
Abstract
Breast cancer risk has both heritable and environment/lifestyle components. The heritable component is a small contribution (5–27 %), leaving the majority of risk to environment (e.g., applied chemicals, food residues, occupational hazards, pharmaceuticals, stress) and lifestyle (e.g., physical activity, cosmetics, water source, alcohol, smoking). However, these factors are not well-defined, primarily due to the enormous number of factors to be considered. In both humans and rodent models, environmental factors that act as endocrine disrupting compounds (EDCs) have been shown to disrupt normal mammary development and lead to adverse lifelong consequences, especially when exposures occur during early life. EDCs can act directly or indirectly on mammary tissue to increase sensitivity to chemical carcinogens or enhance development of hyperplasia, beaded ducts, or tumors. Protective effects have also been reported. The mechanisms for these changes are not well understood. Environmental agents may also act as carcinogens in adult rodent models, directly causing or promoting tumor development, typically in more than one organ. Many of the environmental agents that act as EDCs and are known to affect the breast are discussed. Understanding the mechanism(s) of action for these compounds will be critical to prevent their effects on the breast in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the second most common type of cancer among women in the United States (second to skin cancers) and the second leading cause of death due to cancer, after lung cancer [1]. In fact, the most recent US statistics estimate that 1 in 8 women will be diagnosed with breast cancer in their lifetime [2], a number that has not declined even following a dramatic decrease in the use of postmenopausal hormone treatments shown to be associated with increased breast cancer risk [2]. Breast cancer also affects men, but to a much lesser degree, with only 1 % of the US male population annually diagnosed [3]. As with other cancers the formation of breast tumors has a heritable link; between 5 to 27 % of all breast cancers are attributed to factors such as specific gene mutations, certain traits (i.e., dense breasts, fibrocystic disease), and metabolic issues [4]. The environmental and lifestyle factors that contribute the other 70–95 % of breast cancer risk are largely unknown. Some general risk factors for breast cancer include: sex, age, parity, age at menarche, and age at menopause. The importance of environmental influences and exogenous exposures on some of these factors (parity, age at menarche) have been demonstrated [5, 6], but we have only begun to understand how exogenous chemicals that disrupt the function of the endocrine system may increase susceptibility to breast cancer and other diseases.
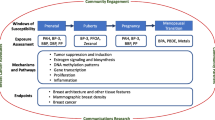
The endocrine system is composed of multiple glandular organs that secrete hormones directly into the bloodstream. Organs of the endocrine system have important functions such as developing and maintaining normal tissue functions, bone health, metabolism, reproduction, lactation, and stress response, among others. For the purposes of this review, we will focus primarily on the effects of the endocrine system on reproduction and metabolism. Hormones secreted from the endocrine system affect nearly every organ and system within the body. Endocrine disrupting chemicals (EDCs or endocrine disruptors), as their name suggests, are chemicals that alter normal actions of the endocrine system. EDCs are defined by the US Environmental Protection Agency as “exogenous agents that interfere with the production, release, transport, metabolism, binding, action, or elimination of the natural hormones in the body responsible for the maintenance of homeostasis and the regulation of developmental processes” [7]. Table 1 lists ways in which chemicals can disrupt the endocrine system.
Normal growth of the mammary gland involves endocrine signaling from the hypothalamic-pituitary-gonadal axis. However, autocrine and paracrine hormones and growth factors also play critical roles in development and regulation of the mammary gland. Some of the many endocrine hormones and growth factors known to modulate mammary gland development include: growth hormone, prolactin, oxytocin, epidermal growth factor, insulin, insulin-like growth factors, adrenal corticosteroids, transforming growth factors, thyroxine, estrogen, progesterone, activin, and inhibin [8]. Figure 1 illustrates the endocrine-mediated influences on mammary gland growth and function. This figure becomes complicated by the numerous possible endocrine-mediated mechanisms/pathways that may be altered by the >84,000 chemicals on the Toxic Substance Control Act Chemical Inventory list [9]. The effects that a small portion of these chemicals have on mammary gland development are known, but the link between early life health effects and later life disease, such as the inability to lactate or breast cancer susceptibility, are less clear.

Endocrine tissues and their corresponding secreted hormone that influence mammary gland growth or function. GH= Growth Hormone; EGF= Epidermal Growth Factor. (Modified and republished with permission of Annual Reviews, Inc., from Ref [8]; permission conveyed through Copyright Clearance Center, Inc.)
Chemical Carcinogenesis and Endocrine Disruptors
Although there is a lack of consensus regarding theories of carcinogenesis, tumor maturation of aberrant cells is thought by some to be regulated by three main events: initiation, promotion, and progression [10]. Carcinogens, or chemicals that cause tumor growth, can affect any one or more of these events and may involve dynamics of the genome related to increased expression of oncogenes, decreased expression of tumor suppressors, disruption of signal transduction, or changes in expression of cell cycle or apoptosis regulator genes. Initiation of tumors may result from chemical-induced DNA damage or inherited mutations of the DNA. Un-checked cellular DNA-damage becomes replicated and leads to mutations within the genome. Formation of radicals from oxidative damage can also lead to DNA-damage. Other heritable, genomic origins of tumors may arise from epigenetic changes. Epigenetic changes do not involve direct changes of the genome, rather it affects how the genome is transcribed or translated. This can involve DNA methylation, histone modification, or RNA mediated modifications. Promotion of tumorigenesis is often described as reversible alteration in gene expression and tumor progression as irreversible alteration in gene expression. Tumor growth is promoted through clonal expansion of mutated cells, concomitant with an increased cellular proliferation rate and decreased apoptotic rate. There is also merit in the idea that cancer originates from aberrant tissue organization and not simply aberrant cellular growth [11]. This theory specifically refers to carcinogenesis as a problem similar to abnormal development—which could explain how fetal exposure to a non-mutagenic compound predisposes the mammary gland to carcinogenesis, supporting the theory of fetal origins of adult disease. Under this theory the unfavorable alterations are reversible under certain conditions.
Some chemicals have been tested for their carcinogenic potential. This is primarily done by chemical manufacturers and government-funded research, such as the 2 year cancer bioassays conducted in rodents by the National Toxicology Program (NTP)—chemicals are nominated by the public, other agencies, or in house for testing [12]- or through other test guidelines, such as those developed and used within the Organisation for Economic Co-operation and Development (http://www.oecd.org/env/chemicalsafetyandbiosafety/testingofchemicals/oecdguidelinesforthetestingofchemicals.htm) and the European Union’s 2007 (Registration, Evaluation, Authorisation and Restriction of Chemical Substances) REACH regulation. The NTP produces the Report on Carcinogens (RoC) that documents chemical data which support cancer causation in animals or humans. Approximately 2,500 chemicals have been evaluated and chronic rodent cancer studies (adult only exposure to test compound for 2 year) have been conducted on just over 600 chemicals. About 250 of those have shown carcinogenic potential in female rodents, and about 60 of those have shown evidence as mammary gland carcinogens. However, many of those mammary gland carcinogens caused cancer in other sites, also. In 2010, the NTP re-instituted experiments designed to expose the fetus and developing offspring, in addition to the adult, to the test compounds of interest. There are six substances in the RoC that cause or may cause breast cancer in humans. These include the drug diethylstilbestrol (DES), steroid hormones used for menopausal therapy, some forms of radiation, alcohol, tobacco smoke (see Reynolds et al., this issue), and the sterilizing agent, ethylene oxide.
Although DES is a classical carcinogen [13, 14], it is also an EDC. It is a synthetic estrogen that interferes in estrogen receptor-mediated signaling to disrupt endocrine-mediated pathways [14] and is covered in detail elsewhere (see Hilakivi-Clarke et. al, this issue). There are few examples of EDCs that act as chemical carcinogens; however EDCs have the potential to influence tumorigenesis by altering lifetime susceptibility to a second ‘hit’ or exposure [15]. In recent years, examples of altered mammary histoarchitecture [16] and spontaneous (no second ‘hit’) ductal carcinoma in situ in mice and rats following prenatal Bisphenol A exposure has been demonstrated (reviewed in [17]). Importantly, these discoveries suggest that a second hit may or may not be needed for some EDCs to influence carcinogenesis. In women, it is estimated that 40 % of all cancers are hormonally regulated [8] and thus there are many potential modes or mechanisms by which EDC exposure could modify cancer risk. Many factors that influence breast cancer are endocrine-regulated processes (e.g., age at menarche, age at 1st pregnancy, and age at menopause) and may be modified by EDCs. EDCs that do not affect DNA are proposed to influence tumorigenesis in a variety of ways, including impaired or altered 1) epithelial growth rate, 2) stromal composition of the gland, 3) immune response, 4) response to endogenous hormones, 5) terminal end bud presence, or 6) cell-cell communication, to name a few. Yet, it is important to note that exposure to EDCs may influence tumorigenesis through both DNA-mediated and non DNA-mediated mechanisms.
Mammary Gland Development and Correlation to Cancer
In the US, more money is spent towards the detection and treatment of breast cancer (~$16.5 billion in 2010 dollars) than any other type of cancer [18]. One of the reasons is that breast cancer is not a single disease, but a heterogeneous and phenotypically diverse set of diseases [19]. Several molecular subtypes exist [20–22], and each respond to treatment differently. Numerous studies have been conducted in an effort to better understand the etiology of the several types of breast cancer. Not only is breast cancer complex, but so is normal breast morphology. Breast tissue consists of multiple cell types, which must remain in close communication. Normal mammary gland growth involves intricate crosstalk between the epithelium and the surrounding stroma, to balance proper proliferation/apoptosis and remodel the gland at the different stages of life.
Rapid mammary gland development occurs in three distinctive life stages: fetal, peri-pubertal, and pregnancy [23]. In girls, the fetal mammary bud begins to form late in the first trimester of pregnancy. During the last few weeks of pregnancy, the nipple and the primary epithelial ducts form and ducts branch outward into the stroma. Relatively little epithelial growth is observed until around the time of puberty when the gland growth is influenced by the release of pituitary and ovarian hormones. During thelarche, one of the earliest signs of puberty, female breast tissue enlarges and grows outward, making it noticeable. At the same time the mammary fat pad enlarges in size, and the rapidly extending epithelium form bulbous or club-like structures at the duct ends, termed terminal ductal lobular units (TDLU). The TDLU are structurally similar to terminal end buds (TEB) in rats and mice, present during the same life stage. These structures are undifferentiated and highly proliferative, and as such they are sensitive to the effects of carcinogens and other chemicals. The breast reaches a static stage some time after first menstruation, changing slightly with each additional menstrual cycle. Figure 2 demonstrates the progressive growth of the mammary epithelium within the fat pad of the mouse until it reaches the adult resting state. Thereafter, the gland remains in a fairly static stage throughout life, until a pregnancy occurs. At this time, morphological maturation is achieved and the gland continues to branch and fill with lobulo-alveoli to support the production and release of milk. EDC-induced disruption of normal development at any and all of these stages can cause permanent developmental abnormalities, impaired lactation, and influence risk for the development of breast cancer. It is important to note that male breast development also occurs in utero, and is halted in boys by the androgen surge that occurs just before birth. Anti-androgenic chemical exposures in rodent models have caused reversal of that gender-specific response [24–26]. It is important to note that EDC-induced changes affect risk for hormonally-responsive mammary tumors, such as the common ER+/PR+ subtype.

