
Abstract
Recombinant tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is currently under clinical trials for cancer, however many tumor cells, including hepatocellular carcinoma (HCC) develop resistance to TRAIL-induced apoptosis. Hence, novel agents that can alleviate TRAIL-induced resistance are urgently needed. In the present report, we investigated the potential of emodin to enhance apoptosis induced by TRAIL in HCC cells. As observed by MTT cytotoxicity assay and the externalization of the membrane phospholipid phosphatidylserine, we found that emodin can significantly potentiate TRAIL-induced apoptosis in HCC cells. When investigated for the mechanism(s), we observed that emodin can downregulate the expression of various cell survival proteins, and induce the cell surface expression of both TRAIL receptors, death receptors (DR) 4 as well as 5. In addition, emodin increased the expression of C/EBP homologous protein (CHOP) in a time-dependent manner. Knockdown of CHOP by siRNA decreased the induction of emodin-induced DR5 expression and apoptosis. Emodin-induced induction of DR5 was mediated through the generation of reactive oxygen species (ROS), as N-acetylcysteine blocked the induction of DR5 and the induction of apoptosis. Also, the knockdown of X-linked inhibitor of apoptosis protein by siRNA significantly reduced the sensitization effect of emodin on TRAIL-induced apoptosis. Overall, our experimental results clearly indicate that emodin can indeed potentiate TRAIL-induced apoptosis through the downregulation of antiapoptotic proteins, increased expression of apoptotic proteins, and ROS mediated upregulation of DR in HCC cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is the most common primary liver cancer, with an estimated incidence of half a million new cases per year around the world [1, 2]. Although surgery remains to be the first choice for HCC treatment, tumor size, hepatic functional reserve and/or portal hypertension may all restrict surgical ablation [3]. First line drugs used for HCC include doxorubicin, fuorouracil, cisplatin, and mitomycin, but most of these are non-selective cytotoxic molecules that exhibit significant adverse effects [4, 5]. Thus, new therapeutic strategies that are safe and effective are urgently needed.
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is currently being tested as therapeutic agent for HCC treatment, but its utility is greatly hampered by the fact tumor cells ultimately develop resistant to the TRAIL-induced apoptosis [6, 7]. The exact mechanism(s) responsible for resistance to TRAIL is still not completely understood; however, susceptibility to TRAIL-induced apoptosis can be regulated at several levels in the apoptotic signaling cascades [8–10]. For instance, dysfunction of DR4 and DR5, overexpression of antiapoptotic proteins, and loss of proapoptotic proteins has all been linked with TRAIL resistance. Activation of mitogen-activated protein kinases [11], PI3 K/AKT [12], nuclear factor-kappa B [13, 14], and signal transducer and activator of transcription 3 [15] are also reported to mediate TRAIL-induced resistance in HCC cells. Thus, identification of novel sensitizers capable of modulating the expression and/or activity levels of these apoptotic regulators may be an effective strategy for overcoming TRAIL resistance in tumor cells.
In the current report, we investigated whether emodin (1,3,8-trihydroxy-6-methylanthraquinone), an active component found in the root and rhizome of Rheum palmatum L. can potentiate TRAIL-induced apoptosis in HCC cells. Emodin has been previously reported to possess antiviral [16], anti-inflammatory [17], antiulcerogenic [18], immunosuppressive [19], and chemopreventive activities [20]. Interestingly, antiproliferative effects of emodin have been observed in many tumor cell lines, including HER2/neu-overexpressing breast cancer [21], lung cancer [22], leukemia [23], HCC [24], and neuroblastoma [25] but not in normal cells, suggesting that emodin may have a significant therapeutic efficacy as an anticancer agent. Emodin exerts its pleiotropic anti-cancer effects through diverse mechanisms including the activation of caspase-3 [23] and upregulation of p53 and p21 [26]. Moreover, emodin has been reported to inhibit the kinase activity/activation of p56lck, HER2/neu [21], casein kinase [27], NF-κB [28], activator protein 1 [29, 30], AKT [30], matrix metalloproteinases [29, 31], and the expression of chemokine receptor CXCR4 [32]. Our findings specifically in HCC cells clearly indicate that this quinone can enhance TRAIL-induced apoptosis through the downregulation of antiapoptotic proteins, upregulation of death receptors and the activation of C/EBP homologous protein (CHOP).
Materials and methods
Reagents and antibodies
A 50 mM solution of emodin (from Aldrich), with purity of 99 %, was prepared in DMSO, stored as small aliquots at −20 °C, and then diluted further in cell culture medium as needed. Soluble recombinant human TRAIL/Apo2L was purchased from PeproTech. Penicillin, streptomycin, Dulbecco’s modified Eagle medium (DMEM), minimum essential modified (MEM) medium, fetal bovine serum and 5-(and-6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) were purchased from Invitrogen. β-Actin antibody, N-acetylcysteine (NAC), and glutathione (GSH) were obtained from Sigma-Aldrich. Antibodies against Bcl-xL, Bcl-2, Bax, cFLIP, survivin, XIAP, DR4, DR5, DcR1, DcR2, CHOP, and annexin V staining kit were purchased from Santa Cruz Biotechnology. Bid antibody was purchased from Cell Signaling Technology (Beverly, MA, USA). High purity control (scrambled RNA), CHOP and XIAP small interfering RNAs (siRNAs) were purchased from Santa Cruz Biotechnology. DR5 plasmid was obtained from Addgene and has been described previously [33].
