
Abstract
Identifying the origin of a biological invasion has important applications to the effective control of the invaders. This is more critical for invasive agricultural pests that cause severe economic losses. The brown marmorated stink bug, Halyomorpha halys, originally from East Asia, has become a principal agricultural pest in the US since its first detection in Pennsylvania in 1996. This species is responsible for crop failures on many mid-Atlantic farms and current control efforts rely on heavy insecticide applications because no other options are available. To examine the genetic diversity and identify the source region of the US introductions, we sequenced portions of the mitochondrial cytochrome c oxidase subunit II gene, 12S ribosomal RNA gene and control region in populations from the US, China, South Korea and Japan. We detected high genetic divergence among native populations and traced the origin of US H. halys to the Beijing area in China. We observed much lower genetic diversity in exotic compared to native populations—two mitochondrial haplotypes in 55 US specimens versus 43 haplotypes in 77 native specimens. A single introduction of small propagule size matches the invasion history in the US. For the effective control of the US population, we suggest that surveys on egg parasitoids and insecticide resistance in natives should focus on the Beijing area in China.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The brown marmorated stink bug, Halyomorpha halys (Stål) (Hemiptera: Pentatomidae), also called the yellow–brown stink bug and referred to as H. picus or H. mista in earlier Asian literature, is native to East Asia (China, Korea and Japan), and was reported in the United States for the first time in 1996 from Allentown, Pennsylvania (Hoebeke and Carter 2003). Since then, this species has also been detected in Switzerland and Canada (Quebec and Ontario), and intercepted in New Zealand (Wermelinger et al. 2008; Harris 2010; Fogain and Graff 2011). In its native range, H. halys is an occasional agricultural pest of tree fruits and soybeans (Hoffman 1931; Kobayashi et al. 1972; Funayama 2004). In the US, it is now a widespread agricultural pest occurring in all states east of the Mississippi River as well as several west coast states (Leskey et al. 2012). H. halys has become the dominant pentatomid species in many mid-Atlantic areas (Nielsen and Hamilton 2009; Nielsen et al. 2011), causing $37 million loss to apples in 2010 (Seetin 2011) as well as unreported losses to a variety of ornamentals, vegetables and field crops (Leskey and Hamilton 2010; Kuhar et al. 2012). Moreover, great potential for similar economic losses exists in southern, western and mid-western states (Holtz and Kamminga 2010).
Pest management programs, especially for invasive agricultural species, are complex programs developed over many years to minimize insecticide use and costs. The severe agricultural damage by H. halys has lead many farmers to rely heavily on insecticides with up to fourfold increase in applications (Leskey et al. 2012), increasing the risk of insecticide resistance and concerns regarding food safety and environmental pollution. Alternative management strategies such as biological control are in urgent need but lacking. Traditionally, natural enemies of a pest, either endemic or purposefully introduced, have been used to control invasive populations (Kidd and Jervis 2007). Several natural enemies of H. halys, primarily hymenopteran egg parasitoids, have been reported in Japan (Kawada and Kitamura 1992; Arakawa and Namura 2002) and China (Yang et al. 2009). The predominant egg parasitoid of H. halys in northern China is Trissolcus halyomorphae (Hymenoptera: Scelionidae), with an average parasitism rate of 50 % (Yang et al. 2009). In the US, endemic enemies have minimal impact on H. halys, thus surveys for egg parasitoids are ongoing in native East Asia. To obtain useful knowledge on insecticide resistance and efficient natural enemies of an invasive pest, a prerequisite is to identify its source region(s), where surveys can be started subsequently.
Molecular genetic markers, both mitochondrial and nuclear ones, are powerful tools to examine diversity and define the source region(s) of an invasive pest, and thus to understand the mechanisms behind invasions (Ficetola et al. 2008). For these purposes, extensive and representative sampling in both native and exotic ranges as well as employing informative genetic markers is the key to success. In the case of invasions originated from divergent and structured native populations, the exact source region(s) and multiple introductions can often be identified (e.g., Kolbe et al. 2004); otherwise, the source region may be defined to a large native area and multiple introductions may be indiscernible.
The present study aims to (1) examine the genetic structure and assess the genetic diversity of H. halys in native East Asia and in the US, (2) identify the source region(s) of the US introductions, and (3) estimate the number of introductions and the likely size of founding populations. We also discuss possible mechanisms underlying the rapid infestation of H. halys in the US. The results of this study have important applications to the efficient control of this invasive agricultural pest.
Materials and methods
Specimens of H. halys
Halyomorpha halys specimens were collected in East Asia and across the US in 2004–2008 (Table 1; Fig. 1). Adults and large nymphs were preferred over small nymphs whenever possible to decrease the possibility of sampling siblings. Native H. halys was sampled from seven locations in China—Beijing (BJCN, Haidian District in Beijing), Xi’an (XACN, Shaanxi Province), Xuzhou (XZCN, Jiangsu Province), Nanjing (NJCN, Jiangsu Province), Hefei (HFCN, Anhui Province), Fuzhou (FZCN, Fujian Province) and Kunming (KMCN, Yunnan Province), three nearby sites combined into one sample in South Korea—Seoul/Suwon area (SWKR, Gyeonggi Province), and one location in Japan—Tsukuba City (TKJP, Honshu), with sample size of 5–21. These samples were collected from urban ornamentals, botanical gardens, agricultural crops and urban structures during the fall when H. halys aggregated prior to overwintering. In locations Xi’an, Fuzhou and Kunming, which are at the edges of its distribution in China (Zhu et al. 2012), all nymphs were included because it was difficult to find H. halys even on the well-known host plants such as Paulownia tomentosa.

