
Abstract
Phagocytosis of dying cells is a complex and dynamic process coordinated by the interaction of many surface molecules, adaptors, and chemotactic molecules, and it is controlled at multiple levels. This well regulated clearance process is of utmost importance for the development and homeostasis of organisms because defective or inefficient phagocytosis may contribute to human pathologies. In this review we discuss recent advances in the knowledge of the molecular interactions involved in recognition and clearance of apoptotic cells and how derangement of these processes can contribute to the pathogenesis of chronic airway diseases such as chronic obstructive pulmonary disease, cystic fibrosis and asthma. We will briefly consider how different types of macrophages are implicated in chronic airway diseases. Finally, we will address possible therapeutic strategies, such as the use of macrolide antibiotics and statins, for modulating apoptotic cell clearance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Chronic obstructive pulmonary disease
Chronic obstructive pulmonary disease (COPD) is a chronic inflammation of the airways accompanied by oxidative stress and imbalance between proteolytic and anti-proteolytic enzymatic activity, which leads to progressive destruction of lung parenchyma. Smoking tobacco is a major risk factor for development of COPD. The chronic inflammation in the lungs of COPD patients is supported by the continuous influx of macrophages, neutrophils and lymphocytes and by the production of tumor necrosis factor-α (TNF-α), IL-1β, IL-6, IL-17, IL-32, IL-18 and thymic stromal lymphopoietin (TSLP), which amplify inflammation [1]. Numerous studies identified apoptosis as a prominent feature of COPD-related changes in the lungs [2–4]. Expression of the apoptosis-associated proteins Bax and Bad has been observed in alveolar epithelial cells in the lungs of patients with emphysema, but not in healthy individuals [5]. Homogenates of emphysematous lung contain cleaved forms of caspase-3 and poly(ADP-ribose) polymerase (PARP), which are not observed in healthy individuals [5]. Moreover, apoptosis was shown to be increased in all stages of the disease. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay of induced sputum shows that patients with more severe symptoms of COPD and in whom forced expiratory volume (FEV1) is <50% tend to have significantly higher apoptosis rates than patients with FEV1 >50% and controls [6]. Rates of apoptosis of endothelial, alveolar epithelial, interstitial and inflammatory cells (neutrophils and lymphocytes) in situ in the lungs were higher in advanced stages of COPD than in healthy controls and asymptomatic smokers [2, 7–10]. TUNEL-positive cells were more frequently detected in the alveolar lumens with the most enlarged walls [11]. Moreover, the rate of apoptosis was higher in COPD patients even after ceasing smoking. In this regard, TUNEL-positive cells were observed throughout the parenchyma of alveolar and mesenchymal cells in lung sections of COPD patients who had ceased smoking for more than 6 months [5].
Increased spontaneous apoptosis of alveolar epithelial cells is accompanied by a higher rate of cell proliferation (analyzed by proliferating cell nuclear antigen) in the lung of COPD patients, whereas in the lungs of patients with terminal stage COPD, no increase in cell proliferation was observed anymore albeit an increased apoptotic index [5, 11, 12]. These observations indicate that an accelerated apoptosis rate in COPD is not always associated with increased proliferation in the lungs.
The increased rate of apoptosis observed in human samples from COPD patients was confirmed in mice exposed to cigarette smoke (CS) for 6 months: they developed emphysema and had more TUNEL-positive endothelial and epithelial cells than mice who were not exposed to CS [13]. Although an increase in the number of apoptotic cells in the lungs might be explained by deficient phagocytosis, an increased rate of apoptosis, or by a combination of both, it is difficult to draw a conclusion based only on these ex vivo studies.
Hodge and co-workers have elegantly shown in vitro that apoptotic cell clearance by alveolar macrophages (AMs) is impaired in COPD patients [14]. The authors established a phagocytosis assay using AMs from bronchoalveolar lavage (BAL) of COPD patients and healthy controls. When BAL cells were cocultured with apoptotic 16HBE airway epithelial cells, phagocytosis by AMs from COPD subjects was found to be significantly reduced as compared with controls (11.6 ± 4.1% vs 25.6 ± 9.2%) [14].
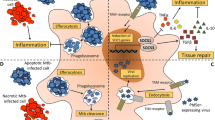
Several mechanisms have been proposed for the defective interaction between phagocytes and dying cells in COPD (Fig. 1). One of them is CS, which was shown to be directly toxic to the epithelial and endothelial cells and to cause DNA damage and induce apoptosis. CS could also alter the tissue repair mechanisms, including proliferation and production and remodeling of extracellular matrix [15–17]. The role of CS in regulation of phagocytosis is supported by the fact that the ability of AM to engulf apoptotic bronchial epithelial cells was significantly decreased in current smokers with COPD and in healthy smokers compared with control subjects who never smoked [18]. The COPD patients showed a 40% reduction in phagocytosis of apoptotic cells compared to healthy controls. Moreover, significant improvement of phagocytosis was observed in COPD patients who had ceased smoking compared with those who continued to smoke. This observation suggests that a smoking-related effect on AM may be partially reversed upon cessation of smoking. However, phagocytosis in COPD patients does not return to normal levels even in those who stop smoking for a long time [18]. This supports the notion that other mechanisms, such as oxidative stress, and protease and anti-protease imbalance, contribute to induction of apoptosis upon smoking cessation [2, 19, 20]. Importantly, CS suppresses the activation status and functional activity of macrophages. In vitro, CS extract was shown to decrease expression of CD44, CD71, CD31 and CD91 by AMs [18]. Interestingly, only COPD patients who still smoke have reduced expression of CD31, CD91, CD44, and CD71 on AMs, whereas COPD patients who ceased smoking for at least 1 year display no changes in the expression of these markers [18]. Blocking CD91 and CD31 reduced the phagocytic capacity of AMs about 20% [18]. This implies that reduced expression of the markers has only a partial negative effect on phagocytosis of apoptotic cells and suggests that other mechanisms are involved. In mice, acute, subacute and chronic regimens of smoking resulted in impaired phagocytosis of apoptotic cells by AMs [21].