Typical trajectory of mammary gland development in the CD-1 mouse from neonate to young adult. PND = Postnatal Day. Carmine-stained whole mount mammary gland samples were prepared using the NTP Animal Studies Protocol; Section XIII, Appendix 6, Section E. http://ntp.niehs.nih.gov/ntp/Test_Info/FinalNTP_ReproSpecsMay2011_508.pdf
The use of rodents in laboratory studies has advanced our knowledge about the mechanisms controlling normal mammary gland growth, as well as fill knowledge gaps concerning spontaneous and chemical-induced carcinogenesis. The 2 year bioassay, involving adult exposure to test chemicals, provides evidence of spontaneous carcinogenesis (in the entire body), whereas the use of a chemical carcinogen such as dimethylbenz-a-anthracene (DMBA) following an early life chemical exposure tests the specific susceptibility of the mammary gland to form tumors. It is important to emphasize that dose levels and timing of exposure to EDCs may affect the severity, or lack thereof, of an effect on mammary gland growth and consequently breast cancer risk [23]. EDC exposure can accelerate rodent mammary gland growth leading to unbalanced or unchecked proliferation of the gland. For example, prenatal exposure to DES accelerates mammary gland growth with increased lobular proliferation resulting in increased hyperplastic nests of epithelium and tumor multiplicity, as well as decreased tumor latency [27, 28]. Compared to controls, a virgin rodent with accelerated development of the mammary gland will have more TEBs compared to terminal ducts at weaning (3 weeks of age), and rat weanlings may exhibit extensive alveolar budding. As development progresses, exposed rodent mammary glands will exhibit a greater number of lobules, possibly with greater complexity, than unexposed animals. Yet, EDCs that cause delays in mammary growth can also increase sensitivity to carcinogens and increase risk for breast cancer. Delayed development of rodent mammary glands often manifests as decreased longitudinal growth of the epithelium and fewer TEBs compared to controls at the time of weaning. These changes may also be present in adolescent animals; fewer duct ends, decreased branching density and smaller gland outgrowth. At puberty, delayed glands may have more TEB than controls due to the slower pace of development. Although somewhat counter-intuitive, mammary gland growth delays can increase cancer risk by prolonging the presence of carcinogen-sensitive structures, such as TDLU or TEBs. Figure 3 demonstrates examples of accelerated and delayed mammary gland development, compared to vehicle-treated controls, in the rat.

Examples of accelerated development (right) and delayed development (left) compared to a representative vehicle-treated control mammary gland at the time of weaning in the Long Evans rat. Carmine stained whole mount mammary gland. Specific examples of EDC effects in Table 2
Although exposure to EDCs can increase breast cancer risk, some EDCs have been reported to have beneficial effects and protect against the development of breast cancer (see phytoestrogens). Exposure to EDCs can stimulate growth so that the gland is mature with a high ratio of fully differentiated structures compared to immature or un-differentiated structures or can reduce the proliferation and apoptosis ratio within the epithelium. As stated before, the beneficial effects of EDCs are highly dependent upon exposure levels and timing of exposure. Inhibition of mammary gland development early in life can be so severe that the mammary epithelial tree does not form past a nascent structure, leaving little proliferative potential [29]. In addition to reduced risk for mammary tumor formation, these delays result in glands that are less responsive to ovarian and pituitary hormones, consequently reducing the functionality of the gland. Severe inhibition of mammary development has only been observed in rodents at high EDC exposures [30, 31]. For humans, these elevated levels of exposure may not be reached except in rare cases of high occupational or non-occupational accidental exposures (i.e. pollution). Yet, in wildlife and domesticated mammals, excessive exposures that result in severely stunted glands diminish sustenance to developing offspring, resulting in reduced postnatal survival because of minimal/no milk production [32].
Although the ductal or lobular epithelia are often the sites of tumor production, mammary stroma growth patterns can also influence breast cancer risk. The communication of the multiple cell types in the mammary gland, i.e. stromal-epithelial interaction, is critical for normal development throughout life [8]. Studies have linked increased stromal density in the breast to later development of epithelial based mammary tumors [33]. When mammary tumor tissues were separated into epithelial and stromal compartments, recombination of normal epithelium with tumor stroma resulted in the growth of epithelial tumors while recombination of tumor epithelium with normal stroma resulted in the growth of normal tissue, indicating that stromal tissues have strong influences on overall gland growth and tumor status [11]. Deleterious modifications to stromal tissues can occur along with accelerated or delayed overall mammary gland growth and is another way EDC exposure can influence the development of mammary tumors [16, 34, 35]. In fact, thickening of the stromal compartment surrounding the epithelium has been found in the case of prenatal Bisphenol A exposure in rats [34] and following prenatal exposure to perfluorooctanoic acid in mice [36]. As expected, exposure to EDCs that alter mammary development may also cause an array of detrimental health effects in other organs. Table 2 demonstrates a multitude of reproductive/hormonal effects from a handful of EDCs that are known to affect the mammary gland.
Endocrine Disruptors and Breast Effects
Timing of exposure to EDCs in rodents and women determines how the mammary gland may react to the exposure. This finding, and our current understanding of the various subtypes of breast cancer, may provide insight to the way researchers address breast cancer prevention. Although we review several EDCs here (and summarize their impact on breast cancer in Table 3), many other examples are given in a review of EDCs by Rudel et al. 2011 [17].
Polycyclic Aromatic Hydrocarbons
Polycyclic aromatic hydrocarbons (PAHs) are a class of chemicals formed during the combustion of many substances including coal, wood, gasoline, and food. Additionally, PAHs are found in cigarette smoke (see Reynolds et al., this issue). These are highly stable organic compounds that often have genotoxic and mutagenic properties. Due to these properties, PAHs are considered carcinogenic, as they cause mutations which initiate and/or promote the growth of tumors.
PAHs also have endocrine-disrupting properties and affect hormone signaling by binding to estrogen receptors [37]. PAH-induced tumors regress in ovariectomized rats [38] indicating these effects are hormonally regulated. One of the most notable PAHs shown to cause breast cancer in animals is DMBA, which has been introduced as a mammary-specific carcinogen. In laboratory studies, DMBA is often administered to rodents to determine the influence of other chemicals on tumor latency and multiplicity.
PAHs are also found in food. 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine (PhIP) is a PAH found in well-done or chargrilled meats. This estrogenic compound is a mammary carcinogen in rodents [39–41], but concerns have risen over the presence of PhIP induced DNA adducts in human mammary glands [42]. Epidemiological studies report that women that ate well done meat were more likely to have breast cancer than women who ate rare to medium-well cooked meats [43]. Due to their mutagenic properties, the effects of PAH exposure is not limited to the mammary gland.
Bisphenol A
Bisphenol A (BPA) is a high production-volume chemical that is found in a variety of consumer products including plastics used to make drinking containers, epoxy resins that line metal food and drink cans, dental sealants and paper receipts [44]. Through physical manipulation or contact, BPA can leach from these products leading to unintended exposures. As a result, BPA contaminates food and water, with oral consumption being the main route of exposure to humans. Following oral exposure, BPA is metabolized to its glucuronidated-conjugate [44, 45]. Recent attention has been paid to BPA and its potential effects on development. It can be found in plastics used to make baby bottles and children’s toys, is found in the serum of infants and children, and has been linked to obesity [46]. It is one of the few EDCs discussed that does not persist in organisms, i.e., it has a short half-life (5.3 h in adults), yet internal serum levels may remain steady due to continual exposure [45]. It also has structural similarity to estradiol and, like DES, it demonstrates estrogenic properties [47, 48].
Since BPA mimics estrogen, exposures to this compound can cause endocrine disruption. In vitro studies indicate BPA binds to estrogen receptors (ERs; α and β) [48], estrogen receptor related gamma (ERRγ), and the G-coupled protein receptor, GPER/GPR30 [49]. BPA elevates testosterone, pregnenolone, and estradiol in rat ovarian cell cultures [50]. Adult male rats orally exposured to 200 mg/kg BPA had elevated relative testicular, seminal vesicles, and prostate weights, with decreased sperm production and motility [51]. These same oral adult exposures had no effect on female reproductive organ or estrous cycle, but did elevate serum estradiol and androstenedione [51]. Developmental exposures to BPA affect neuroendocrine signaling by increasing ERα and ERβ in the hypothalamus [52]. BPA disruption of the endocrine system also plays a part in altered mammary gland growth.
In rodents, prenatal exposure to BPA has been shown to accelerate mammary gland growth. Mice given BPA through subcutaneous implanted osmotic pumps had mammary glands with hyperplasic epithelial ducts and a beaded appearance [47]. Munoz de Toro [53] also found that prenatal exposure to BPA in rats (via minipump) induced mammary gland growth acceleration. In another study, prenatal exposure to 25 or 250 μg BPA/kg in CD-1 mice resulted in glands with increased mature structures (alveolar buds and terminal ducts) and increased stromal cell proliferation compare to controls at 6 months of age [16], indicating that prenatal BPA accelerated MG maturation. Although in the adult gland it was clear that BPA stimulated accelerated development following early life exposure, at 1 month of age, mice in the 250 μg/kg group displayed mammary growth patterns indicative of delayed development, with reduced ductal elongation, reduced stroma proliferation, and fewer TEBs compared to controls. These results are similar to those observed previously for other estrogenic compounds [54]. These findings highlight the importance of examination of multiple time-points following chemical exposure and shows how initial growth delays can eventually result in rampant cellular proliferation for estrogenic compounds.
The relevance of the earlier animal studies using minipumps has been questioned since human exposures are primarily oral (metabolized differently) and circulating blood levels were not provided, limiting cross-species extrapolation. However, fetuses are exposed to BPA through the maternal circulation regardless of mothers’ route of exposure, thus dampening this argument, especially as it pertains to low-dose prenatal exposure studies. To further address these issues, recent animal studies have administered BPA in the water supply or through oral gavage. Using the more translatable route of exposure, neonatal BPA exposures (25 and 250 μg/kg) in rats resulted in numerous TEBs which had a high proliferative index [55]. Similar mammary gland growth accelerations were observed in rhesus monkeys prenatally exposed to oral BPA, creating human relevant unconjugated BPA serum levels [56]. To determine whether or not mammary defects caused by BPA were related to cancer risk, prepubertally exposed Sprague Dawley rats were given DMBA at 50 days of age. This dual exposure increased tumor multiplicity and reduced tumor latency in 250 μg BPA/kg BW-treated animals. BPA treatment increased proliferation and reduced apoptosis compared to controls [55, 57]. Prenatal exposures to BPA also were found to increase mammary tumor multiplicity and decrease tumor latency. However BPA shifted the time at which the mammary gland was most susceptible to DMBA possibly by altering the timing of TEB abundance in the gland [55, 58]. These data indicate that oral prenatal BPA exposure, at low levels, resulted in mammary gland aberrations that affected carcinogen susceptibility, in addition to the already strong data from mini-pump delivery studies in rats and mice. Furthermore, recent evidence suggests that BPA persistent effects are likely due to epigenetic alterations [59].
Exposure to BPA during adulthood has also been found to influence cancer risk in rodent models. Using a transgenic MMTV-erB2 mouse model, female mice were exposed to drinking water with 2.5, 25, 250, 2,500 μg BPA/L from PND 56–122 or PND 56–252. At PND 112, the mammary glands of BPA exposed mice had a higher proliferation index when compared to controls. However, the highest exposure of BPA (2,500 μg/L) resulted in increased apoptotic index [60]. At PND 252, only exposures to the low doses of BPA (2.5, 25) resulted in statistically significant increase in mammary tumor multiplicity and reduced tumor latency, yet there was no increases observed in the 3 higher doses [60]. Other studies have found similar effects that do not follow typical dose response curves, which creates some difficulty in extrapolating data to humans. These data taken together suggest that low doses of BPA have adverse effects on the mammary gland distinct from those of high doses. This could be due to separate/multiple signaling paths being triggered at various dose levels [58].
Organochlorine Pesticides
As the name suggests, organochlorines are chlorinated hydrocarbons and they are typically used as pesticides—either insecticides or herbicides. The insecticides were designed to be neurotoxins, and the herbicides are effective at disrupting photosynthesis, but they have also been found to have effects on the reproductive system, including the mammary gland.