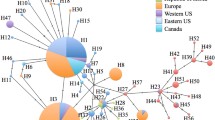
Maps of East Asia and the US showing the sampling locations and mtDNA haplotype distribution of H. halys. Native specimens were collected from China (CN), South Korea (KR) and Japan (JP). The US specimens were sampled from Massachusetts (MA), New York (NY), New Jersey (NJ), Pennsylvania (PA), Maryland (MD), Delaware (DE), West Virginia (WV), Virginia (VA), Mississippi (MS) and California (CA). Two haplotypes identified in the US are shown in white (H1) and black (H2). Haplotypes unique to China, South Korea and Japan are shown in grey, stippled white and stippled black, respectively. No shared haplotypes were detected among the three native countries
The US H. halys, all adults, were obtained from Massachusetts (MA: town of Bridgewater, Plymouth County), New York (NY: New York City, New York; Staten Island, Richmond), New Jersey (NJ: Mt. Royal, Gloucester; Basking Ridge, Somerset; Freehold, Monmouth; Hillsborough, Somerset; Somerset, Somerset), Pennsylvania (PA: Edgeworth, Allegheny; Monaca, Beaver; Allentown, Lehigh), Maryland (MD: Bethesda, Montgomery; Silver Spring, Montgomery; Rockville, Montgomery; Hagerstown, Washington; Williamsport, Washington), Delaware (DE: Dover, Kent; Wilmington, New Castle), West Virginia (WV: Shepherdstown, Jefferson), Virginia (VA: Lynchburg, Lynchburg; Roanoke, Roanoke), Mississippi (MS: Stoneville, Cleveland) and California (CA: San Marino, Los Angeles), with 1–6 specimens from each county and 2–12 from each state (Fig. 1). Most of these specimens were donated by the general public, through a web based reporting system (www.njaes.rutgers.edu/stinkbug) that we developed for documenting the distribution of H. halys. This website derives its usefulness from the fact that in the US, adult H. halys overwinters inside residences. The general public is an excellent source of information regarding the spread and infestation levels of H. halys, reporting thousands of sightings annually and mailing us many of the specimens used in this study. Specimens were either stored at −20 °C or in 95 % ethanol, and deposited in the Rutgers University Insect Museum. Genomic DNA was extracted from thoracic tissue with DNeasy tissue kit (Qiagen, Valencia, CA) following the manufacturer’s procedures. For testing the possible infection with Wolbachia, DNA was also extracted from the ovaries of a few specimens (see below).
Genetic data
We sequenced portions of three mitochondrial DNA genes, the cytochrome c oxidase subunit II (COII), and a fragment starting on the 12S ribosomal RNA and spanning part of the control region (12S/CR). To further narrow down the source region(s), sequences of cytochrome c oxidase subunit I (COI) gene were also collected from several select specimens. Nuclear markers were not included because highly variable markers such as microsatellites are still lacking for the species. Considering the infection of cytoplasmic parasites such as Wolbachia in other Pentatomidae species (Kikuchi et al. 2008), which leads to a decreased mtDNA variability due to selective sweeps (Engelstädter and Telschow 2009), we also tested for the possible presence of Wolbachia in the species, particularly in the ovaries.
Mitochondrial genes
Partial COII (559 bp) and 12S/CR (592 bp) genes were amplified with primers HhalysCO2F2 (5′-TAACCCAAGATGCAAATTCT-3′) and HhalysCO2R2 (5′-CCATATATAATTCCTGGACGA-3′), and HalCRf (5′-TTCCTAATCCTACTATTTAAGC-3′) and HalCRr (5′-GGTAACTTTATAAGAGGTCG-3′), respectively. These primers were designed based on H. halys sequences in GenBank (AY679135 and FJ685650). Complete COI gene (1,542 bp) was amplified with primers HhalCOIF (5′-CGCCTAAAAATTTCAGCCAC-3′; newly designed) and COIR (Xu and Fonseca 2011). PCR was composed of 1× PCR buffer, 2.5 mM of MgCl2, 200 μM of each dNTP, 0.2 mg/ml of BSA, 0.2 μM of each primer, and 1 unit of AmpliTaq ® (Applied Biosystems, Foster City, CA). A two-step PCR program, denaturation for 3 min at 96 °C followed by 10 cycles of 30 s at 94 °C, 30 s at 48 °C and 45 s at 72 °C, then 30 cycles of 30 s at 94 °C, 30 s at 50 °C and 45 s at 72 °C, and a final extension of 10 min at 72 °C, was used to amplify COII. 12S/CR and COI were amplified with a program of denaturation for 3 min at 94 °C, followed by 35 cycles of 30 s at 94 °C, 30 s at 58 °C and 45/90 s at 72 °C (12S/CR 45 s and COI 90 s), and a final extension of 10 min at 72 °C. PCR products were treated with ExoSAP-IT® (USB, Cleveland, OH) and cycle sequenced for analysis on an ABI 3100 automated sequencer (Applied Biosystems). Sequences were cleaned and checked with Sequencher 5.0 (Gene Codes, Ann Harbor, MI), and aligned with MUSCLE (Edgar 2004).