Cell lines
Human HCC cell line HepG2 was obtained from American Type Culture Collection (Manassass, VA, USA). Hep3B cells were kindly provided by Professor Kam Man Hui at National Cancer Centre, Singapore. HepG2 cells were cultured in DMEM containing 1× antibiotic–antimycotic solution with 10 % FBS. Hep3B cells were cultured in MEM containing 1× Antibiotic–antimycotic solution with 10 % FBS. The primary normal liver cells THLE-2 were kindly provided by Dr. Ho Han Kiat, Department of Pharmacy, National University of Singapore. The cells were cultured in LHC-9 media from Invitrogen supplemented with 5 ng/ml epidermal growth factor and 10 % fetal bovine serum and grown in fibronectin coated culture dishes from BD Biosciences (Heidelberg, Germany, Cat. # 354403).
Live/dead assay
Apoptosis of cells was also determined by Live/Dead assay (Molecular Probes, Eugene, OR, USA) that measures intracellular esterase activity and plasma membrane integrity as described previously [34]. Cell viability was quantified under a fluorescence microscope by counting live (green) and dead (red) cells in twenty random fields.
Propidium iodide (PI) staining for flow cytometry
Cells were pretreated with emodin (10 μM) for 16 h and then exposed to TRAIL (50 ng/ml) for 24 h. Thereafter cells were washed, fixed with 70 % ethanol, and incubated for 30 min at 37 °C with 0.1 % RNase A in PBS. Cells were then washed again, resuspended, and stained in PBS containing 25 μg/ml propidium iodide (PI) for 30 min at room temperature. Cell distribution across the cell cycle was analyzed with a (BD FACSCalibur, BD Biosciences, US).
Cytotoxicity assay
The effects of emodin on TRAIL-induced cytotoxicity were determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) uptake method as described previously [34].
Annexin V/PI assay
To identify apoptosis, we stained cells with annexin V antibody conjugated with FITC fluorescence dye. Briefly, 5 × 105 cells were treated with emodin, TRAIL, or combination of both and then stained with Annexin V-FITC conjugate. Cells were washed in PBS, resuspended in 100 μl of binding buffer containing FITC-conjugated anti-annexin V antibody, and then analyzed by flow cytometer (BD FACSCalibur, BD Biosciences, US).
Analysis of cell surface expression of DR4 and DR5
Cells were analyzed for the surface expression of DR4 and DR5 by indirect staining with primary mouse monoclonal anti-human DR4 and DR5 (Santa Cruz Biotechnology) followed by FITC-conjugated mouse anti-goat IgG. Briefly, 4 × 105 cells were stained with 200 μl PBS containing saturating amounts of anti-DR4 or anti-DR5 antibody on ice for 30 min.
After incubation, cells were washed twice and reacted with FITC conjugated mouse anti-goat IgG on ice for 30 min. After washing with PBS, the expressions of these death receptors were analyzed by a FACS sorter (BD FACSCalibur, BD Biosciences, US) with FITC conjugated normal mouse IgG as an isotype control.
Western blot analysis
To determine the levels of protein expression, we prepared whole-cell extracts and analyzed by Western blot as described previously [34].
Transfection experiments with CHOP/XIAP siRNA and DR5 plasmid
HepG2 cells were plated in each well of six-well plates and allowed to adhere for 24 h. On the day of transfection, 4 μl of lipofectamine Invitrogen (Carlsbad, CA, USA) was added to 50 nM siRNA in a final volume of 100 μl of culture medium. After 48 h of transfection, cells were treated with emodin for 16 h and then exposed to TRAIL for 24 h. For DR5 overexpression, HepG2 cells were transfected with DR5 and/or pcDNA (control) plasmids and thereafter incubated for 48 h. The cells were then treated with TRAIL (0, 10 and 50 ng/ml) for 4 h, followed by western blot analysis to analyze PARP cleavage.
RNA isolation and reverse transcription
Total cellular RNA was extracted from untreated and emodin treated cells using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) as described previously [34]. For real time PCR, 100 ng/μl of total RNA was transcribed as described above. For a 50 μl reaction, 10 μl of RT product was mixed with 1× TaqMan® Universal PCR Master mix, 2.5 μl of 20× TaqMan probes for DR4 and DR5 respectively, 2.5 μl of 20× HuGAPDH TaqMan probe as the endogenous control for each targeting gene, and topped up to 50 μl with sterile water. Primers are as follows: DR5 sense 5′-AAGACCCTTGTGCTCGTTGTC-3′, DR5 antisense 5′-GACACATTCGATGTCACTCCA-3′, DR4 sense 5′-CTGAGCAACGCAGACTCGCTGTCCAC-3′, DR4 antisense 5′-TCCAAGGACACGGCAGAGCCTGTGCCAT-3′, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) sense, 5′-GTCTTCACCACCATGGAG-3′, and GAPDH antisense 5′-CCACCCTGTTGCTGTAGC-3′.
Measurement of ROS
Cells were incubated with emodin with or without NAC for 16 h and thereafter treated with 5 μM 5-(and-6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) dye (Invitrogen). After 30 min of incubation, stained samples were then analyzed by flow cytometry (BD FACSCalibur, BD Biosciences, US) with excitation set at 488 nm (FL-1) and cell counter set at 10,000 events. Subsequent data analysis was done by using the WinMDI 2.9 software.
Statistical analysis
Statistical significance was determined using unpaired Student’s t test and the probability (p) value <0.05 were considered statistically significant.
