
Abstract
Objectives
To discover novel feruloyl esterases (FAEs) by the function-driven screening procedure from soil metagenome.
Results
A novel FAE gene bds4 was isolated from a soil metagenomic library and over-expressed in Escherichia coli. The recombinant enzyme BDS4 was purified to homogeneity with a predicted molecular weight of 38.8 kDa. BDS4 exhibited strong activity (57.05 U/mg) toward methyl ferulate under the optimum pH and temperature of 8.0 and 37°C. Based on its amino acid sequence and model substrates specificity, BDS4 was classified as a type-C FAE. The quantity of the releasing ferulic acid can be enhanced significantly in the presence of xylanase compared with BDS4 alone from de-starched wheat bran. In addition, BDS4 can also hydrolyze several phthalates such as diethyl phthalate, dimethyl phthalate and dibutyl phthalate.
Conclusion
The current investigation discovered a novel FAE with phthalate-degrading activity and highlighted the usefulness of metagenomic approaches as a powerful tool for discovery of novel FAEs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Feruloyl esterases (FAEs) (EC 3.1.1.73) are a group of enzymes that catalyze the cleavage of cross-linkages among plant cell wall polysaccharides and phenolic compounds, involving ferulic acid (FA) and p-coumaric acid (Williamson et al. 1998). There has been great interest to find new FAEs for biotechnological use in recent years. FAEs not only play critical roles in the lignocellulosic biomass conversion for renewable energy production (Gopalan et al. 2015; Dilokpimol et al. 2018), but also release phenolic acids, e.g., FA, caffeic acid and sinapic acid, which are valuable by-products as antioxidants, flavor precursors and antimicrobial agents (Shahidi and Chandrasekara 2010).
FAEs are distributed in plants, fungi and bacteria of which most FAEs are of microbial origins, more than 80 FAEs have been characterized from microbial source since it was identified in the 1980s and purification in 1991 (Oliveira et al. 2019). However, the traditional enzyme screening approaches have been confronted with the predicament of microorganisms’ cultivation and the subsequent screen of the pure strains for the desired catalytic activity, as much as 99% bacterial strains are uncultivable by standard methods in the lab, which limit the exploitation for possible novel FAEs in a large untapped gene pool (Amann et al. 1995; Schloss and Handelsman 2005). In addition, few FAEs were able to meet the requirements for a given application due to their low levels of expression and weak catalytic activities (Dilokpimol et al. 2016).
Culture-independent metagenomic method circumvents the culture of microorganisms, the DNA is extracted directly from i.e. a given environmental soil and cloned into a cultured heterologous host, providing an efficient way for systematically exploring plenty of biosynthetic gene clusters from unculturable microbial species (Handelsman 2004). A variety of enzymes have been identified by using metagenomic technology, including lipases, esterases and phospholipases (Reyes-Duarte et al. 2012). Given that soil sample was the largest microorganisms reservoir with a wide range of diversity (Torsvik et al. 1990, 1998), we focused on soil sample to screen, clone and identify novel FAE genes through a metagenomic strategy.
Herein, we described the utility of metagenomics coupled with function-driven screening for mining a novel FAE from the soil sample. The detailed steps were described including construction of metagenomic library, screening of positive clones, molecular cloning and over-expression of a FAE, enzymatic activity assay and application in wheat bran hydrolyzation. Additionally, the degradation effect of FAE on phthalates which are one of the most frequently detected persistent organic pollutants in the environment was investigated as well.
Materials and methods
Bacterial strains, plasmids, enzymes and materials
Escherichia coli EPI300 and pcc2FOS (Epicenter, USA) were used for the construction of the metagenomic library. The pUC118 and E.coli DH5α Competent Cells (Takara, Japan) were taken for subcloning. The pET-28a (+) and E. coli BL21(DE3) (Takara, Japan) were designed for protein expression. The Sau3A I, BamHI and HindIII (Vazyme, China) were used as restriction enzymes. The standard chemicals, reagents and recombinant xylanase from Aspergillus oryzae were purchased from Sigma-Aldrich.