Wolbachia detection
The possible infection with Wolbachia was tested with universal primers for Wolbachia wsp 81F/wsp 691R (Zhou et al. 1998) by standard PCR, and with primers FtsZ-F/FtsZ-R by long PCR (Jeyaprakash and Hoy 2000). Standard PCR conditions were the same as above but with 35 cycles of 60 s at 94 °C, 60 s at 55 °C and 60 s at 72 °C. Long PCR amplification followed the program of Jeyaprakash and Hoy (2000). We used DNA of the mosquito Culex pipiens as a positive control because it is infected with Wolbachia. We tested specimens from native and exotic ranges, and included both thoracic tissues and ovaries of three female individuals.
Data analysis
Phylogeny and network of haplotypes
We examined the relationships among mtDNA haplotypes by creating a Bayesian phylogenetic tree with MrBayes 3.1 (Ronquist and Huelsenbeck 2003). Gene partition was used for COII and 12S/CR, and codon position (cp) partition (cp1+2 and cp3) was further used for COII. The best-fit models of sequence evolution were determined with MrModeltest (Nylander 2004). Analysis was run for 10 million generations using 4 chains with a sampling frequency of 1/1000. The results were visualized and checked using Tracer 1.4 (Rambaut and Drummond, 2003), and a burn-in of 2,500 was discarded. Parsimony network of the haplotypes was constructed with TCS (Clement et al. 2000), using all sequences from native samples. We excluded exotic specimens to avoid sampling bias, because there were only two haplotypes identified across all the US specimens, which were observed and included in native populations (see “Results”). Nested clade analysis (NCA) for populations from China was further conducted to infer possible evolutionary processes that played significant roles in forming the present genetic structure. Associations between haplotypes and geography were tested with GeoDis (Posada et al. 2000).
Population analysis
Population genetic diversity, as indexed by the number of haplotypes (Hn), haplotype diversity (Hd) and nucleotide diversity (θπ), was assessed with DnaSP 5.0 (Librado and Rozas 2009). Genetic differentiation among populations was estimated by the fixation index Φ ST as implemented in Arlequin 3.5 (Excoffier and Lischer 2010). The most appropriate model of sequence evolution for this analysis was estimated with Modeltest (Posada and Crandall 1998), and HKY + I was the selected model. Because this model is not implemented in Arlequin, the more inclusive Tamura-Nei (TrN, Tamura and Nei 1993) model was used for all relevant analyses. We examined the statistical significance of the estimates with 10,000 permutations. Significance levels for multiple tests were adjusted with sequential Bonferroni corrections (Rice 1989). To show the relationships among populations, a principal coordinate analysis (PCA) was created with GenAlEx 6.4 (Peakall and Smouse 2006), based on pairwise genetic distances among populations. Population genetic structure was assessed with an analysis of molecular variance (AMOVA) in Arlequin. The significance of the measures was examined with 1,000 permutations.
We examined the historical demographic expansion in natives with Fu’s Fs neutrality test (Fu 1997) and mismatch distribution based on COII gene, as implemented in DnaSP. Fu’s Fs value is sensitive to demographic expansion, which usually leads to large negative values. Mismatch distribution was used to distinguish between smooth unimodal distribution and multimodal distribution (Rogers and Harpending 1992), which respectively indicate historical expansion and population equilibrium. We estimated population expansion time with τ = 2μt, where τ is the crest of mismatch distribution, μ is nucleotide substitution rate and t is the time in generations. We assumed a mutation rate of 6.2 × 10−8 per site per generation—an experimental estimation for mutation of Drosophila mtDNA (Haag-Liautard et al. 2008)—for the COII gene of H. halys, because no estimation for H. halys is available. To transfer the time in generations to years, we adopted a generation time of two generations a year for native H. halys.
Results
We identified 18 haplotypes (H1–18) defined by 17 polymorphic sites in a 534 bp COII fragment, and 20 haplotypes (H1–20) defined by 20 polymorphic sites in a 550–552 bp 12S/CR fragment (83–84 bp of 12S and 467–468 bp of CR), from a total of 132 H. halys specimens—48 of China, 8 of South Korea, 21 of Japan and 55 of the US (Table S1). In the US, a single COII haplotype (H1) and two 12S/CR haplotypes (H1 and H2) were detected from 55 specimens. Sequences of haplotypes were deposited in GenBank (accession # KF112000–KF112037). Mutations in COII resulted in four amino acid changes, two in Japanese specimens (28Ala → Val and 32Thr → Ser) and two in Chinese specimens (63Met → Thr and 172Ileu → Met). Four indels (insertion and deletion) and a 20 bp highly mutant region (eight mutation sites in four specimens of XACN and one specimen of TKJP) were observed in 12S/CR. The mean nucleotide difference and genetic distance (uncorrected) among haplotypes were 3.3 and 0.0062 for COII, and 4 and 0.0065 for 12S/CR. When the sequences of COII and 12S/CR were combined, we identified 43 haplotypes (H1–43) in the 132 specimens (Table 2).