Results
Emodin enhances TRAIL-induced apoptosis
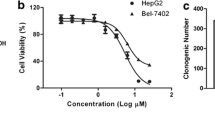
The chemical structure of emodin is shown in Fig. 1a. We first examined the effect of emodin on TRAIL-induced cytotoxicity by using the MTT method, which detects mitochondrial activity. For these experiments, we used HepG2, and Hep3B cell lines. Cells were treated with different doses of emodin and TRAIL alone or in combination for 24 h. As shown in Fig. 1b, after treatment with TRAIL (50 or 100 ng/ml) or emodin (0, 2.5, 5, 10 and 25 μM) for 24 h, cell viability was only slightly decreased. Instead, emodin/TRAIL combined treatments resulted in a marked reduction in cell viability, which was observed under conditions of a fixed emodin concentration and varied TRAIL concentrations or vice versa. Time-dependent experiments indicated that the combination of emodin (10 μM) and TRAIL (50 ng/ml) clearly reduced HepG2 cell viability and after 24 h, cell viability diminished by approximately 60 % (Fig. 1c). When apoptosis was examined using annexin V/PI staining, we found that 7.2 % apoptosis was induced by emodin alone, 9.8 % by TRAIL alone, and 35.2 % by the combination of the two (Fig. 1d). To further confirm the effect of emodin on TRAIL-induced apoptosis, we also analyzed the apoptosis by FACS analysis of the sub-G1 fraction. The results indicated that the emodin and TRAIL treatment alone induced 6.6 and 21.6 % apoptosis, respectively. Combination treatment with both emodin and TRAIL enhanced apoptosis to 50.9 % (Fig. 1e). Interestingly, we also found that emodin neither alone nor in combination with TRAIL had any significant effect on the viability of primary normal liver THLE-2 cells, thereby clearly indicating selective toxicity of emodin plus TRAIL combination towards tumor cells (Fig. 1f). Also, based on the isobologram-illustrated results, we found that emodin synergistically enhanced the cytotoxicity of HCC cells when used in combination with TRAIL (Fig. 1g). Together, these results clearly indicate that emodin can significantly enhance TRAIL-induced apoptosis in HCC cells.


Emodin potentiated TRAIL-induced apoptosis of HCC cells. a Chemical structure of emodin. b The effect of emodin and/or TRAIL on HepG2, and Hep3B cell viability. Cells were treated with emodin and/or TRAIL for 24 h at the indicated concentrations. c Time-dependent effect of emodin and/or TRAIL on HepG2 cell viability. Cell viability was estimated by MTT assay as reported under “Materials and methods” and expressed as the percentage of control value. The data represents the means of three independent experiments involving triplicate assays. ***p < 0.001 compared to control. d HepG2 cells were pretreated with 10 μM emodin for 16 h. The cells were then exposed to TRAIL (50 ng/ml) for 24 h. Cells were stained with PI/annexin V and then analyzed by FACS. e HepG2 cells were pretreated with 10 μM emodin for 16 h. The cells were then exposed to TRAIL (50 ng/ml) for 24 h. Cells were stained with PI and then analyzed by FACS. f The effect of emodin and/or TRAIL on cell viability on the primary normal liver THLE-2 cells. Cells were treated with emodin (10 μM) and/or TRAIL (50 ng/ml) for 48 h and cell viability was determined by MTT assay as reported under “Materials and methods”. g Assessment on the type of combination relationship on the effect of emodin and/or TRAIL on HepG2 viability was done using the method developed by Chou-Talalay multiple drug effect equation. The degree of synergy was assessed based on CI values, where CI values of >1.0 implies antagonism, 1.0 implies additive and <1.0 implies synergistic effect relationships
Emodin down-modulates the expression of antiapoptotic proteins linked to TRAIL resistance
How emodin enhances TRAIL-induced apoptosis was investigated next. First, cells were treated with different concentrations of emodin for 16 h and then examined for expression of antiapoptotic proteins using relevant antibodies. The results indicated that emodin down-regulated the expression of XIAP, cFLIP (both long and short form), survivin, Bcl-2, and Bcl-xL (Fig. 2a).

Emodin modulates antiapoptotic protein expression. a HepG2 cells were treated with the indicated doses of emodin for 16 h. Whole cell extracts were prepared and analyzed by Western blotting using relevant antibodies against antiapoptotic proteins. The blots were also probed with β-actin antibody to verify equal protein loading. b HepG2 cells were treated with the indicated doses of emodin for 16 h; whole cell extracts were prepared, and Western blotting performed using antibodies against pro-apoptotic proteins. The blots were also probed with β-actin antibody to verify equal protein loading
Emodin upregulates the expression of Bax and modulates the expression of Bid
Whether emodin can modulate the expression of pro-apoptotic proteins was also examined. We found that emodin up-regulated the expression of Bax and decreased the expression of full length Bid in a dose-dependent manner in HepG2 cells (Fig. 2b). Also, we noticed that emodin treatment caused an increase in the level of cleaved Bid in HepG2 cells (Fig. 2b).