Metagenomic library construction and screening
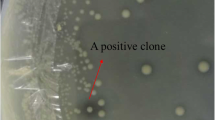
The soil sample was collected from Baoding region (Latitude-38.548913 N, Longitude-115.23515 E), at Hebei Province, China. The crude environment DNA was extracted according to the described protocol (Brady 2007). The DNA fragments of around 40 kb were obtained, and then ligated to the pcc2FOS vector and transfected into E. coli EPI300 for constructing the library according to the manufacturer’s guide (Copy Control™ Fosmid Library Production Kit). The function-driven screening was performed on the Luria–Bertani (LB) agar plates with 1.5 mg/mL methyl ferulate (MFA) (Cheng et al. 2012a). The putative FAE clone was identified by the formation of a clear halo around the colony margin. One single positive clone was picked up and cultured in LB liquid medium (chloramphenicol 12.5 μg/mL, MFA 1.5 mg/mL, IPTG 0.5 mM). The fermented broth was filtrated with a 0.22 μm organic membrane filter and then analyzed by high pressure liquid chromatography (HPLC) equipped with an Agilent Zorbax SB-C18 column (4.6 × 150 mm, 5 Micron). The linear gradient elution was performed from 10% solution A/90% solution B to 0% solution A/100% solution B (solvents solution A, 1% acetic acid; solution B, methanol) with a flow rate of 1 mL/min. The detected peak area of FA at 320 nm was used to calculate the FAE activity.
Subclone library construction
The plasmid from the positive clone was extracted using Plasmid Mini kit I (OMEGA, USA). The obtained plasmid was partially digested by Sau3A I enzyme. The fragments were purified by a 1% agarose gel. The acquired DNA fragments (1-5 kb) were chosen to ligate into the BamHI site of pUC118 vector and then transferred into the competent E. coli DH5α via the heat shock method. The transformants were cultivated on LB agar plates (ampicillin 100 μg/mL, MFA 1.5 mg/mL) at 37°C overnight and the E. coil subclone library was constructed. The desired subclone was rescreened for FAE activity by HPLC and confirmed by DNA sequencing.
Bioinformatics analysis
The sequence was analyzed by online server Open Reading Frame (ORF) Finder (https://www.ncbi.nlm.nih.gov/orffinder/) (Hancock and Bishop 2004). The sequence similarity was retrieved by BLAST program (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (Altschul et al. 2005). The multiple alignment of FAEs sequence was performed on the online software Clustal Omega program (https://www.ebi.ac.uk/Tools/msa/clustalo/) (Li et al. 2015). Theoretical molecular weight was calculated using the ProtParam program (https://web.expasy.org/protparam/). The phylogenic tree was generated by neighbor joining algorithm method with bootstrap test and Poisson model using MEGA 6.0 software (Tamura et al. 2013). The three-dimensional structure was simulated by COACH database (https://zhanglab.ccmb.med.umich.edu/COACH/) (Yang et al. 2013a, b).
Expression and purification of recombinant enzyme
The FAE-encoding gene was amplified by PCR with a pair of specific primers: hFv/BamHI 5′-GCTGGATCCATGCCATATATTTCCACC-3′ and hRv/HindIII 5′-GCTAAGCTTTGGCTTTTGAATTAATTG-3′ (underlined bases are the BamHI and HindIII sites, respectively). The PCR reaction system (50 μl): ddH2O 20 μl, 2 × Taq Master Mix 25 μl, primers (10 μM) 2 μl, DNA template 1 μl. The PCR reaction conditions: 95°C for 5 min; 35 cycles of 94°C for 5 min, 56°C for 30 s, and 72°C for 1 min; 72°C for 10 min. The PCR products were digested with BamHI and HindIII, and then ligated into the corresponding sites of pET-28a (+) expression vector by T4 DNA ligase. The recombinant plasmid (pET-bds4) was yielded and then transformed into E. coli BL21(DE3) for protein expression.
The transformants were incubated at 37°C in the LB liquid medium (kanamycin 50 μg/mL, MFA 1.5 mg/mL, IPTG 0.5 mM) and E. coli BL21(DE3) with an empty vector pET-28a (+) was used as a negative control. The cells were induced with 0.5 mM IPTG when the OD600 value of the culture reached 0.4. The cells were harvested by centrifugation (4°C, 6000 *g, 5 min) and then suspended in the sodium phosphate buffer (20 mM, pH 8). The cells were lysed by sonication and the supernatant was collected as crude enzyme by centrifugation (4°C, 12,000 *g, 20 min). The recombinant enzyme was purified by Ni–NTA-Sefinose Column (Sangon Biotech, China) and eluted with a linear gradient of imidazole buffer solution (20, 50, 100, 250, 500 mM) in a flow of 1 mL/min. The molecular weight of purified BDS4 was examined by SDS-PAGE analysis. The BDS4 was concentrated by an ultrafiltration centrifuge tube with 10 kDa cutoff (Millipore, USA). The content of total protein was quantified by using the Bradford Protein Assay.