In the native range, we detected 26, 7 and 10 haplotypes in China, South Korea and Japan, respectively, with 2–10 haplotypes from each location. There were no shared haplotypes among the three countries (Table 2; Fig. 1). In the 26 haplotypes from China, five were present in more than one location, i.e., H1 in BJCN and FZCN, H2 in BJCN and KMCN, H4 in BJCN, XZCN and NJCN, H12 in XZCN and NJCN, and H13 in XZCN and FZCN. Populations XACN and HFCN contained only private haplotypes and seem to be isolated from others. In the 55 H. halys specimens across 10 US states, we only found two haplotypes—H1 and H2; H2 was much more common (N = 46) than H1 (N = 9). These two haplotypes coexisted in the coastal states MA, NJ, MD, DE and VA; H2 also occurred singly in the inland states PA, WV and MS as well as in NY, a coastal state, and H1 occurred singly in the west coast in CA (Fig. 1). Interestingly, in native regions, H1 and H2 only coexisted in BJCN, but also occurred separately in FZCN and KMCN. This distribution pattern pinpoints BJCN as the source origin, though we cannot completely exclude FZCN and KMCN as possible origins if multiple introductions have occurred. The COI sequences (1,542 bp), however, further excluded FZCN as the origin, because the single individual in FZCN with COII+12S/CR haplotype H1 had a different COI haplotype (H3) from those in the US, and those from BJCN and KMCN (Table S2). Two specimens of BJCN with COII+12S/CR haplotype H1 had different COI haplotypes (H1 and H2). In total, we recovered three COI haplotypes (GenBank accession # KF112038–KF112040) from nine specimens (five from China and four from the US), differing from each other at 1–2 mutation sites. KMCN and FZCN are at the edge of the species’ distribution, and it was difficult to find H. halys there; KMCN is therefore unlikely to be an alternative source region for the US introductions. Haplotypes H1 and H2 in FZCN and KMCN were more likely a result of long distance dispersal from BJCN (possibly the precursor of intra-continental dispersal), rather than indigenous ones (see “Phylogenetic and network analyses”).
Phylogenetic and network analyses
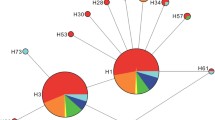
Phylogenetic analysis revealed structuring among haplotypes, but most of the posterior probabilities supporting the topology were lower than 0.95 (not well supported; Fig. 2). Despite the low support, we observed divergence among China, South Korea and Japan, as indicated by the fact that most of their haplotypes were clustered into private lineages (Maximum Likelihood phylogeny resulted in the same topology with low support, not shown). Two lineages were also observed in China—one consisting of haplotypes (H3–19) in north China and the other consisting of haplotypes in both north (H1–2, 7, 10–11 and 13–14) and south China (H20–26). FZCN in south China had two haplotypes both shared with populations in north China. A parsimony network (95 % confidence level, 14 steps) indicated that haplotypes in China, South Korea and Japan each formed a network, and the three networks were linked by one mutation (Fig. 3A). Chinese haplotypes formed a star-like network, with those from BJCN, XZCN and NJCN in the center (Fig. 3), implying north China (BJCN, XZCN and NJCN) as the center of distribution (ancestral clades) and population expansion in its demographic history. NCA for populations in China inferred restricted gene flow but with some long distance dispersal for clade 2-1, which includes all populations except HFCN (Table 3; Fig. 3B). Long distance dispersal, likely facilitated by human movement, could explain why the marginal populations FZCN and KMCN shared haplotypes with the central ones BJCN and XZCN.

Bayesian phylogenetic tree of mtDNA haplotypes of H. halys. Bayesian posterior probabilities >0.95 are shown above branches. Haplotypes of China, South Korea and Japan are respectively labeled with normal, italic and bold fonts, and grouped separately with double, grey and black lines. The two haplotypes found in the US are indicated with lined circles

A Parsimony network of mtDNA haplotypes of H. halys, and B haplotype network with nested clade design for H. halys in China. Each haplotype is indicated with a number in a circle. Numbers correspond to the haplotype numbers in Table 2. Haplotype circle size is proportional to its frequency in native populations (see scale on the right lower corner of A). The haplotype in a square refers to the ancestral haplotype. Black dots represent undetected haplotypes. The two haplotypes found in the US are shown with lined circles
Population analysis
Genetic diversity in the native populations, particularly the haplotype diversity (Hd), was generally high (Hd = 0.81–1) except in populations at the edge of the species’ distribution, i.e., XACN, FZCN and KMCN, which showed much lower diversity (Hd = 0.33–0.7; Table 1). In contrast, genetic diversity in the US populations was extremely low, even zero in half of the populations because only a single haplotype was detected. This indicates that population size of the introductions was quite small, and a population bottleneck has occurred due to a founder effect upon introduction.
