
Abstract
The efficient clearance of apoptotic cells is an evolutionarily conserved process crucial for homeostasis in multicellular organisms. The clearance involves a series of steps that ultimately facilitates the recognition of the apoptotic cell by the phagocytes and the subsequent uptake and processing of the corpse. These steps include the phagocyte sensing of “find-me” signals released by the apoptotic cell, recognizing “eat-me” signals displayed on the apoptotic cell surface, and then intracellular signaling within the phagocyte to mediate phagocytic cup formation around the corpse and corpse internalization, and the processing of the ingested contents. The engulfment of apoptotic cells by phagocytes not only eliminates debris from tissues but also produces an anti-inflammatory response that suppresses local tissue inflammation. Conversely, impaired corpse clearance can result in loss of immune tolerance and the development of various inflammation-associated disorders such as autoimmunity, atherosclerosis, and airway inflammation but can also affect cancer progression. Recent studies suggest that the clearance process can also influence antitumor immune responses. In this review, we will discuss how apoptotic cells interact with their engulfing phagocytes to generate important immune responses, and how modulation of such responses can influence pathology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
2.1 Introduction
Phagocytes are often thought of as the “garbage collectors” of the body, eliminating pathogens, immune complexes, and dying cells. Sensing infection and coordinating an immune response is fundamental in the body’s fight to prevent disease, but discriminating between harmless debris and a true threat is equally vital. Every day, billions of cells in the body undergo apoptotic cell death as part of the normal physiology/homeostasis that is essential for healthy living [1]. The phagocytes that clear them must respond appropriately to prevent an unnecessary and unwanted immune response to such homeostatic cell turnover [2–5].
Apoptosis is often described as “immunologically silent” cell death; however, apoptotic cells are anything but unheard. The homeostatic clearance of apoptotic cells elicits critical immunosuppressive responses in the phagocytes that are often specific to apoptotic cell recognition. This includes the release of anti-inflammatory cytokines, the inhibition of proinflammatory cytokine expression, and the regulation of new immune cell production. Importantly, these are active responses to apoptotic cells and not just the lack of an inflammatory response [6–10]. Showcasing the vital role of apoptotic cell clearance in the maintenance of immune tolerance, uncleared corpses can result in inflammation [11]. This is partly due to the nature of apoptosis, which when left uncompleted by engulfment, progresses to “secondary necrosis,” a state in which the dying cell loses its membrane integrity and releases some or most of its intracellular components [12]. However, while the body may not want to produce an immunogenic response to apoptotic cells that are routinely turned over as part of homeostasis, generating immune responses to tumor-derived antigens could obviously be beneficial. Recent studies suggest that the cell clearance process can also initiate antitumor immunity [13, 14].
In this review, we will discuss how phagocytes sense, engulf, and respond to apoptotic cells. Since the identification of phosphatidylserine (PtdSer) as an eat-me signal on apoptotic cells in the early 1990s, there has been a rapid expansion in our knowledge of how apoptotic cells are recognized and removed. As apoptosis can be immunologically silent only when the corpses are cleared in a timely manner, understanding the mechanisms of cell clearance has provided important insights and tools for the study of cell clearance and its relationship to disease.
2.2 Types of Apoptotic Cells and Phagocytes
It is estimated that every day, we turnover about 200–300 billion cells, or about one million cells/s. The cells can die by many different modalities, including caspase-dependent apoptosis, necroptosis, as well as necrosis [15] (see Chap. 1 for more details). Even within each modality of death, there are multiple subroutines that eventually lead to the death of the cells fated to die [15]. This chapter primarily deals with clearance of cells that undergo caspase-mediated apoptosis as this is the most common and perhaps the best understood mode of cell death in vivo and in vitro.
There are different types of phagocytes that mediate the removal of the dying cells in the various tissues [16]. Phagocytes can be broadly classified into three different types: professional phagocytes, nonprofessional phagocytes, and specialized phagocytes. The professional phagocytes include the macrophages and immature dendritic cells. Given the recent understanding of self-renewing tissue resident, and recruited macrophages, they both seem capable of engulfing apoptotic cells. The professional phagocytes are named as such because in different analyses they tend to have a large capacity to engulf, engulf the targets with faster kinetics, and can also ingest multiple corpses in succession. The nonprofessional phagocytes include cell types such as epithelial cells, fibroblasts, and other tissue-resident cells. Although these nonprofessional phagocytes often display a lower capacity to engulf and slower kinetics in vitro, they do have a numerical supremacy and due to their proximity to the apoptotic cells (e.g., neighbor), these nonprofessional phagocytes likely contribute substantially to the clearance of apoptotic cells in vivo. In fact, disruption of nonprofessional phagocyte engulfment has important immunological consequences.
