions that are still in progress, due to passive dispersal mediated mainly by commercial trading (Haye et al. 2015). The source-areas for these invasions are varied (e.g. multiple Chinese provinces, Korea, or newly colonized areas), further increasing the haplotype diversity in newly colonized territories. After having successfully established in localized areas, invasive specimens can further expand their range by an active, natural, dispersion process and/or by jump dispersal events due to passive transport mediated by human activities. Multiple introductions may have a significant impact on the success of an invasive species if the introductions come from different areas of the native range (Dlugosch et al. 2015). This, together with the different patterns of diffusion of H. halys, may hamper the pest management strategies for its containment.
Population genetic studies provide additional information to identify the invasive pathways, modes of introduction, and original source sites. This information will be useful to set up and improve pest management strategies (Dlugosch and Parker 2008), such as prevention of new introductions by targeting specific source areas and potential use of control agents with the same geographical origin as the invasive H. halys specimens. Obtained data will also be useful for the development of predictive models that can identify territories most at risk under different scenarios, other than obtaining new insights into the evolutionary ecology of introduced pests.
References
Acebes-Doria AL, Leskey TC, Bergh JC (2016) Host plant effects on Halyomorpha halys (Hemiptera: Pentatomidae) nymphal development and survivorship. Environ Entomol 45:663–670
Arnold K (2009) Halyomorpha halys (Stål, 1855), eine für die europäische Fauna neu nachgewiesene Wanzenart (Insecta: Heteroptera, Pentatomidae, Pentatominae, Cappaeini). Mitt Thür Entomologenverbande e 16(1):19
Bariselli M, Bugiani R, Maistrello L (2016) Distribution and damage caused by Halyomorpha halys in Italy. EPPO Bull 46:332–334
Bossdorf O, Auge H, Lafuma L, Rogers WE, Siemann E, Prati D (2005) Phenotypic and genetic differentiation between native and introduced plant populations. Oecologia 144:1–11
Callot H, Brua C (2013) Halyomorpha halys (Stål, 1855), la Punaise diabolique, nouvelle espèce pour la faune de France (Heteroptera Pentatomidae). L’Entomologiste 69(2):69–71
Cesari M, Maistrello L, Ganzerli F, Dioli P, Rebecchi L, Guidetti R (2015) A pest alien invasion in progress: potential pathways of origin of the brown marmorated stink bug Halyomorpha halys populations in Italy. J Pest Sci 88:1–7
Clement M, Posada D, Crandall K (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1660
Corin SE, Lester PJ, Abbott KL, Ritchie PA (2007) Inferring historical introduction pathways with mitochondrial DNA: the case of introduced Argentine ants (Linepithema humile) into New Zealand. Divers Distrib 13:510–518
Dhami MK, Dsouza M, Waite DW, Anderson D, Li D (2016) Real-time PCR assay for the identification of the brown marmorated stink bug (Halyomorpha halys). Front Mol Biosci 3:5
Dioli P, Leo P, Maistrello L (2016) Prime segnalazioni in Spagna e in Sardegna della specie aliena Halyomorpha halys (Stål, 1855) e note sulla sua distribuzione in Europa (Hemiptera, Pentatomidae). Revta gad Entom 7(1):539–548
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17(1):431–449
Dlugosch KM, Anderson SR, Braasch J, Cang FA, Gillette HD (2015) The devil is in the details: genetic variation in introduced populations and its contributions to invasion. Mol Ecol 24:2095–2111
Duthie CD, Michael T, Stephenson B, Yamoah E, McDonald B (2012) Risk analysis of Halyomorpha halys (brown marmorated stink bug) on all pathways. Biosecurity Risk Analysis Group, Ministry of Agriculture and Forestry of New Zealand, Wellington
Ellstrand NC, Schierenbeck KA (2000) Hybridization as a stimulus for the evolution of invasiveness in plants? PNAS 97(13):7043–7050
Estoup A, Guillemaud T (2010) Reconstructing routes of invasion using genetic data: why, how and so what? Mol Ecol 19:4113–4130