Emodin induces the expression of TRAIL receptors DR4 and DR5 in HCC cells
To explore the underlying mechanism that may be responsible for enhancement of TRAIL-induced apoptosis by emodin, we examined the effect of emodin on the expression of death receptors. Emodin induced both DR4 and DR5 in a dose-dependent manner in HepG2 cells (Fig. 3a). Whether this induction of the DRs was dependent on time was also examined. We noticed that emodin induced both DR4 and DR5 in a time-dependent manner in HepG2 cells with maximum induction observed at 16 and 24 h (Fig. 3b). Interestingly, we did not observe a substantial increase in DR4 and DR5 expression upon emodin treatment in Hep3B cells which has p53 mutation (Fig. 3c), thereby indicating that p53 may have a role in the induction of DRs by emodin. Next we determined whether emodin can modulate the expression of decoy receptors (DcR). We found that while emodin had no substantial effect on the expression of DcR1, it decreased the expression of DcR2 (Fig. 3d). Thus inhibition of DcR2 expression by emodin could also contribute to apoptosis by TRAIL. We also investigated whether emodin can induce cell surface expression of TRAIL receptors in HCC cells. We found that emodin increased the cell surface expression of both DR5 and DR4 in HepG2 cells (Fig. 3e). The level of DR4 cell surface expression induced by emodin was almost equal to that for DR5. Whether TRAIL receptors are induced by emodin at the transcriptional level was investigated by real time PCR analysis. Emodin significantly upregulated both DR4 and DR5 mRNA expression in a time-dependent manner in HepG2 cells (Fig. 3f).

Emodin up-regulates DR4 and DR5 expression. a HepG2 cells were treated with the indicated dose of emodin for 16 h. Whole cell extracts were then prepared and analyzed for DR4 DR5 expression by Western blotting. b For time-dependent assessment, HepG2 cells were treated with 25 μM emodin for indicated times and analyzed for DR4 and DR5 expression. c Hep3B cells were treated with the indicated dose of emodin for 16 h. Whole cell extracts were then prepared and analyzed for DR4 and DR5 expression by Western blotting. d HepG2 cells were pretreated with the indicated doses of emodin for 16 h. Whole-cell extracts were prepared and subjected to Western blotting for DcR1, DcR2 and β-actin. e Cell surface expression of DR4 and DR5 was measured by flow cytometry on HepG2 cells following treatment with 25 μM emodin for 16 h using anti-DR4 and anti-DR5 antibodies. f Emodin induces DR4 and DR5 mRNA expression. HepG2 cells were treated with 25 μM emodin for indicated times, total RNA was extracted and examined for expression of DR4 and DR5 using real time PCR analysis. GAPDH was used as an internal control to show equal RNA loading. The experiments were repeated thrice and asterisk indicates p < 0.05
Emodin-induced up-regulation of DR5 is mediated through induction of CHOP
Because CHOP has been linked with the up-regulation of DR5 expression [35]; we next examined the role of CHOP in emodin-induced DR5 up-regulation. Our results showed that emodin induced the expression of CHOP, with optimal induction occurring around 16–24 h (Fig. 4a).

Up-regulation of DR5 by emodin requires CHOP. a HepG2 cells were treated with 25 μM emodin for indicated times, and whole cell lysates were subjected to Western blotting analysis using CHOP and β-actin antibodies. b HepG2 cells were transfected with either CHOP siRNA or control siRNA. After 48 h of transfection, cells were treated with 25 μM emodin. After 16 h, whole-cell extracts were prepared and analyzed by Western blotting using CHOP, DR5, DR4, and β-actin antibodies. c HepG2 cells were pretreated with 10 μM emodin for 16 h. The cells were then exposed to TRAIL (50 ng/ml) for 24 h. Cell death was determined using the live/dead cell viability assay. d HepG2 cells were transfected with pcDNA3-DR5 plasmid or pcDNA (control). After 48 h, the cells were treated with TRAIL (0, 10 and 50 ng/ml) for 4 h, and subjected to western blot analysis was done to analyze PARP cleavage
Knockdown of CHOP using specific siRNA abolishes emodin-induced up-regulation of DR5 expression and its effect on TRAIL-induced apoptosis
To elucidate the functional role of CHOP in emodin-induced up-regulation of death receptors, CHOP siRNA was used. Whereas DR5 was up-regulated by emodin in non-transfected and control-transfected cells, transfection with CHOP siRNA significantly abrogated the up-regulation of DR5 (Fig. 4b). CHOP siRNA had no effect on emodin-induced DR4 expression (Fig. 4b). We also examined whether the suppression of CHOP by siRNA could abrogate the effects of emodin on TRAIL-induced apoptosis using an esterase-staining assay (the live/dead assay). Emodin had substantially reduced effects (from 34 to 22 %) on TRAIL-induced apoptosis in cells transfected with CHOP siRNA, whereas treatment with control siRNA had no effect (Fig. 4c). These results indicate that CHOP-dependent DR5 up-regulation contributes to the sensitizing effect of emodin on TRAIL-induced-apoptosis.
Effect of ectopic expression of DR5 on TRAIL-induced apoptosis in HCC cells
To characterize the functional significance of DR5 upregulation in TRAIL-induced apoptosis, we used a DR5 expression vector to induce ectopic expression of DR5 in HCC cells. HepG2 cells transfected with the control plasmid (pcDNA) or pcDNA3-DR5 were treated with TRAIL (0, 10 and 50 ng/ml) and then subjected to western blot analysis. As shown in Fig. 4d, overexpression of DR5 in HepG2 cells enhanced TRAIL-induced apoptosis compared with cells transfected with pcDNA alone as evident by the cleavage of PARP. Overall, the result clearly indicates that potentiation of TRAIL-induced apoptosis by emodin can be mediated through the upregulation of DR5 in HCC cells.