The third type of phagocytes, specialized phagocytes, refers to cells that generally have a multitude of functions, one of them being phagocytosis of dying cells. The examples include the Sertoli cells of the testis (which provide nurse function for the developing male germ cells while also engulfing those with defective development) [17] and retinal pigment epithelial cells (which remove the “used” photoreceptor outer segments daily in a circadian fashion) [18, 19]. Obviously, the key difference between the three types of phagocytes is that not all of them have to express the same collection of receptors and they may not have similar postengulfment responses. However, some of the responses of professional phagocytes and nonprofessional phagocytes clearly overlap, and could be relevant in tissues where often both of them are involved in apoptotic cell clearance simultaneously.
2.3 Steps in Apoptotic Cell Engulfment
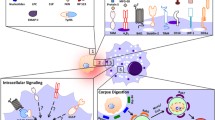
Work from a number of laboratories over the past nearly two decades has detailed a series of distinguishable steps in apoptotic cell recognition and clearance (Fig. 2.1). These have helped us understand how the phagocytes and apoptotic cells get near each other, how the phagocytes specifically recognize the targets, the types of intracellular signaling within phagocytes that leads to the corpse uptake, as well as some of the subsequent responses of phagocytes.

Major steps in apoptotic cell clearance . The key events in recognition and clearance of dying cells can be broadly classified into four steps. Step 1 depicts the recruitment of motile phagocytes (such as tissue-resident macrophages) by apoptotic cells via the release of find-me signals. Step 2 is the specific recognition of the apoptotic cell via “eat-me” ligands on the dying cells engaged by the receptors on phagocytes. Often, the recognition of apoptotic cells alone (even without corpse internalization) is sufficient to trigger some of the key anti-inflammatory mediators from the phagocytes. Step 3 is the intracellular signaling that occurs within the phagocytes leading to physical corpse internalization. The fourth step is the processing/digestion of the internalized targets and the regulation of the metabolic overload within the phagocytes
2.3.1 Find-Me Signals
The apoptotic cell is an active participant in its own clearance. The response of phagocytes to apoptotic cells can be influenced by several actions taken by the apoptotic cell. These include the release of find-me signals that attract phagocytes to the site of death and the exposure of eat-me signals that allow the phagocyte to distinguish the dying cell from its healthy neighbors [20].
For apoptotic cells to be rapidly cleared, which is the case in vivo, they must be rapidly “found” [21]. During homeostatic cell turnover in tissues, a single corpse might be surrounded by a vast number of healthy neighbors, and therefore “calling out” the professional phagocytes such as resident macrophages to come clear the apoptotic cell is important for the prompt removal. Several such “find-me” signals have been identified and may be differentially important depending on the situation. The first find-me signal proposed was lysophosphatidylcholine (LPC); however, the role LPC played to attract phagocytes seemed specific to both the type of apoptotic cell (the MCF-7 breast cancer line) and phagocyte (THP-1 monocyte line) used [22]. Furthermore, in vivo relevance of LPC as a find-me signal remains to be established. Later, an elegant study showed that cleavage of CX3CL1/Fractalkine (FKN) during apoptosis leads to release of a soluble fragment that induces the migration of monocytes to Burkitt lymphoma B-cells in vitro and to germinal centers in vivo [23]. This could also be relevant for attraction of monocytes and the complex interplay between macrophages and tumor cells in a tumor microenvironment. The significance of FKN has been established for locating apoptotic B-cells, but FKN per se as a universal find-me signal in other cell types is at present less defined. Finally, the triphosphate nucleotides ATP and UTP were found to be released in a regulated manner during apoptosis by the caspase-mediated cleavage of Pannexin-1 (PANX1), a transmembrane protein that forms hexameric hemichannels [24]. The nucleotides released by PANX1 cleavage are chemotactic for monocytes in vitro and in vivo by signaling through the nucleotide receptor P2Y2 [24, 25]. Although nucleotides clearly are relevant find-me signals, one of the interesting challenges with such nucleotide find-me signals is how far the nucleotide signal can travel before extracellular nucleotidases convert them into their nonchemotactic diphosphate and monophosphate forms. In addition to attracting phagocytes to the site of death, these find-me signals may also prime the phagocytes for engulfment, although this has only been shown in the case of FKN, which stimulates macrophages to produce the apoptotic cell bridging molecule milk fat globule-EGF factor 8 (MFG-E8, discussed later) [26, 27].