A possible role of deficient clearance of apoptotic cells in pathogenesis of emphysema in COPD. In healthy lungs, alveolar macrophages (M) efficiently engulf apoptotic cells (A). Several mechanisms of altered uptake of apoptotic cells in COPD patients have been proposed. Smoking is a major etiological factor for development of COPD, and downstream effects of cigarette smoke on phagocytosis of apoptotic cells are summarized. In addition, decreased levels of VEGF and TGF-β could contribute to the progression of emphysema [102, 103]. COPD chronic obstructive pulmonary disease, ROS reactive oxygen species, VEGF vascular endothelial growth factor, HGF hepatocyte growth factor, TGF-β tumor growth factor β, PGE2 prostaglandin E2, TNF tumor necrosis factor, 15-HETE 15-hydroxyeicosatetraenoic acid
By using pharmacologic and genetic approaches in vivo and ex vivo in mice, it has been shown that CS can impair phagocytosis of apoptotic cells by an oxidant-dependent mechanism. CS exposure alters uptake of apoptotic thymocytes instilled into trachea of mice exposed to smoke [21]. Consequently, intraperitoneal administration of superoxide dismutase mimetic and the peroxynitrite scavenger, manganese(III) 5,10,15,20-tetrakis(4-benzoic acid)porphyrin (MnTBAP) before and after smoke exposure prevented the phagocytosis defects observed in mice exposed to CS [21]. These authors also showed that CS did not impair phagocytosis in mice overexpressing extracellular superoxide dismutase. They demonstrated that CS inhibits phagocytosis of apoptotic cells through oxidant-dependent activation of the RhoA–Rho kinase pathway [21]. This defect was shown to be specific for apoptotic cell clearance because CS did not inhibit Fcγ receptor-dependent phagocytosis of IgG-coated viable cells [21].
Several studies have shown that the levels of TNFα are increased in the BAL of COPD patients [22–24]. Interestingly, McPhillips and colleagues reported that TNFα suppresses clearance of apoptotic cells by murine J774 macrophages in vitro by oxidant-dependent mechanisms. The authors investigated the link between TNFα, reactive oxygen species (ROS) and Rho signaling in the suppression of phagocytosis of apoptotic cells [25]. They hypothesized that TNFα activates TNFR, which in turn activates phospholipase A2, leading to cleavage and release of arachidonic acid. Oxidants produced in this reaction induce conversion of Rho to its active form, which causes morphologic changes and prevents efficient phagocytosis of apoptotic cells [25]. This hypothesis was confirmed in another study, in which mice deficient in both receptors for TNFα, i.e. TNFR1 and TNFR2, demonstrated normal clearance of apoptotic cells in vivo after CS exposure [21]. The effect of TNFα on phagocytosis was also shown in vivo in mice: AM uptake of apoptotic murine thymocytes instilled intracheally was down-regulated by prior intratracheal instillation of TNFα. This deficient clearance exacerbated lung inflammation, led to recruitment of inflammatory cells, and increased production of IL-6, monocyte chemoattractant protein-1 (MCP-1), and chemokine (C–X–C motif) ligand 1 (CXCL1) [26]. These data suggest that increased TNFα in COPD patients and in mouse models of COPD could contribute to decreased phagocytosis of apoptotic cells in the airways (Fig. 1).
Another possible mechanism for CS downregulation of phagocytosis is modification of extracellular matrix proteins, such as collagen IV [20, 27]. The interaction of apoptotic and nectoric cells with phagocytes is a complex process that involves multiple membrane associated proteins in macrophages and in dead cells, bridging molecules, and intracellular signaling pathways [28, 29]. Surprisingly, in a study by Kazeros et al. [30], analysis of 14 apoptotic cell removal receptors on AMs from smoking COPD patients (stages I and II according to GOLD) showed upregulation of the expression level only of the gene encoding Mer receptor tyrosine kinase (MERTK). These data demonstrate that despite the major defect of phagocytosis in AMs of COPD patients, either expression of recognition receptors is not altered or otherwise these recognition molecules have a redundant function.
Surfactant proteins A and D (SP-A and SP-D) are pattern recognition molecules of the collectin family of C-type lectins, which can promote apoptotic cell clearance by innate immune cells [31, 32]. SP-A and SP-D bind to cell surface receptors, including Toll-like receptors (TLRs) [33, 34], signal inhibitory regulatory protein α (SIRPα), and the calreticulin/CD91 complex [35]. Surfactant proteins are now recognized as critical components in the host defense in lungs. Pulmonary surfactant-associated proteins are SP-A, SP-B, SP-C and SP-D [36]. Hodge et al. [37] demonstrated that SP-D levels in BAL of COPD patients are significantly lower than in controls. However, in the lungs and in the induced sputum of patients with COPD, the levels of SP-A were shown to be increased and no difference was found for SP-B, SP-C, SP-D levels in these lung compartments [38, 39]. In addition, smoke was reported to decrease SP-A and SP-D levels in human BAL [40]. The effect of collectins on phagocytosis of apoptotic cells has been tested in different experimental settings. Evidence from both in vivo and in vitro experiments supports the notion that collectins have a dual role in regulation of apoptotic cell clearance. It has been demonstrated that SP-A and SP-D increase phagocytosis of apoptotic cells by AMs in an opsonization-dependent manner or via interaction with the CD91/calreticulin complex [35, 41]. When macrophages were preincubated with SP-A or SP-D, the clearance of apoptotic cells was inhibited through tonic interaction with SIRPa [42, 43]. Phagocytic activity of AMs from SP-D deficient mice towards apoptotic cells in vitro was reduced [35, 43]. Moreover, mice deficient in SP-D develop emphysema and also exhibit reduced phagocytosis of apoptotic cells instilled intratracheally [35]. In contrast, SP-A deficiency did not alter clearance of exogenously applied apoptotic cells [35]. This points to the fact that SP-A and SP-D have a dual role in regulation of apoptotic cell clearance.