TCDD
One of the most infamous organochlorines is 2,3,7,8-tetrachloridibenzo-p-dioxin (TCDD or dioxin). TCDD is a polycyclic chlorinated hydrocarbon that is a pollution product of combustion and manufacturing chemicals. Agent Orange, the herbicide used by the United States Army in the Vietnam War, was found to be contaminated with TCDD. TCDD can be found in the soil and water, and due to its lipophilic properties and extensive half-life of approximately 7–8 year in humans [61], TCDD bioaccumulates in animals and the environment. TCDD binds to the aromatic hydrocarbon receptor (AhR) [62] and has anti-estrogenic properties. There are several other environmental contaminants that bind to AhR and their hazard levels are based on their effects relative to TCDD or the toxic equivalency factor [63].
TCDD has several endocrine disrupting effects such as reduced estradiol production and elevated ER mRNA expression in uterine, ovarian, and hypothalamic tissues of rats [64]. A single prenatal TCDD exposure of 1 μg/kg by gavage in female rats delayed timing of vaginal opening and disrupted normal estrous cyclicity [65–67]. Adult exposure to dioxin also reduced uterine-ovarian weights [68]. Endocrine disruption in male rats from TCDD exposure has been shown to reduce sperm production and delay preputial separation, a marker of sexual maturation [69]. Although TCDD binds to AhR, dioxin can increase aromatase activity, the enzyme that irreversibly converts androgens into estrogens, and can consequently elicit effects on androgen- and/or estrogen-dependent organs [70].
From animal studies, it has also been found that TCDD exposure delays mammary gland growth. A single prenatal exposure to TCDD via gavage causes severe retardation of the mammary gland resulting in numerous immature undifferentiated structures [65, 66]. However, the timing for that exposure is critical and must be at the time of mammary bud development in utero, for the most deleterious effects to be observed. Prenatal exposure to TCDD via gavage accompanied by DMBA exposure in young adulthood (PND 50) resulted in increased mammary tumor formation and reduced tumor latency compared to controls [66]. Population studies investigating the effects of high TCDD exposure in Seveso, Italy found that women in the area had an increased risk for breast cancer [71] although a follow-up evaluation of that population found no association between TCDD exposure and breast cancer incidence or mortality [72]. Many of the girls that were exposed to high levels of TCDD had not reached postmenopausal ages (age at most risk for breast cancer in normal population) by the time of the data analysis, and it is possible that the reported results did not reflect the actual effects in women. Therefore, it is important that there is a follow-up study to determine whether these women develop breast cancer to accurately assess the health outcome related to TCDD exposure. Occupational exposure to dioxins in a Hamburg, Germany, pesticide plant resulted in significantly increased breast cancer mortality among women workers [73].
PCB
Polychlorinated biphenyls (PCBs) are a class of compounds that are plasticizers, solvents, and other industrial chemicals. These chemicals have high lipophilicity and bioaccumulate in animals and the environment. These chemicals are stored in adipocytes in the body, and as a result are transferred into breast milk. PCBs have long half lives in humans [61]. In addition, PCBs can also bind to the AhR, and have similar effects to that of TCDD. Not all PCBs act alike; some congeners have anti-androgenic and mixed estrogenic/anti-estrogenic effects, while many of them also alter thyroid activity.
One of the most studied PCBs, 3,3,4,4,5-pentachlorobiphenyl (PCB 126), is classified as a “dioxin-like compound” and has a toxic equivalency factor of 0.1, making it 10 times less toxic than TCDD [74]. Prenatal exposure to PCB126 induces mammary developmental delays (designated by decreased alveolar budding) [75] which at a high dose (7.5 μg) has been shown to reduce carcinogen-induced tumors [29] yet increase tumor weight and tumor proliferation index at a lower dose (250 ng). In addition exposures at these levels also reduced serum estradiol [75]. In vitro studies with mammary tumor cell lines demonstrate that PCB 126 and other PCBs inhibit estradiol-stimulated tumor cell growth [76] highlighting the anti-estrogenic effects of these chemicals. In a review of epidemiology studies, only a few case–control studies found a positive association between PCB exposures and elevated odds for breast cancer [77].
DDT and DDE
Dichlorodiphenyltrichloroethane (DDT) is a synthetic industrial and household insecticide whose widespread use as a mosquito repellent and its long half-life made it a prominent environmental contaminant. Although DDT consequently helped lower incidence of malaria and typhoid, its use was banned in the US in 1972 for its effects on the environment and potential effects on human health [78]. Like most insecticides, DDT demonstrated neurotoxic effects, but DDT and its metabolites also caused an array of reproductive and developmental effects and partitioned into breast milk [79].
DDT and its main metabolites dichlorodiphenyldichloroethylene (DDE) and dichlorodiphenyldichloroethane (DDD) have endocrine disrupting properties. Some of the metabolites have estrogenic properties (o,p’-DDT and o,p’-DDE [1,1-dichloro-2-(o-chlorophenyl)-2-(p-chlorophenyl) ethylene), while others have anti-androgenic properties (p,p’-DDE [1,1-dichloro-2,2-bis(p-chlorophenyl) ethylene) [80].
In a 2012 study by Johnson and colleagues, HER2/Neu transgenic mice were implanted with pellets that released p,p’-DDE or o,p’-DDE and while neither metabolite increased mammary tumor multiplicity, both decreased mammary tumor latency [80]. In previous studies, o,p’-DDE has been shown to enhance mammary epithelium growth and increase epithelial cell proliferation index [68], as well as increase mammary tumor cell proliferation [81]. There are few epidemiological studies that have found positive associations between DDT or its metabolites in serum and increased risk for the development of breast cancer. This is primarily due to the fact that those studies evaluated serum levels of DDT/DDE in women around the time of cancer diagnosis which cannot take into consideration exposure levels in conjunction with tumor latency. However, in a prospective case–control study, girls exposed to higher DDT/DDE levels were more likely to develop breast cancer than those with lower exposures, and risk increased with younger age at exposure [82]. For this study, the blood/serum samples were collected during periods of high and unregulated DDT exposures in the US. Moreover, younger girls (age <14) with higher DDT/DDE blood levels were more likely to develop breast cancer than girls exposed at age ≥14 [82] which highlights the sensitivity of the mammary gland around puberty and underscores the need to consider age of exposures for risk assessments.
Atrazine
Atrazine is a high use organochlorine herbicide utilized to control weeds in various farm/food crops. Atrazine and its metabolites are found in water, soil, plants, and animals [83]. Even though atrazine has a short half-life of ≤1 day in rodents, oral exposures are reported to induce multiple reproductive and developmental effects [84].
In the past 30 years, safety regulations have been placed on atrazine use due to its persistence in the environment and its proven role as an endocrine disruptor in rodents and amphibians [85]. In rats, atrazine exposures reduce the normal surge of luteinizing hormones in female adults via altered hypothalamic signaling [86]. Adult exposure in male rats results in increased relative testicular and epididymal organ weights with reduced sperm production and reduced androgen hormones in rats [51]. It is also thought that atrazine-induced alterations in maternal hormone signaling may play a role in prostate inflammation of developmentally exposed male Long Evans and Wistar rat offspring [87–88].
Prenatal exposure to atrazine alone or as a metabolite mixture has also been shown to deter normal mammary gland development in Long Evans female rats, resulting in glands with more TEBs, sparse branching patterns and impaired growth compared to controls at vaginal opening and young adulthood [87, 89]. Atrazine is not classified as a direct carcinogen or mutagen [90, 91], but lifelong dietary exposures have been shown to increase spontaneous mammary tumor development in rats [90, 91]. There are species and strain differences in effects related to atrazine exposure. Chronic dietary atrazine exposure in Sprague Dawley rats increased the incidence of adenocarcinomas in exposed females compared to controls [90] yet there was no effect on tumor incidence in F344 rats in another study [92]. In addition, atrazine exposure following carcinogen-induced tumor formation increased tumor incidence in human c-Ha-ras proto-oncogene transgenic rats [93]. A full review of atrazine carcinogenicity studies can be found in Stevens et al., 1999 [91]. A review of the one known mode of action for atrazine-induced tumor development—hastened reproductive senescence—was not deemed human relevant, therefore atrazine is not currently regarded as a possible human carcinogen by risk assessors [85].
Epidemiological studies that estimate human exposures from agricultural atrazine use found little if any association between atrazine and breast cancer, however it has been difficult to assess actual exposure of women to this chemical over their lifetime or during sensitive windows of breast development due to the lack of sensitive measurement protocols. Longitudinal epidemiological studies are needed in farming communities, especially in those communities raising corn as a major crop, to truly evaluate early life exposures and late life effects of atrazine and its metabolites.
Vinclozolin
Another chemical with both endocrine disrupting properties and reported mammary gland effects is (3-(3,5-dichlorophenyl)-5methyl-5-vinyl-oxa-zolidine-2,4-dione), vinclozolin. This lipophilic pesticide is used on fruits and vegetable crops to protect against the growth of several fungi. It is also used on golf course turf [94]. Exposure to vinclozolin occurs mainly through eating foods with residual contamination and drinking contaminated water. Since vinclozolin is lipophilic, it is likely to bioaccumulate in wildlife and potentially can be found in breast milk. In addition, a recent study has found measurable concentrations in cord blood [95], suggesting that vinclozolin is transferred to the fetus. Vinclozolin is metabolized to 2-[[(3,5-dichlorophenyl)-carbamoyl]oxy]-2-methyl-3-butenoic acid (M1), and 3′,5′-dichloro-2-hydroxy-2-methylbut-3-enanilide (M2) with the metabolites being more biologically active than the parent compound [96].
Vinclozolin’s endocrine disrupting properties were first noted in the male reproductive system [97, 98]. Several studies have documented the anti-androgenic effects of vinclozolin and its metabolites. Prenatal vinclozolin exposure in rats resulted in reduced prostate weight and anogenitial distance, retention of nipples and areola, hypospadias, and undescended testicles, while adult exposures had far less severe effects [24–26, 51]. In male rats, vinclozolin elevated serum androstenedione, testosterone and LH, and reduced estradiol levels [51]. Multigenerational exposure to vinclozolin elevated the prevalence of a myriad of tumors in males including tumors of the prostate, testis, and immune system [99]. In vitro studies indicate that vinclozolin and M2 are antagonists for mineralocorticoid receptor and PR and all three are weak agonists of both ERs [96]. A recent evaluation of pesticides in human cord blood and birth weight found that vinclozolin is statistically associated with reduced birth weights (0.18 ng/ml geometric mean) [95]. Some vinclozolin effects are epigenetic in nature [99].
Vinclozolin also affects the female reproductive system. Adult exposures in female rats lead to elevated uterine and adrenal relative weights, elevated serum LH and androstenedione, and increased ovarian androstenedione and estrone [51]. While early exposure to vinclozolin delays puberty in males [24], it is not associated with precocious or delayed puberty in females in rodents or humans [100, 101]. In addition, a multigenerational rat study using 100 mg/kg vinclozolin found mammary tumors only in females offspring, however the incidence of these tumors was low and it was unclear whether this endpoint was statistically significant compared to controls [99]. Recent studies indicate that vinclozolin has effects on mammary gland development. In a study by Saad et al. (2011), pregnant Wister Han rats were orally given low level vinclozolin (1 mg/kg) daily from GD 1 through PND 21 [100]. Morphological evaluations of whole mounts of mammary glands on PND 35 showed increased epithelial branching density in the vinclozolin-treated group compared to controls, yet there were no statistically significant changes in counted mammary structures (i.e., TEBs, TD, AB). At PND 50, there were increased TEBs in vinclozolin-treated rats compared to controls while there were no differences in TDs or ABs [100]. In addition, vinclozolin exposure increased areas of hyperplastic ducts, Ki67 gene expression, and stromal-ductal density at PND 50. There were no differences in ER or AR gene expression compared to controls when evaluated by PCR.