2.3.2 Eat-Me Signals
Once the phagocyte has been brought to the area of the dying cell, it must identify the specific cell that needs to be cleared, which is achieved by recognition of eat-me signals on the surface of the apoptotic cell. There are many “eat-me” markers identified to date on apoptotic cells that are linked to corpse uptake. The classic eat-me signal is the lipid phosphatidylserine (PtdSer) . It had been known that aged red blood cells lose their phospholipid asymmetry, but Fadok and colleagues demonstrated that PtdSer is also exposed by thymocytes as they undergo apoptosis [28]. Furthermore, they found that apoptotic thymocyte engulfment by macrophages is inhibited by the competitive addition of PtdSer-containing liposomes. Since then, PtdSer exposure has been found to be an evolutionarily conserved general feature of apoptosis from lower organisms to man and is now commonly used to assay the apoptotic status of a cell [29, 30].
Phosphatidylserine (PtdSer) as an eat-me signal has stood the test of time due to a preponderance of evidence of its importance [31]. Exogenous incorporation of PtdSer into the outer leaflet of viable cells in some cases is sufficient to cause their engulfment by macrophages, and PtdSer liposomes alone in certain circumstances can elicit some of the responses induced in the phagocyte [32, 33]. The asymmetric distribution of PtdSer in healthy cells is maintained through flippases that actively mediate the movement of PtdSer from the outer to the inner membrane [31]. In contrast, during apoptosis induction, the flippases appear to be inactivated, while another set of enzymes called “phospholipid scramblases ” become active, and the latter randomize the PtdSer levels between the outer and inner leaflets. The exposed PtdSer is then recognized by specific receptors on the phagocytes, contributing to corpse internalization [31, 34, 35]. The P4-ATPase family member ATP11C and its chaperone CDC50 have been identified as key components for the flippase function seen in healthy cells. With respect to the scramblases, members of the Xkr-family with six transmembrane domains appear to perform this role. Remarkably, both the Xkr8 scramblase and ATP11C flippase have sites that can be cleaved by apoptotic caspases [31, 34, 35]. Thus, in live cells, the flippase remains active while the scramblase is inactive, while this occurs in opposite ways after caspase-mediated cleavage of these proteins during apoptosis. Current evidence based on mutant proteins suggests that the flippase is likely more dominant in maintaining the PtdSer asymmetry and that it has to be inactivated for the scramblase to fully promote the PtdSer exposure.
While PtdSer exposure is clearly central in apoptotic cell recognition and widely studied, unfortunately that has been at the expense of thorough characterization of many other eat-me signals that have been seen in different apoptotic contexts. These include the ER resident protein calreticulin (CRT), which some studies have found to translocate to the cell surface during apoptosis [36, 37]. However, CRT has also been shown to play a role on the surface of the phagocyte in interacting with Mannose Binding Lectin (MBL) and complement C1q bound to the surface of apoptotic cells [38]. In addition to CRT and PtdSer, many modifications to the apoptotic cell surface have been implicated, such as the presentation of oxidized Low Density Lipoprotein (oxLDL)-like sites, changes to glycosylation such as the capping of CD43, the exposure of Annexin I, and the expression of ICAM3 [39–42]. Although these are less well characterized than PtdSer, they indicate that the apoptotic cell has many ways to make itself known to the phagocyte. Recently, DD1α, a p53-inducible protein that mediates homotypic interaction between apoptotic cells and phagocytes has been defined and has been linked to establishment of immune responses to cancer cells [43, 44].