Emodin-induced up-regulation of CHOP and DR5 requires ROS
How emodin induces CHOP-DR5 was further investigated. The critical role of ROS in TRAIL-induced apoptosis and in the induction of DR5 by various agents has been demonstrated previously [36]. To understand the roles of ROS in emodin-induced DR5 expression, we examined whether emodin can induce ROS production. As shown in Fig. 5a, treatment with emodin increased the production of ROS. This induction of ROS generation was markedly reversed by pretreatment with ROS scavenger NAC. Whether ROS production is needed for expression of DR5 by emodin, was determined next. For this, we pretreated cells with various concentrations of NAC and then exposed them to emodin for 16 h. We found that emodin significantly up-regulated DR5 expression, whereas pretreatment of NAC blocked emodin-induced DR5 protein expression substantially (Fig. 5b). Glutathione, a thiol antioxidant also abrogated the effect of emodin on DR5 expression (Fig. 5c). We also determined next whether ROS production mediates emodin-induced CHOP induction. For this, cells were exposed to various concentration of NAC or GSH for 1 h, and then treated with emodin for a further 16 h. As shown in Fig. 5d, emodin markedly up-regulated CHOP expression, whereas the pretreatment with NAC or GSH blocked the emodin-induced expression of CHOP, thus indicating that ROS generation is critical for emodin-elicited CHOP expression.

Emodin-induced ROS is involved in CHOP induction leading to DR5 up-regulation. a HepG2 cells were treated with 25 μM emodin with or without 10 mM NAC. 16 h later, Intracellular ROS levels were measured by flow cytometry using 5-(and-6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA), as described under “Materials and methods”. b, c HepG2 cells were pretreated with various concentrations of NAC or GSH for 1 h and then treated with 25 μM emodin for 16 h. Whole cell extracts were prepared and analyzed by Western blotting using DR5 and β-actin antibodies. d HepG2 cells were pretreated with various concentrations of NAC or GSH for 1 h and then treated with 25 μM emodin for 16 h. Whole cell extracts were prepared and analyzed by Western blotting using CHOP and β-actin antibodies. e HepG2 cells were pretreated with 10 mM NAC for 1 h and then treated with 25 μM emodin for 16 h. The cells were then exposed to TRAIL (50 ng/ml) for 24 h. Cell death was determined using the live/dead cell viability assay
Blockage of ROS production reversed effect of emodin on TRAIL-induced apoptosis
We next examined whether scavenging of ROS could attenuate the TRAIL-induced apoptosis potentiated by emodin. As shown in Fig. 5e, emodin substantially enhanced TRAIL-induced apoptosis in HepG2 cells, and pretreatment of cells with NAC markedly reduced this emodin-induced enhancement from 36 to 25 %. These results suggest that ROS is indeed needed for the sensitization of cells to TRAIL by emodin.
Downregulation of XIAP attenuates sensitization potential of emodin on TRAIL-induced apoptosis in HCC cells
To evaluate the role of XIAP in emodin-mediated TRAIL sensitization, we treated XIAP siRNA-transfected cells with emodin and TRAIL. In control siRNA transfected cells, emodin significantly enhanced TRAIL-induced apoptosis in a time dependent manner. However, in XIAP siRNA-transfected cells, the sensitizing potential of emodin was comparatively less than observed in the control siRNA transfected cells (Fig. 6). These results demonstrate that silencing XIAP dramatically attenuates emodin-mediated sensitization of TRAIL-induced apoptosis, thereby suggesting that XIAP is an important target involved in TRAIL sensitization by emodin in HCC cells.

Downregulation of XIAP attenuated sensitization effect of emodin on TRAIL-induced apoptosis. Cells transfected with either control siRNA or XIAP siRNA were first treated with 10 μM emodin for 16 h and then exposed to TRAIL (50 and 100 ng/ml) for the indicated time intervals. Cell viability was estimated by MTT assay as reported under “Materials and methods” and expressed as the percentage of control value
Discussion
TRAIL ligand is currently considered as a highly promising candidate for cancer therapy, because it induces apoptosis specifically in malignant cells without exhibiting any substantial cytotoxicity towards normal cells [9]. Considerable number of tumor cells, including HCC cells, exhibit resistance to the proapoptotic effect of TRAIL, but their sensitivity to TRAIL can be increased by combining TRAIL with several compounds, including chemotherapeutic agents, proteasome inhibitors and HDAC inhibitors [37–40]. In the present report, we conclusively demonstrate that emodin, a methyl-anthraquinone can sensitize HCC cells to TRAIL-induced apoptosis.
When we investigated the molecular mechanism(s), we found that emodin-induced upregulation of death receptors and downregulation of antiapoptotic proteins. We observed that emodin treatment can induce increase in the protein and mRNA levels of both DR5 and DR4 in a time-dependent manner in HepG2 cells. However, we did not notice a substantial increase in DR4 and DR5 expression upon emodin treatment in Hep3B cells which has p53 mutation, thereby indicating that p53 may have a role in the induction of either DR by emodin. We also demonstrated the upregulation of expression of cell surface death receptors by flow cytometric analysis upon emodin treatment of HCC cells. Moreover, we noticed that the knockdown of DR5 and DR4 by their respective siRNAs effectively inhibited the cell death induced by the combination of emodin and TRAIL, demonstrating the pivotal role of death receptors in the apoptotic process (data not shown). These results are consistent to a previous reported in which another chemopreventive agent, zerumbone was found to enhance TRAIL-induced apoptosis through induction of death receptors in colorectal cancer cells [41]. Our results are also partially in agreement with another study by Kim and coworkers who have previously reported that the inhibition of casein kinase 2 by emodin can sensitize HCC cells to both Fas ligand- and TRAIL-induced apoptosis and also increased the NK cell-mediated tumor cell killing [42]. However, the complete molecular mechanism(s) by which emodin can enhance the apoptotic effects of TRAIL were not elucidated in this report.