Biochemical characterization
The biochemical reactions were initiated by the addition of 20 μg/mL purified BDS4 and incubated in 1 mL buffer with 5-9 mM MFA as substrate with time interval 5 min. The optimal pH for enzyme activities was measured in different buffers: pH 3.0–6.0, 20 mM sodium citrate buffer; pH 6.0–8.0, 20 mM Na2HPO4-NaH2PO4 buffer; pH 7.5–9.0, 20 mM Tris-HCl buffer; pH 9.0–11.0, 20 mM glycine-NaOH buffer. To determine pH stability, the purified enzyme in different buffers was pre-incubated at 4°C for 1 h, and then incubated 37°C for 5 min, the residue enzymatic activity was measured under standard condition (Cheng et al. 2012a). The optimal temperature was determined at 4-75°C. For thermostability, the enzyme was pre-incubated at different temperatures for 1–4 h and then the residual activity was measured. One unit of FAE activity was defined as the amount of enzyme that released 1 μmol of FA per minute. The metal ions and chemicals (Table 1) were added into the reaction mixture to investigate their effects on enzyme activity. In addition, enzymatic kinetic parameters (Km, Vmax, kcat, kcat/Km) were calculated using different concentrations of MFA, ethyl ferulate (EFA), methyl p-coumarate (MpCA), methyl caffeate (MCA), methyl vanillate (MV), methyl sinapate (MSA), chlorogenic acid (CGA) and methyl gallate (MG) from 0.5 to 9 mM to determine substrate binding and specificity of BDS4. All quantitative assays were performed in triplicate for statistical analysis.
Site-directed mutagenesis
Site-directed mutagenesis was performed by using Trelief™ SoSoo Cloning Kit (TsingKe Co, China). The primers were designed to introduce base substitutions (Supplementary Table 1) and then elongated by PCR using pET-bds4 vector as the template. The mutated gene was harvested and then the nicks were ligated based on homologous recombination, followed by transferring into E.coli BL21(DE3). The point mutants were generated and sequenced for further confirmation. The transformants were grown at 37°C in LB screening agar plates and further analyzed by enzymatic activity assay.
Release of FA from DSWB
The ability of releasing FA by the purified BDS4 was examined using de-starched wheat bran (DSWB) as the complex substrate according to the previously reported procedure (Haase-Aschoff et al. 2013). The reaction mixture containing 200 mg DSWB powder was incubated at 37°C for 10 h in the 20 mM Tris–HCl buffer (pH 8.0) with BDS4 (20 U) or combined with commercial xylanase (100 U). The released FA was examined by HPLC.
Phthalate biodegrading experiment
The ability of phthalate degradation by the purified BDS4 was determined using diethyl phthalate, dimethyl phthalate and dibutyl phthalate as substrates. 20 mM Tris–HCl buffer (pH 8) was mixed with a single phthalate at a concentration of 1 mg/mL, then the 40 μg/mL purified BDS4 was added into the 1 mL reaction mixture. The control was carried out as the above procedure without enzyme. All the reactions were accomplished in dark at 37°C for 12 h. The samples were dried by N2 and redissolved in methanol, and filtrated by 0.22 μm organic membrane filters. The products were detected and analyzed by a 7890A gas chromatography-5975C mass spectrometry (GC–MS, Agilent, USA) coupled with a Hp-5MS column. The GC conditions were as follow: initially, the oven temperature was 60°C for 1 min; then ramped to 180°C at a rate of 10°C per min for 10 min; further ramped to 220°C at a rate of 15°C per min for 5 min. The injection volume was 1 μl, the split mode was 20:1, and 7 min was set as the solvent delay time.
Results
Metagenomic library construction and FAE screening
The metagenomic library was constructed with 1.2 × 106 clones approximately with an average DNA insert size of 40 kb, and was predicted to contain about 4.5 Gbp genomic DNA. One positive fosmid clone with the clear halo was isolated which showed strong FAE activity by HPLC analysis (Supplementary Fig. 1). To identify an FAE-encoding ORF, the pUC118 subclone library was generated from Sau3A I-digested fosmid clone. The pUC118 subclone library consisted of around 2000 clones. One positive subclone was obtained after rescreening and further analyzed by sequencing with vector-specific primers.