Strong genetic divergence was observed across all populations (Φ ST = 0.616, P < 0.01) or across native populations (Φ ST = 0.518, P < 0.01, Table 4). Pairwise Φ ST values among populations further supported their significant differentiation; populations XACN, HFCN, FZCN, SWKR, TKJP and US were isolated from each other, and from the other Chinese populations (Table 5). PCA based on population genetic distance clearly showed the existence and divergence of five distinct groups in native range (Fig. 4). We therefore separated the natives into five groups, (1) BJCN, XZCN, NJCN, HFCN and KMCN, (2) XACN, (3) FZCN, (4) SWKR and (5) TKJP, to further explore their differentiation (Table 4). The Φ CT value among the five groups was 0.331 and significant, further supporting the differentiation in native populations. H. halys in the three native countries each formed different groups, with at least three groups observed in China—XACN, FZCN and the others.

Principal coordinate analysis (PCA) on H. halys populations based on population pairwise genetic distances. Populations of China, South Korea, Japan and the US are indicated with empty, grey, black and lined circles, respectively
Demographic expansion was detected in native populations with the neutrality test and mismatch distribution based on COII gene. Fu’s Fs values for all native, Chinese, Korean and Japanese populations were negative, and significant for all natives and Korean population (Table 6). When we combined the COII and 12S/CR sequences for this analysis, the Fu’s Fs ranged from −2.71 to −33.02 and were significant (not shown). Mismatch distributions for all natives, Chinese, Korean and Japanese populations were unimodal and fitted well with the expected distribution under expansion model (Fig. 5). These results indicate demographic expansion in the natives. The expansions in all natives and the Japanese population were estimated to start around 11 thousand years ago (ka), and about 6 and 15 ka for the Chinese populations and the Korean population, within the interglacial time of the Holocene (0–12 ka) following the last glacial maximum (LGM, ~20 ka).

Mismatch distribution of native populations of H. halys. The observed pairwise differences and expected values under growth-decline model are shown with dashed and solid lines, respectively
Wolbachia detection
We found no evidence for the presence of Wolbachia in H. halys, either in thoracic or ovary tissue, by both standard PCR and long PCR.
Discussion
Among native populations we detected significant genetic structure and divergence as well as high genetic diversity within populations except those at the edge of the species’ distribution. In contrast, genetic diversity in the US populations was much lower, with only two haplotypes identified across the continental US as of 2008—approximately 10 years post introduction. Based on private haplotypes and phylogeographic analysis of native populations, we traced the origin of the US H. halys to the Beijing area in China, which has important applications to the effective control of this invasive pest.
Genetic structure of H. halys in East Asia
The mtDNA analysis of H. halys revealed a clear genetic structure in native East Asia. We detected few haplotypes shared among populations and significant genetic divergence among populations of China, South Korea and Japan. Furthermore, we observed genetic isolation in populations of China such as XACN, FZCN, KMCN and HFCN. These results indicate limited gene flow or migration among populations, which is also supported by the NCA inference of ‘restricted gene flow’. Based on phylogeny and network analysis of haplotypes, we inferred north China (BJCN, XZCN and NJCN) as the distributional center in China, which agrees with that predicted from ecological niche modeling (Zhu et al. 2012). The haplotypes shared between marginal populations (FZCN and KMCN) and populations at the center (BJCN, XZCN and NJCN) likely resulted from human mediated dispersals from central to marginal areas, which is consistent with the NCA inference of ‘with some long distance dispersal’. The high genetic diversity in the central populations but low in marginal ones further corroborates this dispersal pattern. Both neutrality test and mismatch distribution indicated demographic expansion in the native populations, which was estimated to have started around 6000–15,000 years ago—a time within the interglacial period after the LGM. This explains why the haplotype phylogeny has low statistical support.
Introduction of H. halys to the US
Halyomorpha halys has invaded the US and expanded to 39 states in the past 15 years, causing unprecedented damages to agriculture (Leskey and Hamilton 2010; Seetin 2011; Kuhar et al. 2012). The first and critical question on its invasion is where did this invasive stink bug come from, followed by questions such as how many introductions have occurred and what was the likely size of the founding populations? Two mtDNA haplotypes (H1 and H2) were identified in the US populations, both coexisting in eastern coastal states and each also occurring singly in eastern inland states (H2) and west coast CA (H1). The unique mtDNA haplotypes in each native population, and the significant genetic differentiation among native populations and native countries, allow us to pinpoint the Beijing area in China (BJCN, the only native location where the two US haplotypes were found to coexist) as the likely source region for the US H. halys. We further excluded Kunming (KMCN) and Fuzhou (FZCN) as alternative origins by combining COI sequence analysis and phylogeographic analysis of the Chinese populations. This result disagrees with the proposed origins in northern Japan or western Korea based on climate matching models (Zhu et al. 2012). The US mid-Atlantic region, particularly eastern Pennsylvania and New Jersey where the exotic H. halys was first reported, is the purported epicenter of current distribution in the US (Hoebeke and Carter 2003). We found two haplotypes coexisting in this area, which implies that a single introduction with the two haplotypes may summarize the US invasion history. Specimens in California and eastern inland states appear more likely the result of long-distance dispersal events from the east coast population facilitated by human transportation (e.g., Jones and Lambdin 2009), rather than new invasions from the native range. The single haplotype observed in these locations may result from genetic drift or selection during and after introduction. Since we detected only one haplotype in eight specimens from Pennsylvania, although it is the believed epicenter, examination of more samples from here will be instructive.