Earlier studies by Schagat et al. [41] showed that other collectins, such as mannose-binding lectin (MBL) and complement protein 1q (C1q), failed to affect uptake of apoptotic neutrophils by AMs in vitro. In addition, it was proven that C1q does not modulate apoptotic cells clearance in vivo [35]. However, Hodge et al. [44] showed that the reduced MBL levels in BAL of COPD patients significantly correlated with deficient phagocytosis of apoptotic cells. In smoke-exposed mice, phagocytosis of apoptotic cells was significantly improved when MBL was supplemented by using a nebulizer [44]. In addition, MBL-deficient mice also showed defective clearance of apoptotic thymocytes injected in the peritoneum in vivo [45]. These data suggest that MBL could be a new target for improving phagocytosis of apoptotic cells in the airways of COPD patients.
In recent years it has become evident that autoimmunity has a role in the pathogenesis of COPD. Indeed, T cell and B cell activation and auto-antibodies production were increased in COPD patients and in mouse models after long-term exposure to CS [46–50]. As discussed above, CS, which is the main risk factor for COPD, could lead to defective clearance of apoptotic cells in the airways (see review in this issue by L. Dini: “Phagocytosis of dying cells: influence of smoking and static magnetic fields”). Apoptotic cells that are not cleared at the appropriate time could be a potential source of autoantigens that can amplify the inflammation and trigger the autoimmune component in COPD [51, 52]. Further investigations are required to ascertain a causative link between defective clearance of apoptotic cells and development of autoimmunity in COPD.
Macrophage phenotypes in COPD
The existence of different macrophage activation phenotypes suggests that they have different functional roles in control of infections and in development of immunopathological disease symptoms. Mantovani et al. [53] proposed classifying macrophage activation phenotypes as M1 and M2 based on receptor expression, effector functions, and cytokine and chemokine production. M1 macrophages are the classically activated macrophages that show increased production of pro-inflammatory cytokines (TNFα, IL-1, IL-6, IL-12), inducible nitric oxide synthase (iNOS), and ROS, as well as enhanced antigen presentation [54]. Most chemokines produced during M1 activation of macrophages could amplify resistance to intracellular pathogens and includes production of CXCL9, CXCL10 and CCL5. Thus, these classically activated M1 macrophages are potent effector (killer) cells that destroy microorganisms and tumor cells and produce plentiful amounts of pro-inflammatory cytokines. By contrast, M2 macrophages result from an alternative form of activation and are characterized by increased expression of mannose receptor (MR), dectin 1 and arginase, and by generation of ornithine and polyamines [54–56]. In addition, M2 macrophages, in contrast to M1, produce chemokines such as CCL24, CCL17, CCL22 and others, which lead to attraction of cells involved in remodeling, tissue repair, allergy and promotion of angiogenesis. Therefore, M2 macrophages are often called pro-resolution macrophages. Alternatively activated macrophages (M2) are found during the resolution phase of acute inflammatory reactions in chronic inflammatory diseases, such as rheumatoid arthritis and psoriasis [57]. Considerable data have been collected on the polarization of human macrophages derived from peripheral blood mononuclear cells and on murine macrophages in vitro, but fewer data are available on the differentiation of macrophages in human diseases such as COPD. Affymetrix microarray analysis of polarization-related genes in AMs collected by BAL from COPD patients demonstrated that only few M2-related genes (MMP2, MMP7, adenosine A3 receptor) were up-regulated compared to non-smoking controls [58]. Expression of M2-related genes on the macrophages was not significantly different between COPD patients and smokers. In contrast, the authors observed that smoking alone down-regulated the expression of M1-related genes. Consequently, genes encoding type I chemokines (CXCL9, CXCL10, CXCL11, CCL5), IL-32, CD69 and IL-1β were suppressed in smokers compared to healthy non-smokers, but there was no significant difference between healthy smokers and COPD patients who smoked [58]. This observation suggests that development of the disease correlates with the down-regulation of M1-related genes rather than with the up-regulation of M2-related genes. In agreement, Reynolds’s group has also shown that the MR (an M2 marker) is downregulated on AMs of COPD patients, which suggests a mixed macrophage phenotype, and this needs further investigation [59]. In line, the data on mice exposed to CS for 8 weeks does not support the notion of M1 polarization of macrophages. Consequently, in macrophages from smoke-exposed mice the levels of TNFα, IL-6 and RANTES were reduced by stimulation in vitro with poly I:C, LPS and CpG [60]. It has also been shown that smoke deregulates activation of the transcription factors NF-kB and AP-1 and thereby inhibits the initiation of innate immune responses and cytokine production [60, 61].
To summarize, the data from humans and mice demonstrates that cigarette smoking induces reprogramming of the AMs polarization towards a mixed phenotype (M1-deactivated and partially M2-polarized) macrophages in vivo. With the development of COPD there is a further progression of suppression of M1 polarization program and only few of M2 related genes are up-regulated. The direct immune suppressive effect of smoke on AMs and various changes in cytokine and cellular microenvironment was demonstrated [62–65] and could explain the CS induced deactivation of macrophages.