Sequence analyses of bds4
The insert of 4185 bp-long DNA fragment in subclone harboured 24 possible functional ORFs as revealed by ORF Finder program, in which one ORF was identified as a putative FAE and named bds4. The homologous search of bds4 was retrieved from Genbank database through Position-Specific Iterated BLAST (PSI-BLAST). The bds4 exhibited the top highest identity with Carboxylesterase NlhH (P9WK86, 35%) from Mycobacterium tuberculosis CDC1551, AB hydrolase (Q9US38, 34%) from Schizosaccharomyces pombe and Arylesterase (B5BLW5, 34%) from Sulfolobus solfataricus. The multiple alignment analysis was performed on the online software Clustal Omega program (Fig. 1). The results suggested that the deduced amino acid sequence of bds4 contained a G-D-S-A-G sequence (position 156 to 160) corresponding well to the conserved G-X-S-X-G motif in many other FAEs, such as G-L-S-M-G and G-H–S-Q-G from rumen (Cheng et al. 2012a; Wong et al. 2013), G-C-S-T-G from A. oryzae (Koseki et al. 2009). It can be deduced that the catalytic triad might consist of a nucleophile serine (S, 158), aspartate (D, 256) and highly conserved histidine (H, 286) residues (Prates et al. 2001).

Multiple sequence alignment of bds4 and similar esterase on the amino acid level. The aligned sequences were derived by the PSI-BLAST program: AB hydrolase (Q9HDX3, Q9US38) from Schizosaccharomyces pombe; Arylesterase (B5BLW5) from Sulfolobus solfataricus; Carboxylesterase NlhH (P9WK86) from Mycobacterium tuberculosis CDC1551. The conserved G-X-S-X-G motif was shaded in gray rectangle and the putative catalytic triad (S158, D256 and H286) was indicated by triangles
The predicted three-dimensional structure of BDS4 was built by COACH online tool using 4RE5 protein sequence as a template which generated automatically with the highest score (Fig. 2). The overall topology of BDS4 protein contains a typical α/β hydrolase fold and a Ser-Asp-His catalytic triad, in which two domains can be identified, one is a catalytic domain that consists of an eight-stranded β-sheet with connecting α-helices, the other is a cap domain developed by α-helices which are located at the upper of the central β-sheets and it can be used as the entrance for the substrate binding (Suzuki et al. 2014).

The predicted three-dimensional structure of BDS4 by COACH sever. BDS4 had a typical α/β hydrolase fold and a Ser-Asp-His catalytic triad (S158, D256 and H286), which consisted of the cap domain (above the line) and catalytic domain (below the line). The α-helices, β-strands and random coils were shown in red, blue, and gray respectively
Phylogenic analysis and classification of bds4
According to the amino acid sequence, substrates specificity for aromatic substrates and biochemical properties, FAEs have been classified into four types (A–D) (Crepin et al. 2004). A phylogenetic tree was constructed by the amino acid sequences of bds4 and other known FAEs (Crepin et al. 2004; Dilokpimol et al. 2016) for further understanding the evolutionary relationship (Fig. 3). It can be observed that the FAE sequences were separated into four major phylogenetic clades and BDS4 belonged to C clade, suggesting that BDS4 was a new member of type-C FAEs, resembling the other identified type-C FAEs with the activity toward model substrates (MFA, MCA, MSA and MpCA).

Phylogenetic tree of BDS4 and other known FAEs using the neighbor-joining method (MEGA 6.0) based on 1000 replicates. Four phylogenetic clades respectively represent functional sub-classes of FAEs (type A, B, C, D) according to the classification (Crepin et al. 2004)
Cloning, expression and purification of BDS4
The bds4 gene was cloned into pET-28a (+) and then transformed into E. coli BL21(DE3) for expression. The recombinant enzyme BDS4 was purified by Ni–NTA affinity resin and its molecular weight was determined by SDS-PAGE (Fig. 4). The BDS4 containing polyhistidine tags showed a molecular weight of 38.8 kDa.