2.3.3 The Engulfment Receptors and Bridging Molecules
Eat-me signals on the surface of apoptotic cells are not useful without cognate receptors on phagocytes to recognize the eat-me signals. This is the role of various engulfment receptors on phagocytes and other soluble bridging molecules. Due to the importance of phosphatidylserine, much work has been done to identify its receptors. Although a PtdSer recognizing membrane receptor (simply termed “PSR”) was first identified using an antibody that blocked apoptotic cell engulfment, this is no longer considered a PtdSer recognition receptor as the knockout of the gene in mice did not impact engulfment and PSR is now thought to be a nuclear protein [45]. Since then, multiple receptors that directly or indirectly bind PtdSer have been identified and play a role in engulfment (Fig. 2.2). In 2007, Brain Angiogenesis Inhibitor 1 (BAI1), T-cell immunoglobulin domain-containing 4 (TIM4), and Stabilin-2 (Stab2) were all identified as receptors that can bind directly to PtdSer [46–48]. Modifying BAI1, TIM4, or Stab2 levels altered the engulfment capacity of phagocytes in vitro. In the years since the identification of these receptors, other receptors have been proposed, including triggering receptor expressed on myeloid cells-like protein 2 (TLT2) and the receptor for advanced glycation end-products (RAGE) [49, 50]. The relative role of each of these engulfment receptors to apoptotic cell engulfment, either independent of the others or in cooperation, still needs to be fully elucidated. Some preliminary studies suggest that TIM-4 and MER proteins can cooperate in the clearance of apoptotic targets by peritoneal macrophages [51]. Moreover, MER homolog appears to phosphorylate ELMO1 proteins that also function downstream of BAI1 [52].

Recognition of apoptotic cells by phagocytes . Phosphatidylserine, one of the key eat-me signals on apoptotic cells, can be recognized either directly via phagocytic receptors or indirectly through bridging molecules. Although phosphatidylserine is a key recognition entity, a number of other eat-me markers can also participate to different degrees in the recognition and uptake of apoptotic cells
PtdSer can also be recognized indirectly by phagocytic receptors via bridging molecules. One of the first engulfment receptors identified was αvβ3 integrin, which has since been shown to bind apoptotic cells via the PtdSer-dependent bridging molecule MFG-E8 [8, 53]. Later, the receptor tyrosine kinase Mer (as well as its homologs Tyro3 and Axl, part of a family of receptors termed TAM receptors) was found to mediate corpse clearance [54, 55]. Mer functions by binding to growth arrest-specific gene 6 (Gas6) or protein S, which recognizes PtdSer [54] (see Chap. 6 for more details).
Other less well understood bridging molecules include C1q, MBL, and Thrombospondin-1 (TSP-1) [9, 38]. C1q is thought to opsonize late apoptotic and early necrotic cells [56]. It has multiple receptors, but its role in cell engulfment is thought to be through LDL-related receptor protein 1 (LRP1 or CD91), a multifunctional receptor that has also been found to mediate engulfment through CRT and MBL [36, 38]. Finally, TSP-1 was found to increase macrophage binding to apoptotic neutrophils and mediate their engulfment via the phagocytic receptor CD36 [9]. Other receptors implicated in cell engulfment include MEGF10, the inflammatory receptor CD14, the C1q receptor CD93 (although surprisingly its mechanism is thought to not be through C1q), class A and B scavenger receptors, and the ATP-binding cassette transporter 7 (ABCA7) [57]. The identification of many receptors that seem to all regulate apoptotic cell engulfment indicates that they are used by distinct cell types, work in concert as an “engulfment synapse,” or provide redundancy to the system. The fact that disruption of many of these receptors often results in a partial reduction in apoptotic cell uptake in vitro, and can lead to somewhat similar disease phenotypes in vivo, suggests that the first two possibilities are at least partially correct.
2.3.4 Intracellular Signaling in the Phagocyte
Once a phagocyte recognizes an apoptotic cell, signaling occurs to rearrange the cytoskeleton and engulf the target. In C. elegans, where much of the early work was done to identify some of the relevant engulfment genes, two phagocytic signaling pathways were discovered that share homology with mammalian engulfment pathways. In both nematodes and mammals, the pathways converge on the Rho family GTPase CED10/Rac1, which in turn signals through WAVE to Arp2/3, initiating actin nucleation and cytoskeletal rearrangement [58, 59]. Actin polymerization forms the phagocytic cup around the apoptotic cell and mediates the physical act of engulfment. The intracellular signals will be discussed briefly here, but more in-depth reviews can be found elsewhere [60–64].