Cystic fibrosis
Cystic fibrosis (CF) is a complex, heritable disease caused by mutation of the gene encoding conductance regulator Cl− channel (CFTR). Pulmonary manifestations are chronic and recurrent infection, airway inflammation, bronchiectasis and progressive lung obstruction lung early in life [66]. Examination of sputa demonstrated that CF patients have more apoptotic cells than patients with chronic bronchitis [67]. This observation suggests that defective clearance of apoptotic cells may contribute to ongoing airway inflammation. In fact, increased numbers of apoptotic cells in CF patients were detected not only in the lungs but also in epithelial surfaces of the duodenum, which indicates defective clearance of apoptotic cells in various CF epithelia [68]. Chronic microbial colonization and repeated acute exacerbations of pulmonary infection caused mostly by a unique spectrum of opportunistic bacteria such as Pseudomonas aeruginosa, Burkholderia cepacia, Staphylococcus aureus, and Haemophilus influenzae contribute to the disease-related damage in the lungs [66]. CFTR deficiency could provide an environment conducive to bacterial replication. It has been shown that AMs from CFTR −/− mice are defective in the killing of internalized bacteria and that lysosomes from CFTR-null macrophages fail to acidify [69]. Moreover, it was recently shown by Vandivier’s group that CFTR −/− epithelial cell lines are less able to clear apoptotic Jurkat cells in vitro [70]. Importantly, the authors observed that ineffective clearance of apoptotic cells by CFTR −/− cells has a proinflammatory consequences due to increased production of IL-8 by these cells [70]. The mechanism of this CFTR-dependent effect is related to a substantial increase in the expression of RhoA, a known negative regulator of phagocytosis. The involvement of the Rho kinase pathway was proven by observations that active RhoA was increased in epithelial cells of CF patients and CFTR −/− mice [70, 71] and that inhibitors of RhoA could normalize phagocytosis of apoptotic cells [70]. But, surprisingly, the CFTR −/− AMs do not have clearance defects, indicating that CFTR does not regulate clearance of apoptotic cells by AMs in the same way that it does for epithelial cells [70]. However, to our knowledge no studies of phagocytosis of apoptotic cells have been performed on human AMs from patients with CF. As mentioned above, patients with CF often suffer from chronic colonization with P. aeruginosa, which is the principal cause of mortality in CF lung disease [72]. Bianchi et al. have shown that a toxic metabolite of P. aeruginosa (pyocyanin) inhibits phagocytosis of apoptotic neutrophils and apoptotic Jurkat cells by HMDM in vitro. Moreover, they showed that in mice infected with pyocyanin-producing P. aeruginosa, clearance of apoptotic cells by BAL macrophages was reduced due to generation of ROS and their effects on Rho GTPase signaling. Pyocyanin treatment of HMDMs activated Rho activity 2 h and inhibited Rac-1 activity 24 h after the treatment [73]. This observation suggests that bacterial metabolites, which have not been considered in this context, have to be taken into account when analyzing the complex phenomenon of phagocytosis deficiency in CF.
Damage associated molecular patterns, which are released from dying cells, were shown to affect phagocyte activation and/or differentiation, whereas their role in the process of phagocyte recruitment is just being elucidated [74, 75]. It has been reported that high-mobility group box 1 (HMGB1) is increased in the BAL of CF patients and in the mouse model of CF [76]. Moreover, HMGB1 could block phagocytosis of apoptotic cells in a PS-dependent manner [77]. These results indicate that HMGB1 content in BAL from CF patients could contribute to the decreased phagocytosis of apoptotic cells in this pathology [77]. As many inflammatory and autoimmune diseases have been linked with deregulated appearance of these damage associated factors, future studies on CF should try to fully understand how the substances released from apoptotic cells affect their phagocytosis and the contribution of these substances to the pathology of CF. However, it is important to emphasize that knowledge on the precise molecular mechanisms involved in the deficient phagocytosis of apoptotic cells in CF patients is very limited, which means that many interesting and challenging findings are expected.
Data on the macrophage activation state of AMs isolated from patients with CF is rather scarce. It has been shown that monocytes from CF patients, when challenged with LPS, exhibit a tolerant IL-12lowIL-23lowIL-10high phenotype with low antigen presentation capacity, which is indicative of an M2 polarization state [78]. In opposite, in the nasal tissue of CF patients we observed accumulation of CD14+ cells, without immunohistochemical evidence of an increase in CD68+, CD206+ or CD163+ cells [79–81], thus M1 macrophages. Further studies are needed to evaluate the phenotype of AMs in CF patients and to understand how it contributes to the pathogenesis of the disease.
Asthma
Asthma is a complex syndrome with a variable degree of airflow obstruction, bronchial hyperresponsiveness, and airway inflammation [82, 83]. Tissue damage and increased apoptotic rates with typical TUNEL and caspase-3 positively stained epithelial cells and smooth muscle cells were observed in the proximal conducting airways and in the central bronchus of patients with severe disease, while in patients with intermittent stage of the disease apoptosis rates were comparable to controls [84]. Furthermore, it has been demonstrated that the increase in apoptotic eosinophils in induced sputum and their phagocytosis by AMs is correlated with reduction in asthma symptoms and resolution of asthma exacerbation [85–87]. Huynh et al. [88] observed that human AMs from normal individuals and patients with mild to moderate asthma had similar numbers of phagocytic bodies, suggesting that clearance of apoptotic cells was not disturbed. In contrast, AMs from patients with severe asthma had fewer phagocytic bodies than patients with mild to moderate asthma which indicates that phagocytosis might have been reduced. The authors strengthened these ex vivo observations by performing an in vitro phagocytosis assay (using apoptotic Jurkat cells) of AMs obtained from asthmatic patients. They confirmed that in vitro phagocytosis of apoptotic Jurkat cells by AMs from severely asthmatic patients was defective both in unstimulated and LPS-stimulated conditions. However, AMs from patients with mild to moderate asthma symptoms exhibited no defects in phagocytosis [88]. It has also been shown that in severe asthmatics the normal process of release of anti-fibrotic and/or anti-inflammatory mediators is distorted. The AMs from patients with severe asthma failed to produce prostaglandin E2 and 15-HETE (15-hydroxyeicosatetraenoic acid) [88], which indeed could contribute to the chronic inflammation and airway remodeling in lungs of patients with asthma. Of interest, the phagocytosis defect in patients with severe asthma was not limited to engulfment of apoptotic cells but extended to removal of infectious agents [89].
In AMs from patients with mild asthma, no difference was observed in the expression of surface markers such as CD11b, CD64, CD16, CD14 and HLA-DR [90], while in a group of patients with more severe disease (eosinophilia in sputum >5%) CD64 and CD11b were up-regulated [90].