The SDS-PAGE analysis of BDS4. Lane M, protein marker; lane 1, crude enzymes; land 2, purified BDS4
Biochemical characterization and substrate specificity
The optimum activity of BDS4 was measured over the pH range of 3.0–11.0 and the temperature range of 4–75°C with MFA as the substrate. The activity of BDS4 increased as pH increased from 3.0 to 8.0, the highest activities was achieved at pH 8.0 and declined when the pH was over 8.0 (Fig. 5a). The pH stability experiment revealed BDS4 still maintained more than 60% residual activity in the pH 7.0–8.5, indicating that BDS4 was more stable under weakly alkaline conditions (Fig. 5b). The activity of BDS4 exhibited the almost same profile as the temperature increased from 4 to 37°C with incubation and without incubation in the corresponding temperature, and displayed the maximum activation at 37°C (Fig. 5c). The thermostability experiment showed that the activity of BDS4 decreased with the increasing of the reaction time, and the higher the temperature, the greater the loss of activity (Fig. 5d).The effects of metal ions and chemical reagents on BDS4 were showed in Table 1. For further characterization of BDS4, enzymatic kinetic parameters were examined with different substrates as shown in Table 2. BDS4 were active toward MFA, EFA, MpCA, MCA, MV and MSA except for MG and CGA. It exhibited the strongest activity to MFA with a value of 57.05 U/mg followed by EFA and MpCA. The lowest Km (0.11 mM) for MV suggested that BDS4 have the best affinity toward MV in the tested substrates, while the maximal kcat/Km (79.04 mM−1s−1) and Vmax (0.019 mM s−1) for MFA indicated that BDS4 displayed the highest catalytic efficiency and reactive velocity toward MFA.

The effects of pH and temperature on BDS4. a The effect of pH on activity. b The effect of pH on stability. c The effect of temperature on activity. The enzymatic activity was assayed in Tris–HCl buffers (pH 8.0) with or without pre-incubation for 1 h in different temperatures. d The effect of temperature on thermal stability assay. Each point presented mean ± standard deviation (n = 3)
Site-directed mutagenesis
To confirm the catalytic residues in bds4, three recombinant mutant plasmids were constructed, in which Ser158, Asp256, His286 were replaced with Cys, Asn, Lys respectively by site-directed mutagenesis (Supplementary Fig. 2). The S158C, H286 K and D256 N mutants could completely abolished the catalytic activity, implying that Ser158, Asp256 and His286 were very important to the activity of BDS4 and they might constitute its catalytic triad.
Synergistic effects of BDS4 with xylanase on DSWB
The possible synergy between BDS4 and xylanase was studied using the substrate DSWB as shown in Fig. 6. BDS4 released a small amount FA from DSWB without the presence of the xylanase, and also a few FA was detected with xylanase alone. But, the amount of released FA increased significantly and reached as high as 28.49 μg/mL when BDS4 and xylanase mixed, suggesting a synergistic effect occurred between BDS4 and xylanase.

Release of FA from de-starched wheat bran by BDS4 and xylanase. The FA was detected by HPLC. Error bars represent standard deviation (n = 3)
Phthalate-biodegrading analysis
The reaction products of diethyl phthalate, dimethyl phthalate and dibutyl phthalate were analyzed by GC–MS. According to authentic standards, degradation products of diethyl phthalate, dimethyl phthalate and dibutyl phthalate were identified as phthalic acid with an obvious peak, indicating that these phthalates with shorter side chains were finally transformed into phthalic acid and the possible enzymatic degradation pathway of BDS4 was proposed as shown in Fig. 7.

The proposed degradation pathways for phthalates by BDS4 using GC–MS method. The main detected degradation product of dimethyl phthalate, diethyl phthalate and dibutyl phthalate was phthalic acid in all cases. a dimethyl phthalate, b diethyl phthalate, c dibutyl phthalate
Discussion
Metagenomics strategy has been effectively applied in the discovery of novel FAE biocatalysts from the environment (Wong et al. 2013; Li et al. 2018). The new FAE gene bds4 was identified from a metagenomic library. This is the first report of the molecular cloning, expression and characterization of bds4 gene. BDS4 was described as a novel type-C feruloyl esterase according to phylogenetic analysis and substrate specificity experiments. Also, the current research provides more information for the function of BDS4.