Compared with the high genetic diversity in native populations such as BJCN (five haplotypes in six individuals, Hd = 0.93, θπ = 0.0015), we observed extremely low diversity in US populations (two haplotypes in 55 individuals, Hd = 0.28, θπ = 0.0005). As we discussed above, Beijing (BJCN) is the likely source region of the US introduction. In BJCN, five haplotypes were identified from six individuals, and frequencies of H1 and H2 were 1/3 and 1/6, respectively. This implies that the current US population may result from a single successful introduction with a small population size (as small as two females; theoretically, in any 18 females from the source region, haplotypes H1 and H2 will be observed, because the frequency of H1 coexisting with H2 is 1/18), rather than multiple introductions or a large population. More haplotypes would be expected if the latter scenarios had happened, unless there existed strong selection on the mitochondrial genome in the exotic range. Nuclear markers such as amplified fragment length polymorphism (AFLP) and microsatellites would provide further insight into the nuclear genetic diversity and selection on the mitochondrial genome.
Identifying the source region has important applications to the control of H. halys. Egg parasitoids of H. halys, especially those in the Trissolcus genus, are the primary candidates for biological control and are expected to have the greatest impact on mitigating population growth. Our results strongly suggest that surveys for egg parasitoids should focus on north China, specifically the Beijing area. Trsissolcus halyomorphae, which has the highest reported parasitism rate for H. halys, was first identified in Beijing, China from natural populations (Yang et al. 2009); it has similar developmental thresholds as H. halys and thus should be a well adapted biological control agent for the US population. As an agricultural pest, H. halys has caused severe damage to crops, especially tree fruits, with mid-Atlantic growers experiencing 20–90 % economic losses. Reacting to an emergency situation, up to 16 insecticide applications are made each season against H. halys. Although insecticide resistance has not been reported in the US, we should be mindful that it might have been introduced with the founding population, or it may arise quickly given that the population has experienced such a strong genetic reduction. Our results suggest that surveys for possible insecticide resistance in source regions, particularly in the Beijing area, are necessary.
Successful invasion with low genetic diversity
A few invasive insects have extremely low mtDNA genetic diversity across a wide exotic range, including Colorado potato beetle Leptinotarsa decemlineata (Grapputo et al. 2005), Argentine ant Linepithema humile (Corin et al. 2007), the red imported fire ant Solenopsis invicta (Caldera et al. 2008), Russian wheat aphid Diuraphis noxia (Shufran et al. 2007) and the kudzu bug Megacopta cribraria (Jenkins and Eaton 2011). L. decemlineata was introduced to Europe in the 1920s, where it has become an important pest of solanaceous crops. Across all European populations, single mtDNA haplotype is matched with low nuclear genetic diversity as revealed by AFLP analysis (Grapputo et al. 2005). Haplodiploid ant species are expected to share maternal mitochondrial haplotypes because of a single queen in one colony; exotic populations of both L. humile and S. invicta appear to have spread from a single introduction (Corin et al. 2007; Caldera et al. 2008). Similarly D. noxia with a single haplotype in the US is consistent with a single introduction from South Africa (Shufran et al. 2007). Yet the single maternal haplotype of exotic M. cribraria may be explained by the single Wolbachia haplotype found in the US and Japanese populations (Jenkins and Eaton 2011).
Many invasive species have experienced a decrease of genetic diversity in their exotic habitats, but low genetic variation does not necessarily endanger fitness, in contrast, it may highlight rare alleles that increase fitness or invasiveness (Dlugosch and Parker 2008). In the blueberry maggot Rhagoletis mendax, for example, low genetic diversity is the direct result of selection on individuals that matched the fruiting schedule of commercial blueberries (Teixeira and Polavarapu 2003). In Argentine ant L. humile, inter-colony competition decreases with relatedness and reduction of genetic variation (Tsutsui et al. 2000). Colorado potato beetle L. decemlineata thrived by finding the same food species in southern Europe as in its native range in the southwestern US, where it had specialized in feeding on commercial potatoes (Grapputo et al. 2005).