Additionally, this study also investigated the effects of vinclozolin with concomitant genistein exposure. Exposures to the antiandrogenic and estrogenic compounds resulted in precocious puberty and accelerated MG growth. These alterations in growth presented as increased epithelial branching, increased hyperplastic ducts, increased proliferation, and increased stromal-ductal density at PND 35. At PND 50, exposure to the mixture increased hyperplastic alveolar structures in treated glands compared to controls. Importantly, the resulting effects from exposure to a mixture of vinclozolin and genistein were more pronounced compared to changes with vinclozolin or genistein alone. These data suggest that co-exposure of vinclozolin and genistein act together to synergistically affect MG growth which may potentially lead to increased mammary tumor formation in rats. While the dose of genistein used in this study is well below levels from consumption of soy-based formula, the vinclozolin levels used are higher than those from dietary exposures. Further studies are needed to fully characterize the effects of vinclozolin on mammary gland development and to determine its effects on breast cancer risk.
Phthalates
Phthalate esters are widely used as plasticizers and are added to polymers such as polyvinyl chloride (PVC) to increase pliability. Concern about phthalate exposure has grown due to their presence in plastic baby bottles, children’s toys, various cosmetics and medical tubing. Phthalates easily leach from plastics and PVC due to normal wear and tear and physical manipulation and residues can be found in household dust [102]. As a result, exposure to phthalates occurs mainly through inhalation and oral exposure. Compared to adults, infants and children have higher exposure levels to phthalates
Phthalates have been found to have an array of effects on both the reproduction and development of humans and rodents, primarily mediated by influences on the endocrine system. Exposure to phthalates in rodents reduces estradiol production, delays puberty, and reduces fecundity with reduced implantation sites, increased fetal resorptions, decreased fetal birth weights, nipple retention in males, and increased malformation in offspring [50]. Epidemiological studies have found that boys born to mothers with higher urinary phthalate metabolites had shorter anogenital distance than boys born to mothers with lower urinary phthalate metabolites [103]. In rats this finding was confirmed [104] and prenatal and preconception exposure to phthalates was associated with developmental and testicular toxicity of offspring [105–108]. In women, exposure to phthalates is associated with endometriosis risk and in animal studies has shown to alter mammary gland development [109].
In rats, prenatal exposure to butyl benzyl phthalate (BBP) causes slightly accelerated mammary gland growth but delays the onset of puberty [110, 111]. Glands from developmentally-exposed female offspring had increased proliferation index in TEBs at PND 21 and 35 [112] suggesting that BBP exposure could increase sensitivity of the mammary gland to chemical carcinogens. A two-year BBP feeding study in adult rats reported decreased mammary fibroadenomas [113]. In Mexican women, higher urinary concentrations of monoethyl phthalate were associated with a two-fold increase in breast cancer risk [114]. In the same study, higher concentration of more bulky phthalates, monobenzyl phthalate and mono(3-carboxypropyl) phthalate, were inversely associated with breast cancer risk [114]. It is proposed that phthalates affects mammary gland growth through epigenetic changes [115] and importantly, phthalate exposure counteracts the effects of tamoxifen on mammary cancer cells [116]. The effects of other EDCs on breast cancer chemoprevention are just beginning to be reported.
PFOA
Perfluorooctanoic acid (PFOA) is a chemical that has endocrine disrupting properties and can affect mammary gland growth. PFOA is a synthetic surfactant that is used in the production of fluorotelomers commonly used in non-stick and stain-resistant products, flame resistant foams, dental products, and lining of popcorn bags. PFOA is detected in all Americans sampled [117, 118], and human exposure occurs through use of aforementioned products, household dust, and contaminated drinking water. Additionally PFOA binds proteins, so is transferred to the offspring via the placenta and breast milk. In adult humans, PFOA is slowly eliminated from the body with a half-life of 3.8 years [119]. PFOA exposure in rodents is commonly associated with liver toxicity, but PFOA also causes endocrine disruption and reproductive and developmental toxicity.
PFOA is considered an EDC due to its affects on the immune system, thyroid, and metabolism. Exposures to PFOA modulate immune response in children and have been linked to altered cholesterol and altered thyroid function [120]. Although this linear halogenated hydrocarbon has little physiochemical properties with other EDCs and no structural similarity with estradiol, PFOA has been shown to bind to ER and estrogen receptor response elements [121]. From in vitro studies, PFOA has been shown to be both estrogenic and conversely anti-estrogenic when incubated with estradiol [121]. In mouse studies, prenatal, neonatal, and peripubertal exposures have shown to delay onset of puberty or vaginal opening in females [122–124]. Uterotrophic studies show increased uterine weights at low doses (0.01 mg/kg)[125]. Peripubertal PFOA exposures indicate there are strain differences in uterine effects [123]. Peripubertal exposure to 1–10 mg/kg PFOA in Balb/C mice resulted in reduced uterine weights, while C57Bl/6 mice had increased uterine weights at 1 mg/kg and reduced uterine weights at 10 mg/kg. One prenatal study has shown that PFOA accelerates puberty in young male mice [122]. Two year exposures studies to PFOA in male rats leads to increased Leydig Cell tumors with reduced circulating testosterone [126], suggesting PFOA has anti-androgenic properties. A full review of the endocrine disrupting properties of PFOA can be found in White et al. 2011 [126].
There has been only one published animal study that has investigated the effects of chronic PFOA and evaluated mammary tumor formation [127]. The significance of this study, and others, has been criticized due to the use of female rats as the animal model. Female rats eliminate PFOA at extremely high rates (a few hours) while female mice and women eliminate PFOA at a much slower rate (21 days and 3.8 years, respectively) [119, 128]. As mice (not rats) and humans exhibit no sex differences in PFOA elimination rates, mice are used as a more appropriate animal model for evaluation of PFOA toxicity. To date, there are no published 2 year PFOA exposure studies nor PFOA and DMBA-induced carcinogenicity studies in mice. Most occupational studies solely report on male health outcomes due to small population of female workers. Epidemiology studies would suggest that PFOA may actually lower breast cancer risk, as it has been positively associated with delayed puberty in girls [129] and early menopause [130]: two factors that are negatively associated with breast cancer risk. In addition, recent studies indicate high serum PFOA is also associated with preeclampsia, pregnancy-induced hypertension, which is negatively associated with breast cancer [131]. However, a recent study of Inuit women found a positive association between perfluorinated compounds along with other persistent organic chemicals and breast cancer risk, suggesting that PFOA and other EDCs may affect disease development in this small, highly exposed population [132].
Animal studies with more detailed examination of effects on mammary gland growth agree with the Inuit data, and suggest that PFOA exposure may increase susceptibility for development of mammary tumors. Prenatal exposure to PFOA has been shown to delay mammary epithelial development [35, 36, 124, 133]. Restricted and cross-foster exposure studies in CD-1 mice (Fig. 4) showed delayed mammary gland growth for all PFOA-treated groups assessed by developmental scoring [124]. These delays were characterized by reduced ductal elongation and side-branching into adulthood. Timing of appearance of TEBs is shifted with PFOA exposure as there are fewer TEB at PND 22, but more at PND 42, when compared to controls. Abnormal mammary gland maturation persisted at 18 months in glands exposed to 5 mg/kg with reduced epithelial density and increased stromal density, a factor associated with increased breast cancer risk [33, 35]. In addition there were areas of epithelial hyperplasia in PFOA-treated mice at 18 months. Similar effects were observed using lower PFOA doses (0.01 −3 mg/kg) [124]. In those studies, full gestational exposures resulted in extended presence of mammary TEBs at doses 0.3–3.0 mg/kg, suggesting the PFOA exposure may increase susceptibility to carcinogens. Late gestational exposure to 0.01–1 mg/kg PFOA also resulted in significant mammary developmental delays [124]. These delays were present at serum PFOA concentrations in mice that overlap with those observed in young children living in highly exposed areas in the US [124, 134]. These studies suggest that in utero and lactational exposure to PFOA delay development and could increase the susceptibility of the mammary gland to carcinogens.

Permanent PFOA-induced mammary gland delays following oral prenatal/neonatal exposures. a PFOA-treated glands have statistically significant reduced developmental scores compared to controls in early life, as indicated by *. b Glands at 18 months. Large arrow indicates area of increased stromal density in whole mounts (left) and H&E-stained tissue (right); small arrow indicates area of ductal hyperplasia in H&E-stained tissue. (Modified from Ref [33], with permission)
Like other EDCs, there are differences in effect based upon timing of exposure. Exposure to PFOA from puberty into young adulthood resulted in reduced ductal elongation in Balb/C (2.5–10 mg/kg) and C57Bl/6 mice (7.5–10 mg/kg), decreased side branching in Balb/C mice (2.5–10 mg/kg) and C57Bl/6 mice (7.5 mg/kg) and increased side branching in C57Bl/6 mice (5.0 mg/kg) [123, 135]. This single stimulatory effect in the 5 mg/kg PFOA-treated C57Bl/6 mice may be caused by a concomitant rise in circulating progesterone [136]. PFOA-treated C57Bl/6 adult glands displayed elevated ERα protein expression in the epithelium compared to controls [136]. Furthermore, PFOA-induced systemic immunomodulation and elevated production of reactive oxygen species from mitochondrial damage [137] may increase susceptibility of mammary tissues to chemical carcinogens and remains to be tested.
Phytoestrogens
The chemicals that have been discussed thus far are synthetic compounds that disrupt the endocrine system by mimicking or inhibiting endogenous hormones. However, there are naturally-occurring chemicals that also mimic or mask effects of endogenous hormones. Phytoestrogens are naturally occurring estrogens that are found in plants. There are some that believe that phytoestrogens have beneficial effects to treat and/or prevent breast cancer. Yet, there are reported negative effects associated with phytoestrogens. As with many of the other compounds, the difference between beneficial and detrimental health effects depends on dose and timing of exposure.
Genistein
In recent years, the perceived health benefits of consuming soy due to the high isoflavones and their estrogenic properties has increased in popularity. Asian women have lower lifetime breast cancer rates compared to Caucasian American women [138, 139], and some speculate that their high-soy based diet has protective effects against mammary tumorigenesis. Genistein is the most abundant isoflavone in soy along with daidzein [140]. As it is a polyphenol, genistein shares structural similarities with estradiol and has been shown to behave like an estrogen. Some studies have shown that a high soy diet/supplementation reduces breast cancer risk in women [141–143]. In rats, exposure to isoflavones beginning in utero to adulthood has been shown to protect again carcinogen-induced mammary tumors [144, 145]. Upon closer evaluation of the influence of timing of exposure on effects of genistein in rodent models, prepubertal exposure appeared to have the most beneficial effects against carcinogen-induced mammary tumorigenesis, compared to perinatal or adult only exposures [146–149]. Glands from peripubertal genistein-treated rats demonstrate accelerated growth compared to controls with fewer TEBs and more differentiated ends. Yet when rats were treated with genistein during prenatal or adult time-points, the induced morphological changes had no protective effects on MNU-induced [146] or DMBA-induced mammary gland tumors [147]. Genistein also appears to have some chemotherapeutic properties when given after tumor initiation and promotion. Genistein given in rats with tumors was shown to stabilize DNA in mammary epithelial cells of the normal tissue. It is thought that genistein protects against tumor growth via upregulation of the tumor-suppressor protein PTEN [147].
Genistein exposure can also leave the mammary gland more susceptible to spontaneous tumorigenesis. In a multigenerational study conducted by the National Toxicology Program (2008), developmental genistein exposures in rats increased incidence of adenomas and adenocarcinomas collectively compared to controls [150]. In addition, low-levels of genistein cancel out therapeutic effects of tamoxifen in breast cancer cells. The increased rat mammary tumor incidence from lifetime genistein exposure is inconsistent with the proposed protective effects of high genistein diets in Asian women and resulting low breast cancer rates. However genistein is just one component of soy and studies have found differences between using a mixture of isoflavones versus genistein exposure alone [151]. These adverse effects are not exclusive to females, as mammary ductal hyperplasia has also been observed in males. In two multi-generational studies, dietary genistein increased ductal hyperplasia in male rat offspring [150, 152]. The mammary gland was reported to be the most sensitive tissue affected by genistein in males. Although this particular effect has not been observed in human males, many fear the impact of additional estrogen-like compounds on the development of young boys, as males appear to be more sensitive to the effects of some EDCs.