The purified BDS4 exhibited specificity against the four methyl esters of hydroxycinnamic acids (MFA, MpCA, MCA and MSA) and other substrates (EFA, MV). BDS4 showed strong activity (57.05 U/mg) toward MFA which was much higher than most of FAEs identified, such as EstF27 (7.3 U/mg) and FAE-Xuan (40 U/mg) from soil samples (Shu et al. 2011; Li et al. 2018), lhFAE (10.3 U/mg) from Lactarius hatsudake (Wang et al. 2016), ScFAE1 (12 U/mg) and ScFAE2 (14 U/mg) from Sorangium cellulosum (Wu et al. 2012). The optimum enzymatic activity for BDS4 was under pH 8.0 and 37◦C. Most FAEs analyzed to date show optimal activity under acidic conditions (Oliveira et al. 2019). However, for applications such as increasing the yield of pulp from alkaline pulping in paper-making processes it is important to have enzymes working in alkaline conditions (Record et al. 2003; Moukouli et al. 2008). This makes BDS4 potentially useful in industry. The activity of BDS4 was strongly inhibited by Co2+ and Fe2+, and abolished in the presence of Cu2+, Zn2+and SDS. The impact of Cu2+, Zn2+ on BDS4 activity was consistent with properties of reported FAEs from the EstF27 (Benoit et al. 2008), Tan410 (Yao et al. 2013), FAE-Xuan (Li et al. 2018). SDS suppressed the activity of BDS4, which was similar to Xyln-SH1 (Cheng et al. 2012b). The presence of EDTA-2Na reduced the enzymatic activity of BDS4, however, the activity of FAE-SH1 could slightly improve by EDTA (Cheng et al. 2012a). In future, some attempts on BDS4 can be used to improve enzymatic activity such as site directed mutagenesis and directed evolution (Ohlhoff et al. 2015; Pereira et al. 2017).
In experiments with DSWB, only low amounts of FA were released when FAE or xylanase were present alone, but more FA was released when both enzymes were present. This indicates that the release of FA can be mostly attributed to the synergistic interaction between BDS4 and xylanase. It suggested that BDS4 required low molecular-weight feruloylated substrates; xylanase can cleave glycosidic linkages to produce short-chain xylooligosaccharides on which FAE can act on (Dilokpimol et al. 2016). It has been demonstrated that FAE can significantly enhance the amount of FA released in the presence of cell wall-degrading enzymes (Yu et al. 2011). Similar experimental results have been found that xylanase can enhance the hydrolytic action of FAEs on plant cell wall materials as reported EstF27 (Shu et al. 2011) and FoFaeC-12213 (Moukouli et al. 2008). It is thus common to find synergism between FAEs and xylanase.
In addition, BDS4 was the first reported FAE with phthalate-biodegrading activity. The phthalates are widely used as the plasticizer, their global consumption grow rapidly and they can easily enter into the environment through the process of manufacturing, using and disposing plastics (Lu et al. 2009; Chen et al. 2015). The safety of phthalates has aroused much public attention because they are suspected of playing an estrogen-like effect in humans and animals, which can interfere with endocrine and cause carcinogenic effects (MacOn and Fenton 2013). Microbial activity is responsible for phthalate degradation in the environment (Benjamin et al. 2015). BDS4 can degrade diethyl phthalate, dimethyl phthalate and dibutyl phthalate into phthalic acid which suggested BDS4 may have potential application value in environmental protection.
In summary, a FAE gene was successfully identified from the soil metagenomic library, which illustrated the advantages of metagenomic approach in mining novel enzyme genes and discovering new biocatalysts with industrial and biotechnological applications. The heterologously produced BDS4 possessed the optimal activity in neutral and weakly alkaline environment (pH8.0, 37°C), that showed its potential application in industrial processes. BDS4 was insensitive to some metal ions and chemicals such as Mg2+, Fe3+, isopropanol, acetone, dimethyl sulfoxide, Triton-100, dimethyl formamide, however it was inhibited significantly by Cu2+, Zn2+ and SDS. Moreover, BDS4 acted synergistically with xylanase to release FA from DSWB. Notably, BDS4 can degrade diethyl phthalate, dimethyl phthalate and dibutyl phthalate into the products phthalic acid. Taken together, these properties make BDS4 as an interesting candidate for biomass degradation and environmental protection.