Although all these studies suggest that decreased removal of apoptotic cells in asthmatic patients might contribute to the development of disease, more work is required to prove this hypothesis by providing a more extensive analysis of molecular mechanisms of the phagocytosis of apoptotic cells that can lead to the deregulated removal of apoptotic cells in asthma patients, and their contribution to airway inflammation; such results should be analyzed in mouse models and human asthma.
Possible strategies for improvement of apoptotic cell clearance
One strategy for dealing with the problem of deficient clearance of apoptotic cells in COPD and CF is to therapeutically enhance the phagocytic capacity of macrophages [59, 85]. Examples of drugs that might promote phagocytic clearance of apoptotic cells are the macrolide antibiotics and statins. It has been shown that azithromycin increases the ability of AMs from COPD patients to phagocytose apoptotic bronchial epithelial cells in vitro [91]. Importantly, no effect on the expression of the recognition molecules was observed, including CD31, CD36, CD91, ανβ3 integrin and CD44. The authors reported that increased phagocytosis could be partially inhibited by phosphatidylserine, suggesting that the pro-phagocytic effect of azithromycin is PS-mediated [91]. However, in another study it was shown that azithromycin increases MR expression by 50%. Down-regulation of this receptor has been implicated in the defective phagocytic capacity of AMs [92]. Consequently, administration of azithromycin to COPD patients for 12 weeks significantly improved AM phagocytosis [37]. Low doses of azithromycin were reported to have not only a beneficial anti-inflammatory effect in COPD, CF and panbronchiolitis [93, 94], but also to improve phagocytosis when given for 12 weeks, possibly by increasing the surface expression of MR [37].
Another class of drugs that could be used to modulate the process of phagocytosis of apoptotic cells is statins. These were discovered as drugs that reduce cholesterol levels by inhibiting the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA), but they were also shown to have an anti-inflammatory action by virtue of their ability to block prenylation of signaling molecules, i.e. Rho GTPases [95–97]. It has been shown that lovastatin increases phagocytosis of apoptotic cells by AMs from COPD patients in an HMG-CoA reductase-dependent manner [97]. Interestingly, lovastatin did not affect the expression of recognition molecules such as CD36, CD44, CD14, CD91, ανβ3, ανβ5 integrins and FcγRIIa on macrophages [97]. Administration of simvastatin to mice sensitized them to ovalbumin and reduced features of allergic airway inflammation, such as eosinophilia in BAL and IL-5 production [98], but no data are available in this study about phagocytosis. Although there is a good indication for the use of statins in asthmatic patients, only one randomized, double-blind clinical study has been performed; that study tested the effect of simvastatin on the clinical outcome of disease [99]. However, based on the results, no major improvements were found after simvastatin administration to asthmatic patients, with exception of reduction of exhaled nitric oxide. It is important to note that in the current study only patients with mild features of asthma were included and new studies are needed to test different statins on patients with severe asthma [85, 99]. More data have to be collected to understand the mechanisms of phagocytosis deficiency in asthmatic patients. In COPD patients, statins reduce neutrophil numbers and T cell differentiation and activation, and increase apoptosis of eosinophils [85]. A study by Morimoto et al. [97] has demonstrated that lovastatin is a potent inducer of apoptotic cell removal in COPD patients. Notably, there is evidence that simvastatin impairs phagocytosis and the oxidative burst in response to opsonized bacteria and at the same time enhances the production of pro-inflammatory mediators [100], which should be assessed to prevent potential side-effects in patients undergoing long-term statin treatment. Only carefully designed and controlled clinical trials can determine whether statins can significantly improve the clinical course of COPD and exert their anti-inflammatory effects in these settings. Future studies are needed to determine whether these drugs could provide the basis for a novel therapeutic strategy, based on modulation of phagocytosis, to prevent the progression of advanced stages of the disease. Glucocorticoids could also improve phagocytosis of apoptotic cells, but their adverse effects render this approach impractical [101]. The study by Hodge et al. [44], provided the evidence that the modulation of the mannose pathway by MBL could be considered as supplementary therapy of COPD.
Conclusions
Precise knowledge of the signals, receptors and intracellular signaling pathways leading to decreased clearance of apoptotic cells in COPD, CF and asthma is limited. It is likely that multiple molecules are involved, and here we sought to briefly overview these potential actors. Many more interesting and challenging findings concerning the molecular basis of deficient clearance of dead cells in these diseases are expected to emerge, with consequent resolution of the controversial question of whether disturbed clearance of apoptotic cells in asthma is important for it development and progression. Furthermore, this knowledge will stimulate the development of new treatment strategies for manipulating phagocytic clearance of apoptotic cells for the treatment of chronic pulmonary disorders.