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Fanfani R, Pieri R (2016) Il sistema agro-alimentare dell’Emilia-Romagna. Rapporto 2015. Unione regionale delle Camere di commercio dell’Emilia-Romagna Regione Emilia-Romagna, Assessorato agricoltura, caccia e pesca, Bologna
Fisher RA (1930) The genetical theory of natural selection. Clarendon, Oxford
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3:294–299
Gariepy TD, Haye T, Fraser H, Zhang J (2014) Occurrence, genetic diversity, and potential pathways of entry of Halyomorpha halys in newly invaded areas of Canada and Switzerland. J Pest Sci 87(1):17–28
Gariepy TD, Bruin A, Haye T, Milonas P, Vétek G (2015) Occurrence and genetic diversity of new populations of Halyomorpha halys in Europe. J Pest Sci 88(3):451–460
Handley LJL, Estoup A, Evans DM, Thomas CE, Lombaert E, Facon B, Aebi A, Roy HE (2011) Ecological genetics of invasive alien species. Biocontrol 56:409–428
Haye T, Gariepy T, Hoelmer K, Rossi JP, Streito JC, Tassus X, Desneux N (2015) Range expansion of the invasive brown marmorated stinkbug, Halyomorpha halys: an increasing threat to field, fruit and vegetable crops worldwide. J Pest Sci 88(4):665–673
Heckmann R (2012) Erster Nachweis von Halyomorpha halys (Stål, 1855) (Heteroptera: Pentatomidae) für Deutschland. Heteropteron 36:17–18
Hoebeke ER, Carter EM (2003) Halyomorpha halys (Stål) (Heteroptera: Pentatomidae): a polyphagous plant pest from Asia newly detected in North America. Proc Entomol Soc Wash 105:225–237
Hulme PE, Bacher S, Kenis M, Klotz S, Kühn I, Minchin D, Nentwig W, Olenin S, Panov V, Pergl J, Pyšek P, Roques A, Sol D, Solarz W, Vilà M (2008) Grasping at the routes of biological invasions: a framework for integrating pathways into policy. J Appl Ecol 45:403–414
Kolar CS, Lodge DM (2001) Progress in invasion biology: predicting invaders. Trends Ecol Evol 16:199–204
Kriticos DJ, Kean JM, Phillips CB, Senay SD, Acosta H, Haye T (2017) The potential global distribution of the brown marmorated stink bug, Halyomorpha halys, a critical threat to plant biosecurity. J Pest Sci 90:1033–1043
Lee DH, Leskey TC (2015) Flight behavior of foraging and overwintering brown marmorated stink bug, Halyomorpha halys (Hemiptera: Pentatomidae). Bull Entomol Res 105:566–573
Lee W, Kang J, Jung C, Hoelmer K, Lee SH, Lee S (2009) Complete mitochondrial genome of brown marmorated stink bug Halyomorpha halys (Hemiptera: Pentatomidae), and phylogenetic relationships of Hemipteran suborders. Mol Cells 28:155–165
Lee DH, Short BD, Joseph SV, Bergh JC, Leskey TC (2013) Review of the Biology, Ecology, and Management of Halyomorpha halys (Hemiptera: Pentatomidae) in China, Japan, and the Republic of Korea. Environ Entomol 42:627–641
Lee DH, Nielsen AL, Leskey TC (2014) Dispersal capacity and behavior of nymphal stages of Halyomorpha halys (Hemiptera: Pentatomidae) evaluated under laboratory and field conditions. J Insect Behav 27:639–651
Lee W, Guidetti R, Cesari M, Gariepy TD, Park YR, Park CG (2017) Genetic diversity of Halyomorpha halys (Hemiptera, Pentatomidae) in Korea, comparing with COI sequence dataset of East Asia, Europe, and North America. Fla Entomol (in press)
Leskey TC, Hamilton GC, Nielsen AL, Polk DF, Rodriguez-Saona C, Bergh JC, Herbert DA, Kuhar TP, Pfeiffer D, Dively GP, Hooks CRR, Raupp MJ, Shrewsbury PM, Krawczyk G, Shearer PW, Whalen J, Koplinka-Loehr C, Myers E, Inkley D, Hoelmer KA, Lee DH, Wright SE (2012) Pest status of the brown marmorated stink bug, Halyomorpha halys in the USA. Outlooks Pest Manag 23:218–226
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Lombaert E, Guillemaud T, Cornuet JM, Malausa T, Facon B, Estoup A (2010) Bridgehead effect in the worldwide invasion of the biocontrol harlequin ladybird. PLoS ONE 5(3):e9743
Macavei LI, Bâeţan R, Oltean I, Florian T, Varga M, Costi E, Maistrello L (2015) First detection of Halyomorpha halys Stål, a new invasive species with a high potential of damage on agricultural crops in Romania. Lucrări Ştiinţifice seria Agronomie 58(1):105–108