References
Altschul SF, Wootton JC, Gertz EM et al (2005) Protein database searches using compositionally adjusted substitution matrices. FEBS J 272(20):5101–5109
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59:143–169
Benjamin S, Pradeep S, Sarath Josh M et al (2015) A monograph on the remediation of hazardous phthalates. J Hazard Mater 298:58–72
Benoit I, Danchin EGJ, Bleichrodt RJ, De Vries RP (2008) Biotechnological applications and potential of fungal feruloyl esterases based on prevalence, classification and biochemical diversity. Biotech Lett 30:387–396. https://doi.org/10.1007/s10529-007-9564-6
Brady SF (2007) Construction of soil environmental DNA cosmid libraries and screening for clones that produce biologically active small molecules. Nat Protoc 2:1297–1305
Chen X, Zhang X, Yang Y et al (2015) Biodegradation of an endocrine-disrupting chemical di-n-butyl phthalate by newly isolated Camelimonas sp. and enzymatic properties of its hydrolase. Biodegradation 26:171–182
Cheng F, Sheng J, Cai T et al (2012a) A protease-insensitive feruloyl esterase from China Holstein cow rumen metagenomic library: expression, characterization, and utilization in ferulic acid release from wheat straw. J Agric Food Chem 60:2546–2553. https://doi.org/10.1021/jf204556u
Cheng F, Sheng J, Dong R et al (2012b) Novel xylanase from a holstein cattle rumen metagenomic library and its application in xylooligosaccharide and ferulic Acid production from wheat straw. J Agric Food Chem 60:12516–12524
Crepin VF, Faulds CB, Connerton IF (2004) Functional classification of the microbial feruloyl esterases. Appl Microbiol Biotechnol 63:647–652. https://doi.org/10.1007/s00253-003-1476-3
Dilokpimol A, Mäkelä MR, Aguilar-Pontes MV et al (2016) Diversity of fungal feruloyl esterases: updated phylogenetic classification, properties, and industrial applications. Biotechnol Biofuels 9:1–18. https://doi.org/10.1186/s13068-016-0651-6
Dilokpimol A, Mäkelä MR, Varriale S et al (2018) Fungal feruloyl esterases: functional validation of genome mining based enzyme discovery including uncharacterized subfamilies. New Biotechnol 41:9–14. https://doi.org/10.1016/j.nbt.2017.11.004
Gopalan N, Rodríguez-Duran LV, Saucedo-Castaneda G, Nampoothiri KM (2015) Review on technological and scientific aspects of feruloyl esterases: a versatile enzyme for biorefining of biomass. Biores Technol 193:534–544. https://doi.org/10.1016/j.biortech.2015.06.117
Haase-Aschoff P, Linke D, Nimtz M et al (2013) An enzyme from Auricularia auricula-judae combining both benzoyl and cinnamoyl esterase activity. Process Biochem 48:1872–1878
Hancock JM, Bishop MJ (2004) Dictionary of bioinformatics and computational biology. ORF Finder (Open Reading Frame Finder). Wiley, New Jersey
Handelsman J (2004) Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 68:669–685
Koseki T, Hori A, Seki S et al (2009) Characterization of two distinct feruloyl esterases, AoFaeB and AoFaeC, from Aspergillus oryzae. Appl Microbiol Biotechnol 83:689–696. https://doi.org/10.1007/s00253-009-1913-z
Li W, Cowley A, Uludag M et al (2015) The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res 43:W580–W584. https://doi.org/10.1093/nar/gkv279
Li X, Guo J, Hu Y et al (2018) Identification of a novel feruloyl esterase by functional screening of a soil metagenomic library. Appl Biochem Biotechnol 187(1):1–14
Lu Y, Tang F, Wang Y et al (2009) Biodegradation of dimethyl phthalate, diethyl phthalate and di-n-butyl phthalate by Rhodococcus sp. L4 isolated from activated sludge. J Hazard Mater 168:938–943
MacOn MB, Fenton SE (2013) Endocrine disruptors and the breast: early life effects and later life disease. J Mammary Gland Biol Neoplasia 18(1):43–61
Moukouli M, Topakas E, Christakopoulos P (2008) Cloning, characterization and functional expression of an alkalitolerant type C feruloyl esterase from Fusarium oxysporum. Appl Microbiol Biotechnol 79:245–254. https://doi.org/10.1007/s00253-008-1432-3
Ohlhoff CW, Kirby BM, Van Zyl L et al (2015) An unusual feruloyl esterase belonging to family VIII esterases and displaying a broad substrate range. J Mol Catal B Enzym 118:79–88