Resveratrol
Resveratrol (RES) is a polyphenol, like genistein, that can be found in grapes, red berries, and red wine. It is estimated that human exposures to RES occur most from the consumption of red wine as there is approximately 1.4 mg RES per glass (calculated from [153]). Numerous studies have linked moderate consumption of red wine to better health outcomes such as: reduced risk of cardiovascular disease, stronger immune system, protection against memory loss, and prevention against the development of several cancers, including breast cancer [154]. These beneficial effects are proposed to be due to the antioxidant properties of RES.
The polyphenol has some structural similarity to estradiol and has endocrine disrupting properties. For instance, RES treatment of ovarian thecal cells in culture caused elevated androstenedione and androsterone production, with no effect on progesterone [155]. Prenatal exposure to RES in Sprague Dawley rats increased ovarian weight in ovariectomized adult females and reduced testicular weight and plasma testosterone in adult males [156] demonstrating that RES can act as both an estrogen receptor agonist and antagonist, depending on the target tissue.
Studies in animals support these findings and have shown that RES is protective against mammary tumorigenesis [147, 157–160]. Lifetime exposure to RES in rats beginning at birth was shown to accelerate mammary gland growth and differentiation compared to controls and reduced susceptibility to carcinogen-induced mammary tumors [147]. RES-treated glands were found to have lower proliferative index and a higher apoptotic index in the mammary epithelial cells compared to controls [147]. Adult only oral exposures to RES, which has better translation to human exposure patterns, in HER-2/neu transgenic mice also demonstrated the beneficial effects of RES exposure [158], as the tumor prone mice treated with RES displayed a longer latency to tumor when compared to controls. RES treatment also reduced the metastatic capacity of these tumors by increasing apoptosis of tumor cells [158]. Moreover, in vitro studies have demonstrated that RES has antitumor initiation, promotion, and anti-mutagenic properties which were attributed to its inhibitory effects on cyclooxegenase [160]. Thus, RES may be one of the only EDCs that has primarily beneficial effects on the mammary gland, as related to breast cancer risk.
Zearalenone
Zearalenone (ZEA) is a mycotoxin that is produced by the fungus Fusarium. This mold often grows on plants and crops such as oat, barley, corn, wheat, and rice. ZEA can also contaminate the soil that the plants grow in, and the animals that eat the plants (livestock) [161, 162]. ZEA and its metabolites have some structural similarity to estrogen and have been shown to bind to estrogen receptors [162]. Since ZEA is found on plants and has estrogenic properties it is often considered a phytoestrogen.
A study of mice exposed to ZEA in utero produced female offspring with accelerated vaginal opening timing, increased estrous cycle length, and early mammary gland maturation [157]. In rats, prepubertal exposure to ZEA was shown to reduce carcinogen-induced mammary tumors [163], while prenatal exposure had no effects on tumor incidence [164]. Mammary glands of rats exposed to ZEA during puberty had fewer TEBs and more mature structures [163] which reduced the susceptibility of ZEA-treated glands to tumorigenesis. In vitro studies have shown biphasic effects of ZEA, with low doses promoting breast cancer cell growth and high doses increasing apoptosis and cell cycle arrest [165].
ZEA contamination of food systems has because a concern for humans. Several early studies have linked ZEA exposure with precocious puberty in girls based on unsubstantiated evidence [166, 167]. A recent study measured urinary mycoestrogens in New Jersey girls and found an inverse association between urinary mycoestrogens and age at thelarche, or the beginning stages of breast development, as well as a negative association between ZEA levels and height [168]. As these findings are inconsistent with the data from animal studies, more population studies are needed to determine the influence of ZEA on human mammary gland growth and potential breast cancer risk.
Concluding Remarks
The dynamic signaling of the endocrine system makes it highly susceptible to influences from exogenous chemicals. Since the endocrine system regulates and controls multiple functions within the body, EDCs have the ability to greatly impact health outcomes. The endocrine system plays an important part in timing of puberty and menopause, obesity, breast density, and immune function, all factors which influence breast cancer risk. Additionally, as presented, EDCs can alter mammary gland development potentially increasing susceptibility to chemical carcinogens or spontaneous tumorigenesis. Therefore it is important to understand the correlation between EDC exposure during different critical windows of mammary gland development and breast cancer risk.
Understanding mechanisms of mammary gland development in laboratory animals has led to significant insights into the general role(s) of EDCs on mammary gland development and thus mammary tumor formation. However, there are still many gaps that can be addressed with further studies. Despite the impact that breast cancer poses to women and our health care system, mammary gland evaluation, breast cells, and lactation assessments are often excluded from routine health hazard assessments in industry and government-based chemical testing studies. There is a need to include mammary gland data in chemical hazard and risk assessments. In order for this to be accomplished, it is important that there are methods developed for evaluation of mammary growth patterns and their relation to health outcomes such as changes in functionally capacity or neoplastic transformations.
To investigate the influence of EDC exposure and avoid waiting for 2 year rodent cancer bioassay results for tumor development, many animal studies follow the experimental design of EDC exposure (acute or chronic) followed by acute carcinogenic exposure. This model is effective for a hazard assessment of these chemicals but chronic exposure to low doses of a carcinogen may be more reflective of human exposure to classical chemical carcinogens. In addition utilizing this type of experimental model would address species specific and interspecies differences in timing of puberty as well as timing of mammary gland maturation. Linking early life adverse outcomes, such as retardation or acceleration of mammary development, to later life cancer risks would strengthen the use of whole mount analyses in test guidelines on new or replacement chemicals.
Population studies on EDCs and other chemicals often correlate internal dosimetry, such as serum concentrations, to health outcomes without the ability to determine causation. Animal studies can dose with specific exposures (thus knowing causation), but often do not include internal dosimetry, making it difficult to interpret data for the characterization of risk to humans. There is need to bridge that transdisciplinary research gap. For this to be achieved, inclusion of more time-points during stages of mammary gland development, along with blood collection for dosimetry will help to understand the transition from normal to tumor development. In addition, relatively little is known about how EDCs act together. It is possible that the negative effects of one EDC are balanced by beneficial effects of another or chemicals may act together to heighten the impact of their effects. Co-exposure with prevalent dietary EDCs such as genistein, or altered fat-content diets, would resemble exposures that likely occur in average human life. A comparison of the effects from mixtures relative to the effects of individual exposures would reveal a glimpse into true human exposures and their resulting effects on health outcomes. Future studies must consider determination of mechanisms of action for EDCs if we are to begin preventing breast cancer.
References
Centers for disease contral and prevention. Cancer among women. 2012. http://www.cdc.gov/cancer/dcpc/data/women.htm. 2012.
American Cancer Society. Breast Cancer Overview. 2012. http://www.cancer.org/Cancer/BreastCancer/OverviewGuide/breast-cancer-overview-key-statistics.
American Cancer Society. Breast cancer facts and figures 2011–2012. 2011.
Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78–85. doi:10.1056/NEJM200007133430201.
Buttke DE, Sircar K, Martin C. Exposures to endocrine-disrupting chemicals and age of menarche in adolescent girls in NHANES (2003–2008). Environ Heal Perspect. doi:10.1289/ehp.1104748.
Diamanti-Kandarakis E, Bourguignon J-P, Giudice LC, Hauser R, Prins GS, Soto AM et al. Endocrine-disrupting chemicals: an endocrine society scientific statement. Endocrine Rev. 2009;30(4):293–342. doi:10.1210/er.2009-0002.
US Environmental Protection Agency. Endocrine Disruptors Research. 2012. http://www.epa.gov/endocrine/#eds.
Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2006;22:287–309. doi:10.1146/annurev.cellbio.22.010305.104315.
US Environmental Protection Agency. TSCA Chemical Substance Inventory. 2011. http://www.epa.gov/oppt/existingchemicals/pubs/tscainventory/index.html.
Vineis P, Schatzkin A, Potter JD. Models of carcinogenesis: an overview. Carcinogenesis. 2010;31(10):1703–9. doi:10.1093/carcin/bgq087.
Soto AM, Sonnenschein C. The tissue organization field theory of cancer: a testable replacement for the somatic mutation theory. BioEssays: News Rev Mol Cell Dev Biol. 2011;33(5):332–40. doi:10.1002/bies.201100025.
National Toxicology Program. Nominations to the Testing Program. 2012. http://ntp.niehs.nih.gov/?objectid=25BC6AF8-BDB7-CEBA-F18554656CC4FCD9.
National Toxicology Program. Report on Carcinogens, 12th, http://ntp.niehs.nih.gov/ntp/roc/twelfth/roc12.pdf.
Diethylstilbestrol [database on the Internet]. Available from: http://monographs.iarc.fr/ENG/Monographs/vol100A/mono100A-16.pdf. Accessed.
Knudson AG. Hereditary cancer: two hits revisited. J Cancer Res Clin Oncol. 1996;122(3):135–40.
Markey CM, Luque EH, Munoz De Toro M, Sonnenschein C, Soto AM. In utero exposure to bisphenol A alters the development and tissue organization of the mouse mammary gland. Biol Reprod. 2001;65(4):1215–23.
Rudel RA, Fenton SE, Ackerman JM, Euling SY, Makris SL. Environmental exposures and mammary gland development: state of the science, public health implications, and research recommendations. Environ Heal Perspect. 2011;119(8):1053–61. doi:10.1289/ehp.1002864.
National Cancer Institute. The Cost of Care. 2011. http://www.cancer.gov/aboutnci/servingpeople/cancer-statistics/costofcancer. 2012.
Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race, breast cancer subtypes, and survival in the Carolina breast cancer study. JAMA. 2006;295(21):2492–502. doi:10.1001/jama.295.21.2492.
Eroles P, Bosch A, Perez-Fidalgo JA, Lluch A. Molecular biology in breast cancer: intrinsic subtypes and signaling pathways. Cancer Treat Rev. 2012;38(6):698–707. doi:10.1016/j.ctrv.2011.11.005.
Rouzier R, Perou CM, Symmans WF, Ibrahim N, Cristofanilli M, Anderson K, et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin Cancer Res. 2005;11(16):5678–85. doi:10.1158/1078-0432.CCR-04-2421.
Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, Bontempi G, et al. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res. 2008;14(16):5158–65. doi:10.1158/1078-0432.CCR-07-4756.
Fenton SE. Endocrine-disrupting compounds and mammary gland development: early exposure and later life consequences. Endocrinology. 2006;147(6 Suppl):S18–24. doi:10.1210/en.2005-1131.
Monosson E, Kelce WR, Lambright C, Ostby J, Gray Jr LE. Peripubertal exposure to the antiandrogenic fungicide, vinclozolin, delays puberty, inhibits the development of androgen-dependent tissues, and alters androgen receptor function in the male rat. Toxicol Ind Health. 1999;15(1–2):65–79.
Buckley J, Willingham E, Agras K, Baskin LS. Embryonic exposure to the fungicide vinclozolin causes virilization of females and alteration of progesterone receptor expression in vivo: an experimental study in mice. Environ Health. 2006;5:4. doi:10.1186/1476-069X-5-4.
Gray LE, Ostby J, Furr J, Wolf CJ, Lambright C, Parks L, et al. Effects of environmental antiandrogens on reproductive development in experimental animals. Hum Reprod Update. 2001;7(3):248–64.
Rothschild TC, Boylan ES, Calhoon RE, Vonderhaar BK. Transplacental effects of diethylstilbestrol on mammary development and tumorigenesis in female ACI rats. Cancer Res. 1987;47(16):4508–16.
Kawaguchi H, Umekita Y, Souda M, Gejima K, Kawashima H, Yoshikawa T, et al. Effects of neonatally administered high-dose diethylstilbestrol on the induction of mammary tumors induced by 7,12-dimethylbenz[a]anthracene in female rats. Vet Pathol. 2009;46(1):142–50. doi:10.1354/vp.46-1-142.