MacLellan R (2013) Brown marmorated stink bug: a potential threat to New Zealand. Surveillance (Wellington NZ) 40:3
Maistrello L, Dioli P, Vaccari G, Nannini R, Bortolotti P, Caruso S, Costi E, Montermini A, Casoli L, Bariselli M (2014) First records in Italy of the Asian stinkbug Halyomorpha halys a new threat to fruit crops. Atti Giornate Fitopatologiche 1:283–288
Maistrello L, Costi E, Caruso S, Vaccari G, Bortolotti P, Nannini R, Casoli L, Montermini A, Bariselli M, Guidetti R (2016a) Halyomorpha halys in Italy: first results of field monitoring in fruit orchards. Integrated protection of fruit crops subgroups “pome fruit arthropods” and “stone fruits”. IOBC-WPRS Bull 112:1–5
Maistrello L, Dioli P, Bariselli M, Mazzoli GL, Giacalone-Forini I (2016b) Citizen science and early detection of invasive species: phenology of first occurrences of Halyomorpha halys in Southern Europe. Biol Invasions 18:3109–3116
Maistrello L, Vaccari G, Caruso S, Costi E, Bortolini S, Macavei L, Foca G, Ulrici A, Bortolotti PP, Nannini R, Casoli L, Fornaciari M, Mazzoli GL, Dioli P (2017) Monitoring of the invasive Halyomorpha halys, a new key pest of fruit orchards in northern Italy. J Pest Sci 90:1231–1244
Malumphy C (2014) Second interception of Halyomorpha halys (Stål) (Hemiptera: Pentatomidae) in Britain. Het News 21:4–5
Maurel JP, Blaye G, Valladares L, Roinel É, Cochard PO (2016) Halyomorpha halys (Stål, 1855), la punaise diabolique en France, à Toulouse (Heteroptera; Pentatomidae). Carnets Nat 3:21–25
Milonas PG, Partsinevelos GK (2014) First report of brown marmorated stink bug Halyomorpha halys Stål (Hemiptera: Pentatomidae) in Greece. EPPO Bull 44(2):183–186
Mityushev IM (2016) First record of Halyomorpha halys detection in Russia. Zashchitai Karantin Rasteniĭ 3:48
Moore A (2014) Brown marmorated stink bug Halyomorpha halys (Stål, 1855) (Hemiptera: Pentatomidae). Guam new invasive species alert No. 2014-01. University of Guam, Guam. http://guaminsects.net/anr/sites/default/files/brownMarmoratedStinkBug.pdf
Nikam KN, More SV (2016) Diversity of Insects from Jangamhatti area, Chandgad, Kolhapur district of Maharashtra. Biolife 4(1):209–212
Novak SJ, Mack RN (2005) Genetic bottlenecks in alien plant species. In: Sax DF, Stachowicz JJ, Gaines SD (eds) Species invasions: insights into ecology, evolution and biogeography. Sinauer Associates Inc, Sunderland, pp 201–228
Rabitsch W, Griebe GJ (2015) From the west and from the east? First records of Halyomorpha halys (Stål, 1855) (Hemiptera: Heteroptera: Pentatomidae) in Vorarlberg and Vienna, Austria. Beiträge zur Entomofaunistik 16:115–139
Rice KB, Bergh CJ, Bergmann EJ, Biddinger DJ, Dieckhoff C, Dively G, Fraser H, Gariepy T, Hamilton G, Haye T (2014) Biology, ecology, and management of brown marmorated stink bug (Hemiptera: Pentatomidae). J Integr Pest Manag 5:A1–A13
Rice KB, Troyer RR, Watrous KM, Tooker JF, Fleischer SJ (2016) Landscape factors influencing stink bug injury in Mid-Atlantic tomato fields. J Econ Entomol 110:94–100
Santos AM, Cabezas MP, Tavares AI, Xavier R, Branco M (2015) tcsBU: a tool to extend TCS network layout and visualization. Bioinformatics 32(4):627–628
Šeat J (2015) Halyomorpha halys (Stål, 1855) (Heteroptera: Pentatomidae) a new invasive species in Serbia. Acta Entomol Serb 20:167–171
Simov N (2016) The invasive brown marmorated stink bug Halyomorpha halys (Stål, 1855) (Heteroptera: Pentatomidae) already in Bulgaria. Ecologica Montenegrina 9:51–53
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 132:619–633
Untergrasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3—new capabilities and interfaces. Nucleic Acids Res 40(15):e115
Vétek G, Papp V, Haltrich A, Redei D (2014) First record of the brown marmorated stink bug, Halyomorpha halys (Hemiptera: Heteroptera: Pentatomidae), in Hungary, with description of the genitalia of both sexes. Zootaxa 3780(1):194–200
Walker K (2009) Brown marmorated stink bug (Halyomorpha halys). Updated on 11/25/2011. http://www.padil.gov.au