In addition to DR4 and DR5, other death receptors called DcR are also involved in the TRAIL-induced apoptotic signaling pathway [43]. While DcR1 prevents the assembly of the death-inducing signaling complex (DISC) by titrating TRAIL within lipid rafts, DcR2 is co-recruited with DR5 within the DISC, where it inhibits initiator caspase activation [44]. We further observed that emodin reduced the expression of DcR2 which makes the availability of ligand for DR4 and DR5 for induction of apoptosis, without affecting the expression level of DcR1 in HCC cells. Another potential mechanism by which emodin can sensitize HCC cells to TRAIL-mediated apoptosis seems to be related to the decrease in the level of various survival proteins. We found that emodin down-regulated the expression of cFLIP, an inhibitor of caspase-8 [45]. Both the short and long forms of cFLIP were equally modulated on emodin treatment. Moreover, the down-regulation of cFLIP by the peroxisome proliferator-activated receptor (PPAR)-γ agonist rosiglitazone has been shown to sensitize human renal cancer cells to TRAIL [46]. We also found that emodin substantially down-regulated the expression of Bcl-2, Bcl-xL and survivin in HCC cells. All these proteins have been linked to TRAIL resistance [47]. Recently it was reported that a synthetic cannabinoid, WIN 55,212-2 can also sensitize HCC cells to apoptosis mediated by TRAIL through downregulation of survival proteins, survivin, c-inhibitor of apoptosis protein 2, and Bcl-2 [40]. Additionally, we found dose-dependent down-regulation in XIAP expression upon emodin treatment and the knockdown of XIAP by siRNA abrogated the sensitization effect of emodin on TRAIL-induced apoptosis in HCC cells. Similarly embelin, a natural XIAP inhibitor [48] has been found to enhance TRAIL-induced apoptosis in pancreatic cancer cells [49].
However, how these survival proteins are down-regulated by emodin is not clearly understood. Interestingly, most of the antiapoptotic proteins described above are regulated by pro-inflammatory transcription factor NF-κB [50]. Because emodin has been previously reported to inhibit NF-κB activation [28], it is possible that down-regulation of expression of these proteins is probably due to suppression of NF-κB activation. Also, we have observed that emodin can exert its anticancer effects through the suppression of signal transducer and activator of transcription 3 in HCC cells (unpublished findings) which may also account for its inhibitory effects on various cell survival proteins. When we investigated for other potential mechanisms, we found that emodin substantially up-regulated the expression of Bax and modulated the expression of Bid. The former has been implicated to play a key role in TRAIL-induced apoptosis [51]. Thus the modulation of Bax and Bid expression by emodin could also possibly contribute to TRAIL-induced apoptosis.
Our data further revealed that induction of TRAIL receptors by emodin is mediated through the induction of CHOP. CHOP is a pivotal transcription factor involved in apoptosis induced by diverse stimuli including, DNA damage and chemotherapeutic agents [35]. We observed that emodin treatment induced the expression of CHOP and the knockdown of CHOP gene using the siRNA inhibited the effect of emodin on the induction of death receptors and on the TRAIL-induced apoptosis, thereby implicating the critical role of CHOP in mediating the apoptotic effects of emodin. It has also been reported that oxidative stress plays a key role as a mediator of cell death [52]. ROS generation has been proposed to be involved in the upregulation of DR5 by numerous chemopreventive agents, including curcumin [53] and guggulsterone [54]. In the current study, we also observed that emodin induces the upregulation of DR5 through the production of ROS. The antioxidant NAC abolished the upregulation of DR5 induced by emodin. Therefore, ROS generation is important for emodin-induced DR5-mediated apoptosis in cancer cells. Also, the pivotal role of DR5 upregulation by emodin in TRAIL-induced apoptosis was confirmed by our data that ectopic overexpression of DR5 can substantially augment cell death induced by TRAIL.
Overall, our data provides the mechanistic evidence that emodin treatment results in potentiation of TRAIL-induced apoptosis through CHOP and ROS-mediated upregulation of DR5, thus rendering HCC cells more sensitive to the cytotoxic effects of TRAIL. Considering that there are only limited treatment options available for HCC and emodin exhibits potent anticancer activities against a wide variety of tumors, additional pre-clinical studies are needed to realize the full potential of this novel drug combination.