Muto T, Wakui S, Imano N, Nakaaki K, Hano H, Furusato K, et al. In utero and lactational exposure of 3,3′, 4,4′, 5- pentachlorobiphenyl modulate dimenthlben[a]anthracene-induced rat mammary carcinogenesis. J Toxicologica Pathol. 2001;14:213–24.
Rayner JL, Enoch RR, Fenton SE. Adverse effects of prenatal exposure to atrazine during a critical period of mammary gland growth. Toxicol Sci. 2005;87(1):255–66. doi:10.1093/toxsci/kfi213.
White SS, Fenton SE, Hines EP. Endocrine disrupting properties of perfluorooctanoic acid. J Steroid Biochem Mol Biol. 2011. doi:10.1016/j.jsbmb.2011.03.011.
Restum JC, Bursian SJ, Giesy JP, Render JA, Helferich WG, Shipp EB, et al. Multigenerational study of the effects of consumption of PCB-contaminated carp from Saginaw Bay, Lake Huron, on mink. 1. Effects on mink reproduction, kit growth and survival, and selected biological parameters. J Toxicol Environ Health A. 1998;54(5):343–75.
Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, et al. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi:10.1186/1741-7015-6-11.
Maffini MV, Soto AM, Calabro JM, Ucci AA, Sonnenschein C. The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci. 2004;117(Pt 8):1495–502. doi:10.1242/jcs.01000.
White SE, Kato K, Jia LT, Basden BJ, Calafat AM, Hines EP, et al. Effect of perfluorooctanoic acid on mouse mammary gland development and differention resulting from cross-foster and restricted gestational exposure. Reprod Toxicol. 2009;27:289–98. doi:10.1016/j.reprotox.2008.11.054.
White SE, Calafat AM, Kuklenyik Z, Villanueva L, Zehr RD, Helfant L, et al. Gestational PFOA exposure of mice is associated with altered mammary gland development in dams and female offspring. Toxicol Sci. 2007;96(1):133–44. doi:10.1093/toxsci/kfl177.
Fertuck KC, Kumar S, Sikka HC, Matthews JB, Zacharewski TR. Interaction of PAH-related compounds with the alpha and beta isoforms of the estrogen receptor. Toxicol Lett. 2001;121(3):167–77.
Archer FL, Orlando R. Morphology, natural history, and enzyme patterns in mammary tumors of the rat induced by 7,12-dimethylbenz(a)anthracene. Cancer Res. 1968;28:217–23.
Ito N, Hasegawa R, Sano M, Tamano S, Esumi H, Takayama S, et al. A new colon and mammary carcinogen in cooked food, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP). Carcinogenesis. 1991;12(8):1503–6.
Lauber SN, Ali S, Gooderham NJ. The cooked food derived carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b] pyridine is a potent oestrogen: a mechanistic basis for its tissue-specific carcinogenicity. Carcinogenesis. 2004;25(12):2509–17.
Gooderham NJ, Zhu H, Lauber S, Boyce A, Creton S. Molecular and genetic toxicology of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP). Mutat Res. 2002;507:91–9.
Lightfoot TJ, Coxhead JM, Cupid BC, Nicholson S, Garner RC. Analysis of DNA adducts by accelerator mass spectrometry in human breast tissue after administration of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and benzo[a]pyrene. Mutat Res. 2000;472(1–2):119–27.
Zheng W, Gustafson DR, Sinha R, Cerhan JR, Moore D, Hong CP, et al. Well-done meat intake and the risk of breast cancer. J Natl Canc Inst. 1998;90(22):1724–9.
Pottenger LH, Domoradzki JY, Markham DA, Hansen SC, Cagen SZ, Waechter Jr JM. The relative bioavailability and metabolism of bisphenol A in rats is dependent upon the route of administration. Toxicol Sci. 2000;54(1):3–18.
Völkel W, Colnot T, Csanady GA, Filser JG, Dekant W. Metabolism and kinetics of bisphenol a in humans at low doses following oral administration. Chem Res Toxicol. 2002;15(10):1281–7.
Trasande L, Attina TM, Blustein J. Association between urinary bisphenol A concentration and obesity prevalence in children and adolescents. JAMA. 2012;308(11):1113–21. doi:10.1001/2012.jama.11461.
Vandenberg LN, Maffini MV, Schaeberle CM, Ucci AA, Sonnenschein C, Rubin BS, et al. Perinatal exposure to the xenoestrogen bisphenol-A induces mammary intraductal hyperplasias in adult CD-1 mice. Reprod Toxicol. 2008;26(3–4):210–9. doi:10.1016/j.reprotox.2008.09.015.
Naciff JM, Jump ML, Torontali SM, Carr GJ, Tiesman JP, Overmann GJ, et al. Gene expression profile induced by 17alpha-ethynyl estradiol, bisphenol A, and genistein in the developing female reproductive system of the rat. Toxicol Sci. 2002;68(1):184–99.
Thayer KA, Belcher S. Mechanisms of action of bisphenol A and other biochemical/molecular interactions, http://www.who.int/foodsafety/chem/chemicals/5_biological_activities_of_bpa.pdf.
Bhattacharya P, Keating AF. Impact of environmental exposures on ovarian function and role of xenobiotic metabolism during ovotoxicity. Toxicol Appl Pharmacol. 2012;261(3):227–35. doi:10.1016/j.taap.2012.04.009.
Quignot N, Arnaud M, Robidel F, Lecomte A, Tournier M, Cren-Olive C, et al. Characterization of endocrine-disrupting chemicals based on hormonal balance disruption in male and female adult rats. Reprod Toxicol. 2012;33(3):339–52. doi:10.1016/j.reprotox.2012.01.004.
Richter CA, Birnbaum LS, Farabollini F, Newbold RR, Rubin BS, Talsness CE, et al. In vivo effects of bisphenol A in laboratory rodent studies. Reprod Toxicol. 2007;24(2):199–224. doi:10.1016/j.reprotox.2007.06.004.
Munoz-de-Toro M, Markey CM, Wadia PR, Luque EH, Rubin BS, Sonnenschein C, et al. Perinatal exposure to bisphenol-A alters peripubertal mammary gland development in mice. Endocrinology. 2005;146(9):4138–47. doi:10.1210/en.2005-0340.
Mori T, Bern HA, Mills KT, Young PN. Long-term effects of neonatal steroid exposure on mammary gland development and tumorigenesis in mice. J Natl Cancer Inst. 1976;57(5):1057–62.
Lamartiniere CA, Jenkins S, Betancourt AM, Wang J, Russo J. Exposure to the endocrine disruptor bisphenol A alters susceptibility for mammary cancer. Horm Mol Biol Clin Investig. 2011;5(2):45–52. doi:10.1515/HMBCI.2010.075.
Tharp AP, Maffini MV, Hunt PA, VandeVoort CA, Sonnenschein C, Soto AM. Bisphenol A alters the development of the rhesus monkey mammary gland. Proc Natl Acad Sci U S A. 2012;109(21):8190–5. doi:10.1073/pnas.1120488109.
Jenkins S, Raghuraman N, Eltoum I, Carpenter M, Russo J, Lamartiniere CA. Oral exposure to bisphenol a increases dimethylbenzanthracene-induced mammary cancer in rats. Environ Heal Perspect. 2009;117(6):910–5. doi:10.1289/ehp.11751.
Betancourt AM, Eltoum IA, Desmond RA, Russo J, Lamartiniere CA. In utero exposure to bisphenol A shifts the window of susceptibility for mammary carcinogenesis in the rat. Environ Heal Perspect. 2010;118(11):1614–9. doi:10.1289/ehp.1002148.
Doherty LF, Bromer JG, Zhou Y, Aldad TS, Taylor HS. In utero exposure to diethylstilbestrol (DES) or bisphenol-A (BPA) increases EZH2 expression in the mammary gland: an epigenetic mechanism linking endocrine disruptors to breast cancer. Horm Cancer. 2010;1(3):146–55. doi:10.1007/s12672-010-0015-9.
Jenkins S, Wang J, Eltoum I, Desmond R, Lamartiniere CA. Chronic oral exposure to bisphenol A results in a nonmonotonic dose response in mammary carcinogenesis and metastasis in MMTV-erbB2 mice. Environ Heal Perspect. 2011;119(11):1604–9. doi:10.1289/ehp.1103850.
Ogura I, Masunaga S, Nakanishi J. Quantitative source identification of dioxin-like PCBs in Yokohama, Japan, by temperature dependence of their atmospheric concentrations. Environ Sci Technol. 2004;38(12):3279–85.
Safe SH. Modulation of gene expression and endocrine response pathways by 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds. Pharmacol Ther. 1995;67(2):247–81.
Safe SH. Hazard and risk assessment of chemical mixtures using the toxic equivalency factor approach. Environ Heal Perspect. 1998;106 Suppl 4:1051–8.
Chaffin CL, Peterson RE, Hutz RJ. In utero and lactational exposure of female Holtzman rats to 2,3,7,8-tetrachlorodibenzo-p-dioxin: modulation of the estrogen signal. Biol Reprod. 1996;55(1):62–7.
Fenton SE, Hamm JT, Birnbaum LS, Youngblood GL. Persistent abnormalities in the rat mammary gland following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol Sci. 2002;67(63):63–74.
Brown NM, Manzolillo PA, Zhang JX, Wang J, Lamartiniere CA. Prenatal TCDD and predisposition to mammary cancer in the rat. Carcinogenesis. 1998;19(9):1623–9.
Franczak A, Nynca A, Valdez KE, Mizinga KM, Petroff BK. Effects of acute and chronic exposure to the aryl hydrocarbon receptor agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin on the transition to reproductive senescence in female Sprague–Dawley rats. Biol Reprod. 2006;74(1):125–30. doi:10.1095/biolreprod.105.044396.
Brown NM, Lamartiniere CA. Xenoestrogens alter mammary gland differentiation and cell proliferation in the rat. Environ Heal Perspect. 1995;103(7–8):708–13.
Gray Jr LE, Kelce WR, Monosson E, Ostby JS, Birnbaum LS. Exposure to TCDD during development permanently alters reproductive function in male Long Evans rats and hamsters: reduced ejaculated and epididymal sperm numbers and sex accessory gland weights in offspring with normal androgenic status. Toxicol Appl Pharmacol. 1995;131(1):108–18. doi:10.1006/taap.1995.1052.
Chan MY, Huang H, Leung LK. 2,3,7,8-Tetrachlorodibenzo-para-dioxin increases aromatase (CYP19) mRNA stability in MCF-7 cells. Mol Cell Endocrinol. 2010;317(1–2):8–13. doi:10.1016/j.mce.2009.11.012.
Warner M, Eskenazi B, Mocarelli P, Gerthoux PM, Samuels S, Needham L, et al. Serum dioxin concentrations and breast cancer risk in the Seveso Women’s Health Study. Environ Heal Perspect. 2002;110(7):625–8.
Boffetta P, Mundt KA, Adami HO, Cole P, Mandel JS. TCDD and cancer: a critical review of epidemiologic studies. Crit Rev Toxicol. 2011;41(7):622–36. doi:10.3109/10408444.2011.560141.
Manuwald U, Velasco Garrido M, Berger J, Manz A, Baur X. Mortality study of chemical workers exposed to dioxins: follow-up 23 years after chemical plant closure. Occup Environ Med. 2012;69(9):636–42. doi:10.1136/oemed-2012-100682.
Van den Berg M, Birnbaum LS, Denison M, De Vito M, Farland W, Feeley M, et al. The 2005 World Health Organization reevaluation of human and Mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol Sci. 2006;93(2):223–41. doi:10.1093/toxsci/kfl055.