Wallner AM, Hamilton GC, Nielsen AL, Hahn N, Green EJ, Rodriguez-Saona CRR (2014) Landscape factors facilitating the invasive dynamics and distribution of the Brown Marmorated Stink Bug, Halyomorpha halys (Hemiptera: Pentatomidae), after arrival in the United States. PLoS ONE 9(5):e95691
Ward N (2013) Plants and environment investigation. Surveillance (Wellington NZ) 40:1
Wermelinger B, Wyniger D, Forster B (2008) First records of an invasive bug in Europe: Halyomorpha halys Stål (Heteroptera: Pentatomidae), a new pest on woody ornamentals and fruit trees? Mitt Schweiz Entomol Ges 81:1–8
Wiman NG, Walton VM, Shearer PW, Rondon SI, Lee JC (2015) Factors affecting flight capacity of brown marmorated stink bug, Halyomorpha halys (Hemiptera: Pentatomidae). J Pest Sci 88:37–47
Wright S (1969) Evolution and genetics of natural populations. The theory of gene frequencies, vol 2. University Chicago Press, Chicago
Xia X, Xie Z (2001) DAMBE: data analysis in molecular biology and evolution. J Hered 92:371–373
Xu J, Fonseca DM, Hamilton GC, Hoelmer KA, Nielsen AL (2014) Tracing the origin of US brown marmorated stink bugs, Halyomorpha halys. Biol Invasions 16:153–166
Zhu GP, Bu WJ, Gao Y, Liu G (2012) Potential geographic distribution of brown marmorated stink bug invasion (Halyomorpha halys). PLoS ONE 7:e31246
Zhu GP, Ye Z, Du J, Zhang DL, Zhen Y, Zheng C, Zhao L, Li M, Bu WJ (2016) Range wide molecular data and niche modeling revealed the Pleistocene history of a global invader (Halyomorpha halys). Sci Rep 6:23192
Acknowledgements
The authors wish to thank Wen-Jun Bu and Zhen Ye (Nankai University, China) for kindly supplying the geographical information for Chinese specimens. We are also indebted to Prof. Aparna Palmer (Colorado Mesa University) for the English revision and her valuable suggestions. The authors wish to thank two anonymous reviewers, whose suggestions helped to improve the manuscript. We are grateful to the following people/groups for providing H. halys specimens: Ronni Paolinelli, Gruppo Modenese Scienze Naturali, M. Agosti, L. Mola, M. Salvetti, Moreno Dutto, researchers, students and trainees of UNIMORE-UNIMI-UNITO-UNIPD, technicians and employees of regional and provincial Plant Protection Services and other institutions (SF-Emilia Romagna, Consorzi Fitosanitari MO-RE-PR-PC, Consorzio Agrario di Ravenna, ASTRA, SF-Bolzano, FEM, SF-Veneto, ERSAF Friuli Venezia Giulia, APOFRUIT, SF-Ticino, CREA), Martino Salvetti (Fondazione Fojanini di Studi Superiori, Sondrio, Italy), Maurizio Pavesi (Museo Civico di Storia Naturale, Milano, Italy), Piero Leo (Cagliari, Italy), Livio Mola (Brescia, Italy)), Moreno Dutto (Azienda sanitaria, Cuneo), Stefano Turolo (Monza, Italy), Alida Piglia (Milano, Italy) for providing specimens. Funding for the research was supported by the ‘Fondazione Cassa di Risparmio di Modena’, grant 2013.0665 “Innovative tools and protocols for monitoring and sustainable control of the alien stink bug H. halys, a new phytosanitary threat, and of other harmful heteropterans for the fruit crops of the territory of Modena”.
Authors’ contributions
MC and RG conceived and designed the research. MC, LP and RB conducted the experiments. LM, PD, GKP collected and identified the specimens. WL and PC provided the sequences and geographical information of Korean and U.S. specimens. MC and LP analysed the data. RG, LR and LM provided the research funds. RG, LR, LP, LM and MC wrote the manuscript. All authors read and approved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animals rights
This research did not contain any studies with human participants or animals (vertebrates) performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants involved in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cesari, M., Maistrello, L., Piemontese, L. et al. Genetic diversity of the brown marmorated stink bug Halyomorpha halys in the invaded territories of Europe and its patterns of diffusion in Italy. Biol Invasions 20, 1073–1092 (2018). https://doi.org/10.1007/s10530-017-1611-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-017-1611-1