References
Barnes PJ (2009) The cytokine network in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 41:631–638
Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG (2006) Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res 7:53
Henson PM, Tuder RM (2008) Apoptosis in the lung: induction, clearance and detection. Am J Physiol 294:601–611
Tuder RM, Petrache I, Elias JA, Voelkel NF, Henson PM (2003) Apoptosis and emphysema: the missing link. Am J Respir Cell Mol Biol 28:551–554
Imai K, Mercer BA, Schulman LL, Sonett JR, D’Armiento JM (2005) Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J 25:250–258
Makris D, Vrekoussis T, Izoldi M et al (2009) Increased apoptosis of neutrophils in induced sputum of COPD patients. Respir Med 103:1130–1135
Segura-Valdez L, Pardo A, Gaxiola M, Uhal BD, Becerril C, Selman M (2000) Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest 117:684–694
Uhal BD, Joshi I, Hughes WF, Ramos C, Pardo A, Selman M (1998) Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am J Physiol 275:L1192–L1199
Hodge S, Hodge G, Holmes M, Reynolds PN (2005) Increased peripheral blood T-cell apoptosis and decreased Bcl-2 in chronic obstructive pulmonary disease. Immunol Cell Biol 83:160–166
Kasahara Y, Tuder RM, Cool CD, Voelkel NF (2000) Expression of 15-lipoxygenase and evidence for apoptosis in the lungs from patients with COPD. Chest 117:260S
Calabrese F, Giacometti C, Beghe B, et al (2005) Marked alveolar apoptosis/proliferation imbalance in end-stage emphysema. Respir Res 6:14–26
Yokohori N, Aoshiba K, Nagai A (2004) Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest 125:626–632
Rangasamy T, Misra V, Zhen L, Tankersley CG, Tuder RM, Biswal S (2009) Cigarette smoke-induced emphysema in A/J mice is associated with pulmonary oxidative stress, apoptosis of lung cells, and global alterations in gene expression. Am J Physiol 296:L888–L900
Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M (2003) Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol 81:289–296
Nakamura Y, Romberger DJ, Tate L et al (1995) Cigarette smoke inhibits lung fibroblast proliferation and chemotaxis. Am J Respir Crit Care Med 151:1497–1503
Rennard SI, Togo S, Holz O (2006) Cigarette smoke inhibits alveolar repair: a mechanism for the development of emphysema. Proc Am Thorac Soc 3:703–708
Hoshino S, Yoshida M, Inoue K et al (2005) Cigarette smoke extract induces endothelial cell injury via JNK pathway. Biochem Biophys Res Commun 329:58–63
Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, Reynolds PN (2007) Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 37:748–755
Powell WC, Fingleton B, Wilson CL, Boothby M, Matrisian LM (1999) The metalloproteinase matrilysin proteolytically generates active soluble Fas ligand and potentiates epithelial cell apoptosis. Curr Biol 9:1441–1447
Dini L, Vergallo C (2009) Environmental factors affecting phagocytosis of dying cells: smoking and static magnetic fields. In: Krysko DV, Vandenabeele P (eds) Phagocytosis of dying cells. Springer, Dordrecht, pp 409–438
Richens TR, Linderman DJ, Horstmann SA et al (2009) Cigarette smoke impairs clearance of apoptotic cells through oxidant-dependent activation of RhoA. Am J Respir Crit Care Med 179:1011–1021
D’Hulst AI, Bracke KR, Maes T et al (2006) Role of tumour necrosis factor-alpha receptor p75 in cigarette smoke-induced pulmonary inflammation and emphysema. Eur Respir J 28:102–112
Churg A, Dai J, Tai H, Xie C, Wright JL (2002) Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am J Respir Crit Care Med 166:849–854
Keatings VM, Collins PD, Scott DM, Barnes PJ (1996) Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 153:530–534
McPhillips K, Janssen WJ, Ghosh M et al (2007) TNF-alpha inhibits macrophage clearance of apoptotic cells via cytosolic phospholipase A2 and oxidant-dependent mechanisms. J Immunol 178:8117–8126
Borges VM, Vandivier RW, McPhillips KA et al (2009) TNFalpha inhibits apoptotic cell clearance in the lung, exacerbating acute inflammation. Am J Physiol 297:586–595
Kirkham PA, Spooner G, Rahman I, Rossi AG (2004) Macrophage phagocytosis of apoptotic neutrophils is compromised by matrix proteins modified by cigarette smoke and lipid peroxidation products. Biochem Biophys Res Commun 318:32–37
Krysko DV, D’Herde K, Vandenabeele P (2006) Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis 11:1709–1726
Krysko DV, Vandenabeele P (2008) From regulation of dying cell engulfment to development of anti-cancer therapy. Cell Death Differ 15:29–38
Kazeros A, Harvey BG, Carolan BJ, Vanni H, Krause A, Crystal RG (2008) Overexpression of apoptotic cell removal receptor MERTK in alveolar macrophages of cigarette smokers. Am J Respir Cell Mol Biol 39:747–757
Pastva AM, Wright JR, Williams KL (2007) Immunomodulatory roles of surfactant proteins A and D: implications in lung disease. Proc Am Thorac Soc 4:252–257
Wright JR (2005) Immunoregulatory functions of surfactant proteins. Nat Rev 5:58–68
Ohya M, Nishitani C, Sano H et al (2006) Human pulmonary surfactant protein D binds the extracellular domains of Toll-like receptors 2 and 4 through the carbohydrate recognition domain by a mechanism different from its binding to phosphatidylinositol and lipopolysaccharide. Biochemistry 45:8657–8664
Yamada C, Sano H, Shimizu T et al (2006) Surfactant protein A directly interacts with TLR4 and MD-2 and regulates inflammatory cellular response. Importance of supratrimeric oligomerization. J Biol Chem 281:21771–21780
Vandivier RW, Ogden CA, Fadok VA et al (2002) Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol 169:3978–3986
Kishore U, Greenhough TJ, Waters P et al (2006) Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol 43:1293–1315