Abbreviations
- TRAIL:
-
Tumor necrosis factor-related apoptosis-inducing ligand
- HCC:
-
Hepatocellular carcinoma
- DR:
-
Death receptors
- GSH:
-
Glutathione
- ROS:
-
Reactive oxygen species
- CHOP:
-
C/EBP homologous transcription factor
- NAC:
-
N-acetylcysteine
References
Yang JD, Roberts LR (2010) Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol 7:448–458
Siegel R, Naishadham D, Jemal A (2012) Cancer statistics, 2012. CA Cancer J Clin 62:10–29
Llovet JM, Burroughs A, Bruix J (2003) Hepatocellular carcinoma. Lancet 362:1907–1917
Avila MA, Berasain C, Sangro B, Prieto J (2006) New therapies for hepatocellular carcinoma. Oncogene 25:3866–3884
Del Pozo AC, Lopez P (2007) Management of hepatocellular carcinoma. Clin Liver Dis 11:305–321
Chen KF, Yeh PY, Hsu C et al (2009) Bortezomib overcomes tumor necrosis factor-related apoptosis-inducing ligand resistance in hepatocellular carcinoma cells in part through the inhibition of the phosphatidylinositol 3-kinase/Akt pathway. J Biol Chem 284:11121–11133
Newsom-Davis T, Prieske S, Walczak H (2009) Is TRAIL the holy grail of cancer therapy? Apoptosis 14:607–623
Wang S (2008) The promise of cancer therapeutics targeting the TNF-related apoptosis-inducing ligand and TRAIL receptor pathway. Oncogene 27:6207–6215
Abdulghani J, El-Deiry WS (2010) TRAIL receptor signaling and therapeutics. Expert Opin Ther Targets 14:1091–1108
Jacquemin G, Shirley S, Micheau O (2010) Combining naturally occurring polyphenols with TNF-related apoptosis-inducing ligand: a promising approach to kill resistant cancer cells? Cell Mol Life Sci 67:3115–3130
Moon DO, Kim MO, Choi YH, Kim GY (2010) Butein sensitizes human hepatoma cells to TRAIL-induced apoptosis via extracellular signal-regulated kinase/Sp1-dependent DR5 upregulation and NF-kappaB inactivation. Mol Cancer Ther 9:1583–1595
Lalaoui N, Morle A, Merino D et al (2011) TRAIL-R4 promotes tumor growth and resistance to apoptosis in cervical carcinoma HeLa cells through AKT. PLoS One 6:e19679
Plantivaux A, Szegezdi E, Samali A, Egan L (2009) Is there a role for nuclear factor kappaB in tumor necrosis factor-related apoptosis-inducing ligand resistance? Ann NY Acad Sci 1171:38–49
Kim JY, Lee JY, Kim DG, Koo GB, Yu JW, Kim YS (2011) TRADD is critical for resistance to TRAIL-induced cell death through NF-kappaB activation. FEBS Lett 585:2144–2150
Chen KF, Chen HL, Liu CY et al (2012) Dovitinib sensitizes hepatocellular carcinoma cells to TRAIL and tigatuzumab, a novel anti-DR5 antibody, through SHP-1-dependent inhibition of STAT3. Biochem Pharmacol 83:769–777
Lin CW, Wu CF, Hsiao NW et al (2008) Aloe-emodin is an interferon-inducing agent with antiviral activity against Japanese encephalitis virus and enterovirus 71. Int J Antimicrob Agents 32:355–359
Park MY, Kwon HJ, Sung MK (2009) Evaluation of aloin and aloe-emodin as anti-inflammatory agents in aloe by using murine macrophages. Biosci Biotechnol Biochem 73:828–832
Goel RK, Das Gupta G, Ram SN, Pandey VB (1991) Antiulcerogenic and anti-inflammatory effects of emodin, isolated from Rhamnus triquerta wall. Indian J Exp Biol 29:230–232
Liu YX, Shen NY, Liu C, Lv Y (2009) Immunosuppressive effects of emodin: an in vivo and in vitro study. Transpl Proc 41:1837–1839
Koyama J, Morita I, Tagahara K et al (2002) Chemopreventive effects of emodin and cassiamin B in mouse skin carcinogenesis. Cancer Lett 182:135–139
Wang SC, Zhang L, Hortobagyi GN, Hung MC (2001) Targeting HER2: recent developments and future directions for breast cancer patients. Semin Oncol 28:21–29
Su YT, Chang HL, Shyue SK, Hsu SL (2005) Emodin induces apoptosis in human lung adenocarcinoma cells through a reactive oxygen species-dependent mitochondrial signaling pathway. Biochem Pharmacol 70:229–241
Chen YC, Shen SC, Lee WR et al (2002) Emodin induces apoptosis in human promyeloleukemic HL-60 cells accompanied by activation of caspase 3 cascade but independent of reactive oxygen species production. Biochem Pharmacol 64:1713–1724
Wang XD, Gu LQ, Wu JY (2007) Apoptosis-inducing activity of new pyrazole emodin derivatives in human hepatocellular carcinoma HepG2 cells. Biol Pharm Bull 30:1113–1116
Lu HF, Lai KC, Hsu SC et al (2009) Involvement of matrix metalloproteinases on the inhibition of cells invasion and migration by emodin in human neuroblastoma SH-SY5Y cells. Neurochem Res 34:1575–1583
Shieh DE, Chen YY, Yen MH, Chiang LC, Lin CC (2004) Emodin-induced apoptosis through p53-dependent pathway in human hepatoma cells. Life Sci 74:2279–2290
Battistutta R, Sarno S, De Moliner E, Papinutto E, Zanotti G, Pinna LA (2000) The replacement of ATP by the competitive inhibitor emodin induces conformational modifications in the catalytic site of protein kinase CK2. J Biol Chem 275:29618–29622
Kumar A, Dhawan S, Aggarwal BB (1998) Emodin (3-methyl-1,6,8-trihydroxyanthraquinone) inhibits TNF-induced NF-kappaB activation, IkappaB degradation, and expression of cell surface adhesion proteins in human vascular endothelial cells. Oncogene 17:913–918
Huang Q, Shen HM, Ong CN (2004) Inhibitory effect of emodin on tumor invasion through suppression of activator protein-1 and nuclear factor-kappaB. Biochem Pharmacol 68:361–371
Brown M, Bellon M, Nicot C (2007) Emodin and DHA potently increase arsenic trioxide interferon-alpha-induced cell death of HTLV-I-transformed cells by generation of reactive oxygen species and inhibition of Akt and AP-1. Blood 109:1653–1659