Muto T, Wakui S, Imano N, Nakaaki K, Takahashi H, Hano H, et al. Mammary gland differentiation in female rats after prenatal exposure to 3,3′,4,4′,5-pentachlorobiphenyl. Toxicology. 2002;177(2–3):197–205.
Oenga GN, Spink DC, Carpenter DO. TCDD and PCBs inhibit breast cancer cell proliferation in vitro. Toxicol In Vitro. 2004;18(6):811–9. doi:10.1016/j.tiv.2004.04.004.
Brody JG, Moysich KB, Humblet O, Attfield KR, Beehler GP, Rudel RA. Environmental pollutants and breast cancer: epidemiologic studies. Cancer. 2007;109(12 Suppl):2667–711. doi:10.1002/cncr.22655.
LeBlanc GA. Endocrine system. In: Hodgson E, editor. A textbook of modern toxicology. 3rd ed. Hoboken: John Wiley & Sons, Inc; 2004. p. 299–315.
Registry AfTSaD, Toxicological profile for DDT, DDE, and DDD, http://www.atsdr.cdc.gov/toxprofiles/tp35.pdf.
Johnson NA, Ho A, Cline JM, Hughes CL, Foster WG, Davis VL. Accelerated mammary tumor onset in a HER2/Neu mouse model exposed to DDT metabolites locally delivered to the mammary gland. Environ Heal Perspect. 2012;120(8):1170–6. doi:10.1289/ehp.1104327.
Robison AK, Sirbasku DA, Stancel GM. DDT supports the growth of an estrogen-responsive tumor. Toxicol Lett. 1985;27(1–3):109–13.
Cohn BA, Wolff MS, Cirillo PM, Sholtz RI. DDT and breast cancer in young women: new data on the significance of age at exposure. Environ Health Perspect. 2007;115(10):1406–14. doi:10.1289/ehp.10260.
Agency for Toxic Substances and Disease Registry, Toxicological Profile for Atrazine, http://www.atsdr.cdc.gov/ToxProfiles/tp153.pdf.
Rayner JL, Fenton SE. In: Russo J, editor. Atrazine: an environmental endocrine disruptor that alters mammary gland development and tumor susceptibility environment and breast cancer. New York: Springer; 2011. p. 167–83.
US Environmental Protection Agency. Atrazine: hazard and dose–response assessment and characterization federal insecticide F, and Rodenticide Act Science Advisory Report, http://www.epa.gov/scipoly/sap/meetings/2000/june27/finalatrazine.pdf.
Foradori CD, Hinds LR, Hanneman WH, Handa RJ. Effects of atrazine and its withdrawal on gonadotropin-releasing hormone neuroendocrine function in the adult female Wistar rat. Biol Reprod. 2009;81(6):1099–105. doi:10.1095/biolreprod.109.077453.
Stoker TE, Laws SC, Guidici DL, Cooper RL. The effect of atrazine on puberty in male wistar rats: an evaluation in the protocol for the assessment of pubertal development and thyroid function. Toxicol Sci. 2000;58(1):50–9.
Stanko JP, Enoch RR, Rayner JL, Davis CC, Wolf DC, Malarkey DE, et al. Effects of prenatal exposure to a low dose atrazine metabolite mixture on pubertal timing and prostate development of male Long-Evans rats. Reprod Toxicol. 2010;30(4):540–9. doi:10.1016/j.reprotox.2010.07.006.
Enoch RR, Stanko JP, Greiner SN, Youngblood GL, Rayner JL, Fenton SE. Mammary gland development as a sensitive end point after acute prenatal exposure to an atrazine metabolite mixture in female Long-Evans rats. Environ Heal Perspect. 2007;115(4):541–7. doi:10.1289/ehp.9612.
Gammon DW, Aldous CN, Carr Jr WC, Sanborn JR, Pfeifer KF. A risk assessment of atrazine use in California: human health and ecological aspects. Pest Manag Sci. 2005;61(4):331–55. doi:10.1002/ps.1000.
Stevens JT, Breckenridge CB, Wetzel L. A risk characterization for atrazine: oncogenicity profile. J Toxicol Environ Health A. 1999;56(2):69–109.
Eldridge JC, Wetzel LT, Stevens JT, Simpkins JW. The mammary tumor response in triazine-treated female rats: a threshold-mediated interaction with strain and species-specific reproductive senescence. Steroids. 1999;64(9):672–8.
Fukamachi K, Han BS, Kim CK, Takasuka N, Matsuoka Y, Matsuda E, et al. Possible enhancing effects of atrazine and nonylphenol on 7,12-dimethylbenz[a]anthracene-induced mammary tumor development in human c-Ha-ras proto-oncogene transgenic rats. Cancer Sci. 2004;95(5):404–10.
US Environmental Protection Agency, RED Facts: Vinclozolin http://www.epa.gov/oppsrrd1/REDs/factsheets/2740fact.pdf.
Wickerham EL, Lozoff B, Shao J, Kaciroti N, Xia Y, Meeker JD. Reduced birth weight in relation to pesticide mixtures detected in cord blood of full-term infants. Environ Int. 2012;47:80–5. doi:10.1016/j.envint.2012.06.007.
Molina-Molina JM, Hillenweck A, Jouanin I, Zalko D, Cravedi JP, Fernandez MF, et al. Steroid receptor profiling of vinclozolin and its primary metabolites. Toxicol Appl Pharmacol. 2006;216(1):44–54. doi:10.1016/j.taap.2006.04.005.
Gray Jr LE, Ostby JS, Kelce WR. Developmental effects of an environmental antiandrogen: the fungicide vinclozolin alters sex differentiation of the male rat. Toxicol Appl Pharmacol. 1994;129(1):46–52. doi:10.1006/taap.1994.1227.
Kelce WR, Monosson E, Gamcsik MP, Laws SC, Gray Jr LE. Environmental hormone disruptors: evidence that vinclozolin developmental toxicity is mediated by antiandrogenic metabolites. Toxicol Appl Pharmacol. 1994;126(2):276–85. doi:10.1006/taap.1994.1117.
Anway MD, Leathers C, Skinner MK. Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology. 2006;147(12):5515–23.
El Sheikh Saad H, Meduri G, Phrakonkham P, Berges R, Vacher S, Djallali M, et al. Abnormal peripubertal development of the rat mammary gland following exposure in utero and during lactation to a mixture of genistein and the food contaminant vinclozolin. Reprod Toxicol. 2011;32(1):15–25. doi:10.1016/j.reprotox.2011.03.001.
Ozen S, Darcan S, Bayindir P, Karasulu E, Simsek DG, Gurler T. Effects of pesticides used in agriculture on the development of precocious puberty. Environ Monit Assess. 2012;184(7):4223–32. doi:10.1007/s10661-011-2257-6.
Kamrin MA. Phthalate risks, phthalate regulation, and public health: a review. J Toxicol Environ Health Part B, Crit Rev. 2009;12(2):157–74. doi:10.1080/10937400902729226.
Swan SH, Main KM, Liu F, Stewart SL, Kruse RL, Calafat AM, et al. Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ Heal Perspect. 2005;113(8):1056–61.
Rider CV, Wilson VS, Howdeshell KL, Hotchkiss AK, Furr JR, Lambright CR, et al. Cumulative effects of in utero administration of mixtures of “antiandrogens” on male rat reproductive development. Toxicol Pathol. 2009;37(1):100–13. doi:10.1177/0192623308329478.
Mylchreest E, Cattley RC, Foster PM. Male reproductive tract malformations in rats following gestational and lactational exposure to Di(n-butyl) phthalate: an antiandrogenic mechanism? Toxicol Sci. 1998;43(1):47–60. doi:10.1006/toxs.1998.2436.
Gray Jr LE, Ostby J, Furr J, Price M, Veeramachaneni DN, Parks L. Perinatal exposure to the phthalates DEHP, BBP, and DINP, but not DEP, DMP, or DOTP, alters sexual differentiation of the male rat. Toxicol Sci. 2000;58(2):350–65.
Agas D, Sabbieti MG, Capacchietti M, Materazzi S, Menghi G, Materazzi G, et al. Benzyl butyl phthalate influences actin distribution and cell proliferation in rat Py1a osteoblasts. J Cell Biochem. 2007;101(3):543–51. doi:10.1002/jcb.21212.
Dobrzynska MM, Tyrkiel EJ, Pachocki KA. Developmental toxicity in mice following paternal exposure to Di-N-butyl-phthalate (DBP). Biomed Environ Sci. 2011;24(5):569–78. doi:10.3967/0895-3988.2011.05.017.
Crinnion WJ. Toxic effects of the easily avoidable phthalates and parabens. Alternative Med Rev: J Clin Ther. 2010;15(3):190–6.
Moral R, Wang R, Russo IH, Mailo DA, Lamartiniere CA, Russo J. The plasticizer butyl benzyl phthalate induces genomic changes in rat mammary gland after neonatal/prepubertal exposure. BMC Genom. 2007;8:453. doi:10.1186/1471-2164-8-453.
Moral R, Santucci-Pereira J, Wang R, Russo IH, Lamartiniere CA, Russo J. In utero exposure to butyl benzyl phthalate induces modifications in the morphology and the gene expression profile of the mammary gland: an experimental study in rats. Environ Health. 2011;10(1):5. doi:10.1186/1476-069X-10-5.
Dewitt JC, Copeland CB, Strynar MJ, Luebke RW. Perfluorooctanoic acid-induced immunomodulation in adult C57BL/6J or C57BL/6N female mice. Environ Heal Perspect. 2008;116(5):644–50. doi:10.1289/ehp.10896.
National Toxicology Program, NTP Toxicology and Carcinogenesis Studies of Butyl Benzyl Phthalate (CAS No. 85-68-7) in F344/N Rats (Feed Studies).
Lopez-Carrillo L, Hernandez-Ramirez RU, Calafat AM, Torres-Sanchez L, Galvan-Portillo M, Needham LL, et al. Exposure to phthalates and breast cancer risk in northern Mexico. Environ Health Perspect. 2010;118(4):539–44. doi:10.1289/ehp.0901091.
Kang SC, Lee BM. DNA methylation of estrogen receptor alpha gene by phthalates. J Toxi Environ Health A. 2005;68(23–24):1995–2003. doi:10.1080/15287390491008913.
Kim IY, Han SY, Moon A. Phthalates inhibit tamoxifen-induced apoptosis in MCF-7 human breast cancer cells. J Toxic Environ Health A. 2004;67(23–24):2025–35. doi:10.1080/15287390490514750.
Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Tully JS, Needham LL. Serum concentrations of 11 polyfluoroalkyl compounds in the US population: data from the National Health and Nutrition Examination Survey (NHANES) 1999–2000. Environ Sci Technol. 2007;41:2237–42.
Calafat AM, Wong LY, Kuklenyik Z, Reidy JA, Needham LL. Polyfluoroalkyl chemicals in the U.S. population: data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000. Environ Health Perspect. 2007;115(11):1596–602.
Olsen GW, Burris JM, Ehresman DJ, Froehlich JW, Seacat AM, Butenhoff JL, et al. Half-life of serum elimination of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ Health Perspect. 2007;115(9):1298–305.
Post GB, Cohn PD, Cooper KR. Perfluorooctanoic acid (PFOA), an emerging drinking water contaminant: a critical review of recent literature. Environ Res. 2012;116:93–117. doi:10.1016/j.envres.2012.03.007.
Henry ND, Fair PA. Comparison of in vitro cytotoxicity, estrogenicity and anti-estrogenicity of triclosan, perfluorooctane sulfonate and perfluorooctanoic acid. J Appl Toxicol. 2011. doi:10.1002/jat.1736.
Lau C, Thibodeaux JR, Hanson RG, Narotsky MG, Rogers JM, Lindstrom AB, et al. Effects of perfluorooctanoic acid exposure during pregnancy in the mouse. Toxicol Sci. 2006;90(2):510–8. doi:10.1093/toxsci/kfj105.