Lack of genetic diversity in the US H. halys cannot be explained by a Wolbachia sweep, since we found no evidence for infestation with this intracellular parasite. As a distributional center in the US, eastern Pennsylvania and New Jersey share similar climatic conditions with the native distributional center (Zhu et al. 2012). In the mid-Atlantic area, egg parasitism rates of H. halys are lower than those of native pentatomid species (e.g., Koppel et al. 2009), as well as those of H. halys in native China (Yang et al. 2009). Local climatic conditions and lack of effective natural enemies possibly have played a major role in the explosive growth of H. halys in the US mid-Atlantic region. In addition, the association of an agricultural pest like H. halys with human-made constructions, which remain warm during winter seasons, may be an important adaptation that enabled it to successfully establish and thrive in the mid-Atlantic region. Like H. halys, the invasive kudzu bug M. cribraria also exploits the agriculture/urban structures for overwintering, and its population densities have increased rapidly in the invaded areas. We hypothesize that proximity between homes and crops in the small farms in Pennsylvania and New Jersey is a critical ecological factor driving the expansion of H. halys in the US. Breaking this link, by targeting populations indoors or when they enter or exit homes, may be an effective way to control this pest.
References
Arakawa R, Namura Y (2002) Effects of temperature on development of three Trissolcus spp. (Hymenoptera: Scelionidae), egg parasitoids of the brown marmorated stink bug, Halyomorpha halys (Hemiptera: Pentatomidae). Entomol Sci 5:215–218
Caldera EJ, Ross KG, DeHeer CJ, Shoemaker DD (2008) Putative native source of the invasive fire ant Solenopsis invicta in the USA. Biol Invasions 10:1457–1479
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Corin SE, Lester PJ, Abbott KL, Ritchie PA (2007) Inferring historical introduction pathways with mitochondrial DNA: the case of introduced Argentine ants (Linepithema humile) into New Zealand. Divers Distrib 13:510–518
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17:431–449
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Engelstädter J, Telschow A (2009) Cytoplasmic incompatibility and host population structure. Heredity 103:196–207
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resources 10:564–567
Ficetola GF, Bonin A, Miaud C (2008) Population genetics reveals origin and number of founders in a biological invasion. Mol Ecol 17:773–782
Fogain R, Graff S (2011) First records of the invasive pest, Halyomorpha halys (Hemiptera: Pentatomidae) in Ontario and Quebec. J Entomol Soc Ont 142:45–48
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Funayama K (2004) Importance of apple fruits as food for the brown marmorated stink bug, Halyomorpha halys (Stål) (Heteroptera: Pentatomidae). Appl Entomol Zool 39:617–623
Grapputo A, Boman S, Lindström L, Lyytinen A, Mappes J (2005) The voyage of an invasive species across continents: genetic diversity of North American and European Colorado potato beetle populations. Mol Ecol 14:4207–4219
Haag-Liautard C, Coffey N, Houle D, Lynch M, Charlesworth B, Keightley PD (2008) Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster. PLoS Biol 6:e204. doi:10.1371/journal.pbio.0060204
Harris AC (2010) Halyomorpha halys (Hemiptera: Pentatomidae) and Protaetia brevitarsis (Coleoptera: Scarabaeidae: Cetoniinae) intercepted in Dunedin. The Weta 40:42–44
Hoebeke ER, Carter ME (2003) Halyomorpha halys (Stål) (Heteroptera: Pentatomidae): a polyphagous plant pest from Asia newly detected in North America. Proc Entomol Soc Washington 105:225–237
Hoffman WE (1931) A pentatomid pest of growing beans in South China. Peking Natural History Bull 5:25–26
Holtz T, Kamminga K (2010) Qualitative analysis of the pest risk potential of the brown marmorated stink bug (BMSB), Halyomorpha halys (Stål), in the United States. USDA APHIS PPQ. http://www.michigan.gov/documents/mda/BMSB_Pest_Risk_Potential_-_USDA_APHIS_Nov_2011_344862_7.pdf. Accessed 14 June 2013
Jenkins TM, Eaton TD (2011) Population genetic baseline of the first plataspid stink bug symbiosis (Hemiptera: Heteroptera Plataspidae) reported in North America. Insects 2:264–272
Jeyaprakash A, Hoy MA (2000) Long PCR improves Wolbachia DNA amplification: wsp sequences found in 76% of sixty-three arthropod species. Insect Mol Bio 9:393–405
Jones JR, Lambdin PL (2009) New county and state records for Tennessee of an exotic pest, Halyomorpha halys (Hemiptera: Pentatomidae), with potential economic and ecological implications. Fla Entomol 92:177–178
Kawada H, Kitamura C (1992) The tachinid fly, Bogosia sp. (Diptera: Tachinidae), as a parasitoid of the brown marmorated stink bug, Halyomorpha mista Uhler (Heteroptera: Pentatomidae). Jpn J Environ Entomol Zool 4:65–70
Kidd NAC, Jervis MA (2007) Population dynamics. In: Jervis MA (ed) Insects as natural enemies: a practical perspective. Springer, Dordrecht, pp 435–523
Kikuchi Y, Hosokawa T, Fukatsu T (2008) Diversity of bacterial symbiosis in stinkbugs. In: Dijk TV (ed) Microbial ecology research trends. Nova Publishers, New Yok, pp 39–64
Kobayashi T, Hasegawa T, Kegasawa K (1972) Major insect pests of leguminous crops in Japan. Trop Agric Res Ser 6:109–126
Kolbe JJ, Glor RE, Rodriguez Schettino L, Lara AC, Larson A, Losos JB (2004) Genetic variation increases during biological invasion by a Cuban lizard. Nature 431:177–181
Koppel AL, Herbert DA Jr, Kuhar TP, Kamminga K (2009) Survey of stink bug (Hemiptera: Pentatomidae) egg parasitoids in wheat, soybean, and vegetable crops in southeast Virginia. Environ Entomol 38:375–379
Kuhar TP, Kamminga KL, Whalen J, Dively GP, Brust G, Hooks CRR, Hamilton G, Herbert DA (2012) The pest potential of brown marmorated stink bug on vegetable crops. Online. Plant Health Prog. doi:10.1094/PHP-2012-0523-01-BR
Leskey TC, Hamilton GC (2010) Brown marmorated stink bug working group progress report. http://projects.ipmcenters.org/Northeastern/FundedProjects/ReportFiles/Pship2010/Pship2010_Lesky_ProgressReport_237195_meeting2010_11_17.pdf. Accessed 27 July 2011
Leskey TC, Hamilton GC, Nielsen AL et al (2012) Pest status of the brown marmorated stink bug, Halyomorpha halys in the USA. Outlooks Pest Manag 23:218–226.