Suboj P, Babykutty S, Valiyaparambil Gopi DR, Nair RS, Srinivas P, Gopala S (2012) Aloe emodin inhibits colon cancer cell migration/angiogenesis by downregulating MMP-2/9, RhoB and VEGF via reduced DNA binding activity of NF-kappaB. Eur J Pharm Sci 45:581–591
Manu KA, Shanmugam MK, Ong TH et al (2013) Emodin suppresses migration and invasion through the modulation of CXCR4 expression in an orthotopic model of human hepatocellular carcinoma. PLoS One 8:e57015
Cho YW, Hong T, Hong S et al (2007) PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem 282:20395–20406
Rajendran P, Ong TH, Chen L et al (2011) Suppression of signal transducer and activator of transcription 3 activation by butein inhibits growth of human hepatocellular carcinoma in vivo. Clin Cancer Res 17:1425–1439
Yoshida T, Shiraishi T, Nakata S et al (2005) Proteasome inhibitor MG132 induces death receptor 5 through CCAAT/enhancer-binding protein homologous protein. Cancer Res 65:5662–5667
Kim H, Kim EH, Eom YW et al (2006) Sulforaphane sensitizes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-resistant hepatoma cells to TRAIL-induced apoptosis through reactive oxygen species-mediated up-regulation of DR5. Cancer Res 66:1740–1750
Yamanaka T, Shiraki K, Sugimoto K et al (2000) Chemotherapeutic agents augment TRAIL-induced apoptosis in human hepatocellular carcinoma cell lines. Hepatology 32:482–490
Zhang L, Fang B (2005) Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther 12:228–237
Dzieran J, Beck JF, Sonnemann J (2008) Differential responsiveness of human hepatoma cells versus normal hepatocytes to TRAIL in combination with either histone deacetylase inhibitors or conventional cytostatics. Cancer Sci 99:1685–1692
Pellerito O, Calvaruso G, Portanova P et al (2010) The synthetic cannabinoid WIN 55,212-2 sensitizes hepatocellular carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by activating p8/CCAAT/enhancer binding protein homologous protein (CHOP)/death receptor 5 (DR5) axis. Mol Pharmacol 77:854–863
Yodkeeree S, Sung B, Limtrakul P, Aggarwal BB (2009) Zerumbone enhances TRAIL-induced apoptosis through the induction of death receptors in human colon cancer cells: evidence for an essential role of reactive oxygen species. Cancer Res 69:6581–6589
Kim HR, Kim K, Lee KH, Kim SJ, Kim J (2008) Inhibition of casein kinase 2 enhances the death ligand- and natural kiler cell-induced hepatocellular carcinoma cell death. Clin Exp Immunol 152:336–344
Wang S (2010) TRAIL: a sword for killing tumors. Curr Med Chem 17:3309–3317
Merino D, Lalaoui N, Morizot A, Schneider P, Solary E, Micheau O (2006) Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol Cell Biol 26:7046–7055
Irmler M, Thome M, Hahne M et al (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388:190–195
Kim YH, Jung EM, Lee TJ et al (2008) Rosiglitazone promotes tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by reactive oxygen species-mediated up-regulation of death receptor 5 and down-regulation of c-FLIP. Free Radic Biol Med 44:1055–1068
Mellier G, Huang S, Shenoy K, Pervaiz S (2010) TRAILing death in cancer. Mol Aspects Med 31:93–112
Nikolovska-Coleska Z, Xu L, Hu Z et al (2004) Discovery of embelin as a cell-permeable, small-molecular weight inhibitor of XIAP through structure-based computational screening of a traditional herbal medicine three-dimensional structure database. J Med Chem 47:2430–2440
Naumann U, Bahr O, Wolburg H et al (2007) Adenoviral expression of XIAP antisense RNA induces apoptosis in glioma cells and suppresses the growth of xenografts in nude mice. Gene Ther 14:147–161
Sethi G, Tergaonkar V (2009) Potential pharmacological control of the NF-kappaB pathway. Trends Pharmacol Sci 30:313–321
Ravi R, Bedi A (2002) Requirement of BAX for TRAIL/Apo2L-induced apoptosis of colorectal cancers: synergism with sulindac-mediated inhibition of Bcl-x(L). Cancer Res 62:1583–1587
Jacobson MD (1996) Reactive oxygen species and programmed cell death. Trends Biochem Sci 21:83–86
Jung EM, Lim JH, Lee TJ, Park JW, Choi KS, Kwon TK (2005) Curcumin sensitizes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis through reactive oxygen species-mediated upregulation of death receptor 5 (DR5). Carcinogenesis 26:1905–1913
Moon DO, Park SY, Choi YH, Ahn JS, Kim GY (2011) Guggulsterone sensitizes hepatoma cells to TRAIL-induced apoptosis through the induction of CHOP-dependent DR5: involvement of ROS-dependent ER-stress. Biochem Pharmacol 82:1641–1650
Acknowledgments
This research work was supported by grant from Singapore Ministry of Health’s National Medical Research Council under its Exploratory/Developmental Grant (EDG) funding scheme to GS. KMH was supported by grants from the National Medical Research Council of Singapore, Biomedical Research Council of Singapore, and the Singapore Millennium Foundation. APK was supported by grants from Singapore Ministry of Education Tier 2 [MOE2012-T2-2-139], Academic Research Fund Tier 1 [R-184-000-228-112] and Cancer Science Institute of Singapore, Experimental Therapeutics I Program [Grant R-713-001-011-271].
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Aruljothi Subramaniam and Ser Yue Loo contributed equally to this work.
Rights and permissions
About this article
Cite this article
Subramaniam, A., Loo, S.Y., Rajendran, P. et al. An anthraquinone derivative, emodin sensitizes hepatocellular carcinoma cells to TRAIL induced apoptosis through the induction of death receptors and downregulation of cell survival proteins. Apoptosis 18, 1175–1187 (2013). https://doi.org/10.1007/s10495-013-0851-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-013-0851-5