Hodge S, Hodge G, Jersmann H et al (2008) Azithromycin improves macrophage phagocytic function and expression of mannose receptor in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 178:139–148
Ohlmeier S, Vuolanto M, Toljamo T et al (2008) Proteomics of human lung tissue identifies surfactant protein A as a marker of chronic obstructive pulmonary disease. J Proteome Res 7:5125–5132
Vlachaki EM, Koutsopoulos AV, Tzanakis N et al (2010) Altered surfactant protein-a (Sp-a) expression in type Ii pneumocytes in copd. Chest 137:37–45
Honda Y, Takahashi H, Kuroki Y, Akino T, Abe S (1996) Decreased contents of surfactant proteins A and D in BAL fluids of healthy smokers. Chest 109:1006–1009
Schagat TL, Wofford JA, Wright JR (2001) Surfactant protein A enhances alveolar macrophage phagocytosis of apoptotic neutrophils. J Immunol 166:2727–2733
Reidy MF, Wright JR (2003) Surfactant protein A enhances apoptotic cell uptake and TGF-beta1 release by inflammatory alveolar macrophages. Am J Physiol 285:L854–L861
Janssen WJ, McPhillips KA, Dickinson MG et al (2008) Surfactant proteins A and D suppress alveolar macrophage phagocytosis via interaction with SIRP alpha. Am J Respir Crit Care Med 178:158–167
Hodge S, Matthews G, Dean MM et al (2010) Therapeutic role for mannose-binding lectin in cigarette smoke-induced lung inflammation? Evidence from a murine model. Am J Respir Cell Mol Biol 42:235–242
Stuart LM, Takahashi K, Shi L, Savill J, Ezekowitz RA (2005) Mannose-binding lectin-deficient mice display defective apoptotic cell clearance but no autoimmune phenotype. J Immunol 174:3220–3226
Feghali-Bostwick CA, Gadgil AS, Otterbein LE et al (2008) Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 177:156–163
Lee SH, Goswami S, Grudo A et al (2007) Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med 13:567–569
Gadgil A, Zhu X, Sciurba FC, Duncan SR (2006) Altered T-cell phenotypes in chronic obstructive pulmonary disease. Proc Am Thorac Soc 3:487–488
van der Strate BW, Postma DS, Brandsma CA et al (2006) Cigarette smoke-induced emphysema: A role for the B cell? Am J Respir Crit Care Med 173:751–758
Cosio MG, Saetta M, Agusti A (2009) Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med 360:2445–2454
Eggleton P, Haigh R, Winyard PG (2008) Consequence of neo-antigenicity of the ‘altered self’. Rheumatology 47:567–571
Agusti A, MacNee W, Donaldson K, Cosio M (2003) Hypothesis: does COPD have an autoimmune component? Thorax 58:832–834
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23:549–555
Gordon S, Taylor PR (2005) Monocyte and macrophage heterogeneity. Nat Rev 5:953–964
Mantovani A, Sica A, Locati M (2005) Macrophage polarization comes of age. Immunity 23:344–346
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686
Gratchev A, Schledzewski K, Guillot P, Goerdt S (2001) Alternatively activated antigen-presenting cells: molecular repertoire, immune regulation, and healing. Skin Pharmacol Appl Skin Physiol 14:272–279
Shaykhiev R, Krause A, Salit J et al (2009) Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol 183:2867–2883
Reynolds PN, Hodge SJ (2009) Chapter 14. The impact of defective clearance of apoptotic cells in the pathogenesis of chronic lung diseases: chronic obstructive pulmonary disease, asthma and cystic fibrosis. In: Krysko DV, Vandenabeele P (eds) Phagocytosis of dying cells from molecular mechanisms to human diseases. Springer, Dordrecht, pp 393–407
Gaschler GJ, Zavitz CC, Bauer CM et al (2008) Cigarette smoke exposure attenuates cytokine production by mouse alveolar macrophages. Am J Respir Cell Mol Biol 38:218–226
Laan M, Bozinovski S, Anderson GP (2004) Cigarette smoke inhibits lipopolysaccharide-induced production of inflammatory cytokines by suppressing the activation of activator protein-1 in bronchial epithelial cells. J Immunol 173:4164–4170
Sopori M (2002) Effects of cigarette smoke on the immune system. Nat Rev 2:372–377
Li L, Hamilton RF Jr, Holian A (1999) Effect of acrolein on human alveolar macrophage NF-kappaB activity. Am J Physiol 277:L550–L557
Hagiwara E, Takahashi KI, Okubo T et al (2001) Cigarette smoking depletes cells spontaneously secreting Th(1) cytokines in the human airway. Cytokine 14:121–126
Sakaguchi S, Yamaguchi T, Nomura T, Ono M (2008) Regulatory T cells and immune tolerance. Cell 133:775–787
Rowe SM, Miller S, Sorscher EJ (2005) Cystic fibrosis. N Engl J Med 352:1992–2001
Vandivier RW, Fadok VA, Ogden CA et al (2002) Impaired clearance of apoptotic cells from cystic fibrosis airways. Chest 121:89S
Maiuri L, Raia V, De Marco G et al (1997) DNA fragmentation is a feature of cystic fibrosis epithelial cells: a disease with inappropriate apoptosis? FEBS Lett 408:225–231
Di A, Brown ME, Deriy LV et al (2006) CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol 8:933–944
Vandivier RW, Richens TR, Horstmann SA et al (2009) Dysfunctional cystic fibrosis transmembrane conductance regulator inhibits phagocytosis of apoptotic cells with proinflammatory consequences. Am J Physiol 297:677–686
Kreiselmeier NE, Kraynack NC, Corey DA, Kelley TJ (2003) Statin-mediated correction of STAT1 signaling and inducible nitric oxide synthase expression in cystic fibrosis epithelial cells. Am J Physiol 285:L1286–L1295
Elkin S, Geddes D (2003) Pseudomonal infection in cystic fibrosis: the battle continues. Expert Rev Anti Infect Ther 1:609–618
Bianchi SM, Prince LR, McPhillips K et al (2008) Impairment of apoptotic cell engulfment by pyocyanin, a toxic metabolite of Pseudomonas aeruginosa. Am J Respir Crit Care Med 177:35–43
Peter C, Wesselborg S, Lauber K (2009) Role of attraction and danger signals in the uptake of apoptotic and necrotic cells and its immunological outcome. In: Krysko DV, Vandenabeele P (eds) Phagocytosis of dying cells: from molecular mechanisms to human disease. Springer, Dordrecht, pp 63–101