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Nielsen AL, Hamilton GC (2009) Life history of the invasive species Halyomorpha halys (Hemiptera: Pentatomidae) in northeastern United States. Ann Entomol Soc Am 102:608–616
Nielsen AL, Hamilton GC, Shearer PW (2011) Seasonal phenology and monitoring of the non-native Halyomorpha halys (Hemiptera: Pentatomidae) in soybean. Env Entomol 40:231–238
Nylander JAA (2004) MrModeltest v2. Program distributed by the author. Evolutionary Biology Centre, Uppsala University, Sweden
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Posada D, Crandall KA, Templeton AR (2000) GeoDis: a program for the cladistics nested analysis of the geographical distribution of genetic haplotypes. Mol Ecol 9:487–488
Rambaut A, Drummond AJ (2003). TRACER: MCMC trace analysis tool. Available from: http://evolve.zoo.ox.ac.uk. University of Oxford, Oxford
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 19:552–569
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Seetin M (2011) News release: losses to mid-Atlantic apple growers at $37 million from brown marmorated stink bug. http://www.growingproduce.com/article/21057/brown-marmorated-stink-bug-causes-37-million-in-losses-to-mid-atlantic-apple-growers. Accessed 30 Apr 2012
Shufran KA, Kirkman LR, Puterka GJ (2007) Absence of mitochondrial DNA sequence variation in Russian wheat aphid (Hemiptera: Aphididae) populations consistent with a single introduction into the United States. J Kansas Entomol Soc 80:319–326
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Teixeira LAF, Polavarapu S (2003) Evolution of phenologically distinct populations of Rhagoletis mendax (Diptera: Tephritidae) in highbush blueberry fields. Annals Entomol Soc Am 96:818–827
Tsutsui ND, Suarez AV, Holway DA, Case TJ (2000) Reduced genetic variation and the success of an invasive species. Proc Natl Acad Sci 97:5948–5953
Wermelinger B, Wyniger D, Forster B (2008) First records of an invasive bug in Europe: Halyomorpha halys (Stål) (Heteroptera: Pentatomidae), a new pest on woody ornamentals and fruit trees? Bull Entomol Suisse 81:1–8
Xu J, Fonseca DM (2011) One-way sequencing of multiple amplicons from tandem repetitive mitochondrial DNA control region. Mitochondrial DNA 22:155–158
Yang Z-Q, Yao Y-X, Qiu L-F, Li Z-X (2009) A new species of Trissolcus (Hymenoptera: Scelionidae) parasitizing eggs of Halyomorpha halys (Heteroptera: Pentatomidae) in China with comments on its biology. Ann Entomol Soc Am 102:39–47
Zhou W, Rousset F, O’Neill S (1998) Phylogeny and PCR-based classification of Wolbachia strains using wsp gene sequences. Proc R Soc Lond B 265:509–515
Zhu G, Bu W, Gao Y, Liu G (2012) Potential geographic distribution of brown marmorated stink bug invasion (Halyomorpha halys). PLoS One 7:e31246
Acknowledgments
We are greatly appreciative of the assistance of A. Koppel, L. Zhang, M.-Z. Lin, F. Chen, S. H. Lee, H. J. Kim, S. H. Jung and M. Toyama in collecting specimens from the native range. We are indebted to all the homeowners who enthusiastically provided us with specimens and information. Citizen Science is an important resource in our fight against invasive species. This study was funded in part by NJAES Hatch projects 08191 and 0211785.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Xu, J., Fonseca, D.M., Hamilton, G.C. et al. Tracing the origin of US brown marmorated stink bugs, Halyomorpha halys . Biol Invasions 16, 153–166 (2014). https://doi.org/10.1007/s10530-013-0510-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-013-0510-3