Oliveira DM, Mota TR, Oliva B et al (2019) Feruloyl esterases: biocatalysts to overcome biomass recalcitrance and for the production of bioactive compounds. Biores Technol 278:408–423. https://doi.org/10.1016/j.biortech.2019.01.064
Pereira MR, Maester TC, Mercaldi GF et al (2017) From a metagenomic source to a high-resolution structure of a novel alkaline esterase. Appl Microbiol Biotechnol 101:4935–4949. https://doi.org/10.1007/s00253-017-8226-4
Prates JA, Tarbouriech N, Charnock SJ et al (2001) The structure of the feruloyl esterase module of xylanase 10B from Clostridium thermocellum provides insights into substrate recognition. Structure 9(12):1183–1190
Record E, Asther M, Sigoillot C et al (2003) Overproduction of the Aspergillus niger feruloyl esterase for pulp bleaching application. Appl Microbiol Biotechnol 62(4):349–355
Reyes-Duarte D, Ferrer M, García-Arellano H (2012) Functional-based screening methods for lipases, esterases, and phospholipases in metagenomic libraries. Methods Mol Biol 861:101
Schloss PD, Handelsman J (2005) Metagenomics for studying unculturable microorganisms: cutting the Gordian knot. Genome Biol 6:1–4
Shahidi F, Chandrasekara A (2010) Hydroxycinnamates and their in vitro and in vivo antioxidant activities. Phytochem Rev 9:147–170. https://doi.org/10.1007/s11101-009-9142-8
Shu LS, Gang L, Xiao PH, Yu HL (2011) Molecular cloning, overexpression and characterization of a novel feruloyl esterase from a soil metagenomic library. J Mol Microbiol Biotechnol 20:196–203
Suzuki K, Hori A, Kawamoto K et al (2014) Crystal structure of a feruloyl esterase belonging to the tannase family: a disulfide bond near a catalytic triad. Proteins: structure. Funct Bioinform 82:2857–2867. https://doi.org/10.1002/prot.24649
Tamura K, Stecher G, Peterson D et al (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729
Torsvik V, Goksøyr J, Daae FL (1990) High diversity in DNA of soil bacteria. Appl Environ Microbiol 56:782–787
Torsvik V, Daae FL, Sandaa RA, Ovreås L (1998) Novel techniques for analysing microbial diversity in natural and perturbed environments. J Biotechnol 64:53–62
Wang L, Li Z, Zhu M et al (2016) An acidic feruloyl esterase from the mushroom Lactarius hatsudake: a potential animal feed supplement. Int J Biol Macromol 93:290–295
Williamson G, Kroon PA, Faulds CB (1998) Hairy plant polysaccharides: a close shave with microbial esterases. Microbiology 144:2011–2023
Wong DWS, Chan VJ, Liao H, Zidwick MJ (2013) Cloning of a novel feruloyl esterase gene from rumen microbial metagenome and enzyme characterization in synergism with endoxylanases. J Ind Microbiol Biotechnol 40:287–295. https://doi.org/10.1007/s10295-013-1234-1
Wu M, Abokitse K, Grosse S et al (2012) New feruloyl esterases to access phenolic acids from grass biomass. Appl Biochem Biotechnol 168(1):129–143
Yang J, Roy A, Zhang Y (2013a) BioLiP: a semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res 41:1096–1103
Yang J, Roy A, Zhang Y (2013b) Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 29:2588–2595
Yao J, Chen QL, Shen AX et al (2013) A novel feruloyl esterase from a soil metagenomic library with tannase activity. J Mol Catal B Enzym 95:55–61. https://doi.org/10.1016/j.molcatb.2013.05.026
Yu P, McKinnon JJ, Christensen DA (2011) Hydroxycinnamic acids and ferulic acid esterase in relation to biodegradation of complex plant cell walls. Can J Anim Sci. 85(3):255–267
Acknowledgements
This research was supported by the Fundamental Research Funds for the Central Universities (KYYJ201708) and Qing Lan Project of Jiangsu Province.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Accession Number
The bds4 gene was deposited in the GenBank database with the accession no. MH445495.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wu, S., Nan, F., Jiang, J. et al. Molecular cloning, expression and characterization of a novel feruloyl esterase from a soil metagenomic library with phthalate-degrading activity. Biotechnol Lett 41, 995–1006 (2019). https://doi.org/10.1007/s10529-019-02693-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-019-02693-3