Peter C, Wesselborg S, Herrmann M, Lauber K (2010) Dangerous attraction: phagocyte recruitment and danger signals of apoptotic and necrotic cells. Apoptosis, Feb 6 [Epub ahead of print]
Rowe SM, Jackson PL, Liu G et al (2008) Potential role of high-mobility group box 1 in cystic fibrosis airway disease. Am J Respir Crit Care Med 178:822–831
Liu G, Wang J, Park YJ et al (2008) High mobility group protein-1 inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J Immunol 181:4240–4246
del Fresno C, Garcia-Rio F, Gomez-Pina V et al (2009) Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: demonstration in isolated monocytes from cystic fibrosis patients. J Immunol 182:6494–6507
Claeys S, Van Hoecke H, Holtappels G et al (2005) Nasal polyps in patients with and without cystic fibrosis: a differentiation by innate markers and inflammatory mediators. Clin Exp Allergy 35:467–472
Van Zele T, Claeys S, Gevaert P et al (2006) Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy 61:1280–1289
Krysko O, Van Zele T, Claeys S, Bachert C (2009) Comment on “potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: demonstration in isolated monocytes from cystic fibrosis patients”. J Immunol 183:4831; author reply 4831–4832
Van Hove CL, Maes T, Joos GF, Tournoy KG (2008) Chronic inflammation in asthma: a contest of persistence vs resolution. Allergy 63:1095–1109
Busse WW, Lemanske RF Jr (2001) Asthma. N Engl J Med 344:350–362
Solarewicz-Madejek K, Basinski TM, Crameri R et al (2009) T cells and eosinophils in bronchial smooth muscle cell death in asthma. Clin Exp Allergy 39:845–855
Walsh GM (2008) Defective apoptotic cell clearance in asthma and COPD—a new drug target for statins? Trends Pharmacol Sci 29:6–11
Duncan CJ, Lawrie A, Blaylock MG, Douglas JG, Walsh GM (2003) Reduced eosinophil apoptosis in induced sputum correlates with asthma severity. Eur Respir J 22:484–490
Woolley KL, Gibson PG, Carty K, Wilson AJ, Twaddell SH, Woolley MJ (1996) Eosinophil apoptosis and the resolution of airway inflammation in asthma. Am J Respir Crit Care Med 154:237–243
Huynh ML, Malcolm KC, Kotaru C et al (2005) Defective apoptotic cell phagocytosis attenuates prostaglandin E2 and 15-hydroxyeicosatetraenoic acid in severe asthma alveolar macrophages. Am J Respir Crit Care Med 172:972–979
Fitzpatrick AM, Holguin F, Teague WG, Brown LA (2008) Alveolar macrophage phagocytosis is impaired in children with poorly controlled asthma. J Allergy Clin Immunol 121:1372–1378, 1378 e1371–1373
Alexis NE, Soukup J, Nierkens S, Becker S (2001) Association between airway hyperreactivity and bronchial macrophage dysfunction in individuals with mild asthma. Am J Physiol 280:L369–L375
Hodge S, Hodge G, Brozyna S, Jersmann H, Holmes M, Reynolds PN (2006) Azithromycin increases phagocytosis of apoptotic bronchial epithelial cells by alveolar macrophages. Eur Respir J 28:486–495
Allavena P, Chieppa M, Monti P, Piemonti L (2004) From pattern recognition receptor to regulator of homeostasis: the double-faced macrophage mannose receptor. Crit Rev Immunol 24:179–192
Kobayashi H, Takeda H, Sakayori S et al (1995) Study on azithromycin in treatment of diffuse panbronchiolitis. Kansenshogaku Zasshi 69:711–722
Baumann U, King M, App EM et al (2004) Long term azithromycin therapy in cystic fibrosis patients: a study on drug levels and sputum properties. Can Respir J 11:151–155
Nakaya M, Tanaka M, Okabe Y, Hanayama R, Nagata S (2006) Opposite effects of rho family GTPases on engulfment of apoptotic cells by macrophages. J Biol Chem 281:8836–8842
Eberlein M, Heusinger-Ribeiro J, Goppelt-Struebe M (2001) Rho-dependent inhibition of the induction of connective tissue growth factor (CTGF) by HMG CoA reductase inhibitors (statins). Br J Pharmacol 133:1172–1180
Morimoto K, Janssen WJ, Fessler MB et al (2006) Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J Immunol 176:7657–7665
McKay A, Leung BP, McInnes IB, Thomson NC, Liew FY (2004) A novel anti-inflammatory role of simvastatin in a murine model of allergic asthma. J Immunol 172:2903–2908
Menzies D, Nair A, Meldrum KT, Fleming D, Barnes M, Lipworth BJ (2007) Simvastatin does not exhibit therapeutic anti-inflammatory effects in asthma. J Allergy Clin Immunol 119:328–335
Benati D, Ferro M, Savino MT et al (2009) Opposite effects of simvastatin on the bactericidal and inflammatory response of macrophages to opsonized S. aureus. J Leukoc Biol 87:433–442
Maderna P, Godson C (2003) Phagocytosis of apoptotic cells and the resolution of inflammation. Biochim Biophys Acta 1639:141–151
Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM (1998) Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 101:890–898
Golpon HA, Fadok VA, Taraseviciene-Stewart L et al (2004) Life after corpse engulfment: phagocytosis of apoptotic cells leads to VEGF secretion and cell growth. FASEB J 18:1716–1718
Acknowledgements
This work was supported by the Fund for Scientific Research Flanders (FWO-Vlaanderen) (3G064210 to C.B., O.K. and 3G072810 to D.V.K.), by the Interuniversity Attraction Poles Programme (IUAP)—Belgian state—Belgian Science Policy (P6/35 to C.B.) and “krediet aan navorsers” from Fund for Scientific Research Flanders (FWO-Vlaanderen, 31507110 to D.V.K.). D.V.K. is a postdoctoral research fellow of the Fund for Scientific Research Flanders (FWO-Vlaanderen), Belgium. We thank Dr. A. Bredan for proofreading the manuscript and W. Drijvers for the art work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Olga Krysko and Dmitri V. Krysko contributed equally to this work.
Rights and permissions
About this article
Cite this article
Krysko, O., Vandenabeele, P., Krysko, D.V. et al. Impairment of phagocytosis of apoptotic cells and its role in chronic airway diseases. Apoptosis 15, 1137–1146 (2010). https://doi.org/10.1007/s10495-010-0504-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-010-0504-x