Yang C, Tan YS, Harkema JR, Haslam SZ. Differential effect of peripubertal exposure to perfluorooctanoic acid on mammary gland development in C57Bl/6 and Balb/c mouse strains. Reprod Toxicol. 2009;27(3–4):299–306. doi:10.1016/j.reprotox.2008.10.003.
Macon MB, Villanueva LR, Tatum-Gibbs K, Zehr RD, Strynar M, Stanko JP, et al. Prenatal perfluorooctanoic acid exposure in CD-1 mice: low dose developmental effects and internal dosimetry. Toxicol Sci. 2011;122(1):134–45.
Dixon D, Reed CE, Moore AB, Gibbs-Flournoy EA, Hines EP, Wallace EA, et al. Histopathologic changes in the uterus, cervix and vagina of immature CD-1 mice exposed to low doses of perfluorooctanoic acid (PFOA) in a uterotrophic assay. Reprod Toxicol. 2012;33(4):506–12.
Biegel LB, Liu RC, Hurtt ME, Cook JC. Effects of ammonium perfluorooctanoate on Leydig cell function: in vitro, in vivo, and ex vivo studies. Toxicol Appl Pharmacol. 1995;134(1):18–25. doi:10.1006/taap.1995.1164.
Sibinski L. Two-year oral (diet) toxicity/oncogenicity study of fluorochemical FC-143 in rats.: Riker Laboratories Inc/3m Company1987.
Lou I, Wambaugh JF, Lau C, Hanson RG, Lindstrom AB, Strynar MJ, et al. Modeling single and repeated dose pharmacokinetics of PFOA in mice. Toxicol Sci. 2009;107(2):331–41. doi:10.1093/toxsci/kfn234.
Lopez-Espinosa M, Fletcher T, Armstrong B, Genser B, Dhatariya K, Mondal D, et al. Association of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS) with age of puberty among children living near a chemical plant. Environ Sci Technol. 2011. doi:10.1021/es1038694.
Knox SS, Jackson T, Javins B, Frisbee SJ, Shankar A, Ducatman AM. Implications of early menopause in women exposed to perfluorocarbons. J Clin Endocrinol Metab. 2011;96(6):1747–53. doi:10.1210/jc.2010-2401.
Innes KE, Byers TE. Preeclampsia and breast cancer risk. Epidemiology. 1999;10(6):722–32.
Bonefeld-Jorgensen EC, Long M, Bossi R, Ayotte P, Asmund G, Kruger T, et al. Perfluorinated compounds are related to breast cancer risk in Greenlandic Inuit: a case control study. Environ Health. 2011;10:88. doi:10.1186/1476-069X-10-88.
White SS, Stanko JP, Kato K, Calafat AM, Hines EP, Fenton SE. Gestational and chronic low-dose PFOA exposures and mammary gland growth and differentiation in three generations of CD-1 mice. Environ Heal Perspect. 2011;119(8):1070–6. doi:10.1289/ehp.1002741.
Frisbee SJ, Brooks ABJ, Maher A, Flensborg P, Arnold S, Fletcher T, et al. The C8 health project: design, methods, and participants. Environ Heal Perspect. 2009;117:1873–82.
Zhao Y, Tan YS, Strynar MJ, Perez G, Haslam SZ, Yang C. Perfluorooctanoic acid effects on ovaries mediate its inhibition of peripubertal mammary gland development in Balb/c and C57Bl/6 mice. Reprod Toxicol. 2012;33(4):563–76. doi:10.1016/j.reprotox.2012.02.004.
Zhao Y, Tan YS, Haslam SZ, Yang C. Perfluorooctanoic acid effects on steroid hormone and growth factor levels mediate stimulation of peripubertal mammary gland development in C57Bl/6 mice. Toxicol Sci. 2010;115(1):214–24. doi:10.1093/toxsci/kfq030.
Zhao G, Wang J, Wang X, Chen S, Zhao Y, Gu F, et al. Mutegenicity of PFOA in mammalian cells: role of mitochondtia-dependent reactive oxygem species. Environ Sci Technol. 2011;45:1638–44. doi:10.1021/es1026129.
Lipworth L, Hsieh CC, Wide L, Ekbom A, Yu SZ, Yu GP, et al. Maternal pregnancy hormone levels in an area with a high incidence (Boston, USA) and in an area with a low incidence (Shanghai, China) of breast cancer. Br J Cancer. 1999;79(1):7–12. doi:10.1038/sj.bjc.6690003.
Bray F, McCarron P, Parkin DM. The changing global patterns of female breast cancer incidence and mortality. Breast Cancer Res. 2004;6(6):229–39. doi:10.1186/bcr932.
Jefferson WN, Padilla-Banks E, Newbold RR. Disruption of the female reproductive system by the phytoestrogen genistein. Reprod Toxicol. 2007;23(3):308–16. doi:10.1016/j.reprotox.2006.11.012.
Trock BJ, Hilakivi-Clarke L, Clarke R. Meta-analysis of soy intake and breast cancer risk. J Natl Cancer Inst. 2006;98(7):459–71. doi:10.1093/jnci/djj102.
Wu AH, Wan P, Hankin J, Tseng CC, Yu MC, Pike MC. Adolescent and adult soy intake and risk of breast cancer in Asian-Americans. Carcinogenesis. 2002;23(9):1491–6.
Wu AH, Yu MC, Tseng CC, Pike MC. Epidemiology of soy exposures and breast cancer risk. Br J Cancer. 2008;98(1):9–14. doi:10.1038/sj.bjc.6604145.
Su Y, Eason RR, Geng Y, Till SR, Badger TM, Simmen RC. In utero exposure to maternal diets containing soy protein isolate, but not genistein alone, protects young adult rat offspring from NMU-induced mammary tumorigenesis. Carcinogenesis. 2007;28(5):1046–51. doi:10.1093/carcin/bgl240.
Hakkak R, Korourian S, Shelnutt SR, Lensing S, Ronis MJ, Badger TM. Diets containing whey proteins or soy protein isolate protect against 7,12-dimethylbenz(a)anthracene-induced mammary tumors in female rats. Cancer Epidemiol Biomarkers Prev. 2000;9(1):113–7.
Pei RJ, Sato M, Yuri T, Danbara N, Nikaido Y, Tsubura A. Effect of prenatal and prepubertal genistein exposure on N-methyl-N-nitrosourea-induced mammary tumorigenesis in female Sprague–Dawley rats. In Vivo. 2003;17(4):349–57.
Whitsett Jr TG, Lamartiniere CA. Genistein and resveratrol: mammary cancer chemoprevention and mechanisms of action in the rat. Expert Rev Anticancer Ther. 2006;6(12):1699–706. doi:10.1586/14737140.6.12.1699.
Fritz WA, Coward L, Wang J, Lamartiniere CA. Dietary genistein: perinatal mammary cancer prevention, bioavailability and toxicity testing in the rat. Carcinogenesis. 1998;19(12):2151–8.
Lamartiniere CA. Timing of exposure and mammary cancer risk. J Mammary Gland Biol Neoplasia. 2002;7(1):67–76.
National Toxicology Program, Multigenerational reproductive study of genistein (Cas No. 446-72-0) in Sprague–Dawley rats (feed study), 2008/08/08, http://www.ncbi.nlm.nih.gov/pubmed/18685713.
Molzberger AF, Vollmer G, Hertrampf T, Moller FJ, Kulling S, Diel P. In utero and postnatal exposure to isoflavones results in a reduced responsivity of the mammary gland towards estradiol. Mol Nutr food Res. 2012;56(3):399–409. doi:10.1002/mnfr.201100371.
Latendresse JR, Bucci TJ, Olson G, Mellick P, Weis CC, Thorn B, et al. Genistein and ethinyl estradiol dietary exposure in multigenerational and chronic studies induce similar proliferative lesions in mammary gland of male Sprague–Dawley rats. Reprod Toxicol. 2009;28(3):342–53. doi:10.1016/j.reprotox.2009.04.006.
Juan ME, Vinardell MP, Planas JM. The daily oral administration of high doses of trans-resveratrol to rats for 28 days is not harmful. J Nutr. 2002;132(2):257–60.
Levi F, Pasche C, Lucchini F, Ghidoni R, Ferraroni M, La Vecchia C. Resveratrol and breast cancer risk. Eur J Canc Prev. 2005;14(2):139–42.
Ortega I, Wong DH, Villanueva JA, Cress AB, Sokalska A, Stanley SD, et al. Effects of resveratrol on growth and function of rat ovarian granulosa cells. Fertil Steril. 2012. doi:10.1016/j.fertnstert.2012.08.004.
Henry LA, Witt DM. Effects of neonatal resveratrol exposure on adult male and female reproductive physiology and behavior. Dev Neurosci. 2006;28(3):186–95. doi:10.1159/000091916.
Nikaido Y, Yoshizawa K, Danbara N, Tsujita-Kyutoku M, Yuri T, Uehara N, et al. Effects of maternal xenoestrogen exposure on development of the reproductive tract and mammary gland in female CD-1 mouse offspring. Reprod Toxicol. 2004;18(6):803–11. doi:10.1016/j.reprotox.2004.05.002.
Provinciali M, Re F, Donnini A, Orlando F, Bartozzi B, Di Stasio G, et al. Effect of resveratrol on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Int J Cancer. 2005;115(1):36–45. doi:10.1002/ijc.20874.
Bhat KP, Lantvit D, Christov K, Mehta RG, Moon RC, Pezzuto JM. Estrogenic and antiestrogenic properties of resveratrol in mammary tumor models. Cancer Res. 2001;61(20):7456–63.
Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275(5297):218–20.
Massart F, Saggese G. Oestrogenic mycotoxin exposures and precocious pubertal development. Int J Androl. 2010;33(2):369–76. doi:10.1111/j.1365-2605.2009.01009.x.
Kuiper-Goodman T, Scott PM, Watanabe H. Risk assessment of the mycotoxin zearalenone. Regul Toxicol Pharmacol: RTP. 1987;7(3):253–306.
Hilakivi-Clarke L, Onojafe I, Raygada M, Cho E, Skaar T, Russo I, et al. Prepubertal exposure to zearalenone or genistein reduces mammary tumorigenesis. Br J Cancer. 1999;80(11):1682–8. doi:10.1038/sj.bjc.6690584.
Hilakivi-Clarke L, Cho E, Onojafe I, Raygada M, Clarke R. Maternal exposure to genistein during pregnancy increases carcinogen-induced mammary tumorigenesis in female rat offspring. Oncol Rep. 1999;6(5):1089–95.
Yuri T, Tsukamoto R, Miki K, Uehara N, Matsuoka Y, Tsubura A. Biphasic effects of zeranol on the growth of estrogen receptor-positive human breast carcinoma cells. Oncol Rep. 2006;16(6):1307–12.
Saenz de Rodriguez CA, Bongiovanni AM, Conde de Borrego L. An epidemic of precocious development in Puerto Rican children. J Pediatr. 1985;107(3):393–6.
Fara GM, Del Corvo G, Bernuzzi S, Bigatello A, Di Pietro C, Scaglioni S, et al. Epidemic of breast enlargement in an Italian school. Lancet. 1979;2(8137):295–7.
Bandera EV, Chandran U, Buckley B, Lin Y, Isukapalli S, Marshall I, et al. Urinary mycoestrogens, body size and breast development in New Jersey girls. Sci Total Environ. 2011;409(24):5221–7. doi:10.1016/j.scitotenv.2011.09.029.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclaimer
This review may be the work product of an employee or group of employees of the National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH), however, the statements, opinions or conclusions contained therein do not necessarily represent the statements, opinions or conclusions of NIEHS, NIH or the United States government
Rights and permissions
About this article
Cite this article
Macon, M.B., Fenton, S.E. Endocrine Disruptors and the Breast: Early Life Effects and Later Life Disease. J Mammary Gland Biol Neoplasia 18, 43–61 (2013). https://doi.org/10.1007/s10911-013-9275-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10911-013-9275-7