The first evolutionarily conserved engulfment pathway contains the nemotode genes cell death defective-1 (CED-1), CED-6, and CED-7. In mammals, the orthologous pathway members, respectively, are LRP1 or MEGF10, engulfment adaptor GULP1, and ABCA1 or ABCA7 [61, 62]. In mammals, GULP1 has been shown to be downstream of LRP1 as well as Stab2, whereas the direct functions of ABCA1 and ABCA7 in engulfment have been controversial and may not play the same role as CED-7 in the nematode [65, 66]. Although this pathway requires Rac1 for engulfment, the mechanism by which GULP connects to Rac1 is currently unknown.
The second pathway that is shared between nematodes and mammals is the CED-2, CED-5, CED-12 pathway, corresponding to the mammalian proteins CrkII, Dock180, and engulfment and cell motility (ELMO), respectively. In this pathway, ELMO and Dock180 act together as a bipartite guanine nucleotide-exchange factor (GEF) for Rac1 activation [67]. The phagocytic receptor BAI1 has been shown to signal directly to ELMO, but other unknown receptors may feed in to the pathway through the TRIO protein activating the GTPase RhoG, which can activate ELMO to promote engulfment [47, 68]. In fact, it has been shown that the TAM family receptor MER can lead to phosphorylation of ELMO1. Finally, while CED-2/CrkII is associated with this pathway and has been found in complex with ELMO/Dock180 [69], its actual role is unclear, as ELMO and Dock180 can act without binding to CrkII [70]. This suggests that there may be subpathways within this group of genes regulating engulfment.
In addition to these known canonical signaling pathways, there are alternate mediators of cell engulfment. For example, it was proposed that RAGE acts as an engulfment receptor in mice by activating Rac1 through Diaphenous-1 (mDia1) [50]. The signaling pathways for many of the other phagocytic receptors linked to apoptotic cell clearance remain to be defined. TIM4 has a very short intracellular domain, which has been shown to be dispensable for its function in engulfment [71]. TIM4 appears to function cooperatively with the MER receptor in clearance of apoptotic cells in the peritoneum. Others have suggested that TIM4 works as a tether in conjunction with αvβ3/MFG-E8 to mediate engulfment; however, TIM4-mediated engulfment in the peritoneum is benefited by sequestration of MFG-E8 to prevent its binding to apoptotic cells, suggesting that TIM4 and αvβ3/MFG-E8 do not work in a single engulfment pathway in vivo [72, 73]. Interestingly, the sequestration of MFG-E8 is accomplished by the oxidation of PE on the surface of resident noninflammatory macrophages by 12/15-lipoxygenase as a way to prevent inflammatory infiltrating monocytes from recognizing and clearing the apoptotic cells and initiating an unintended immune response [73]. This highlights the importance of the correct engulfment signals occurring in the correct cell type for the right output, which is the generalized suppression of inflammation characteristic of apoptotic cell engulfment.
2.4 Effects of Apoptotic Cell Clearance
2.4.1 Induction of an Anti-inflammatory Program
One of the first recognized effects of apoptotic cells was their ability to induce the production of anti-inflammatory cytokines by engulfing phagocytes [6]. While the phagocyte encounter with bacterial lipopolysaccharide (LPS) induces production of inflammatory cytokines, phagocyte interaction with apoptotic cells instead stimulates the release of anti-inflammatory cytokines [6]. This finding changed the assumption that apoptotic cells were “immunologically inert” and that it is not just the lack of proinflammatory signals, rather apoptotic cells carry ligands that can actively induce an anti-inflammatory signaling within the phagocytes (Fig. 2.3). It was also shown that engulfment of apoptotic cells uniquely caused the release of anti-inflammatory signals compared to other forms of uptake, such as engulfment of zymosan or IgG-opsonized apoptotic cells [74]. Although it has been reported that PtdSer signaling alone is sufficient to induce these signals, such as the anti-inflammatory effects of PtdSer liposomes [33, 75], or the administration of PtdSer liposomes in mouse models of inflammation to reduce disease [76], the effect of isolated PtdSer liposomes is very variable and they are never as potent as whole apoptotic cells. Either the conformation of the PtdSer exposure on the apoptotic cells, or more likely, one or more additional signals on the apoptotic cells are necessary for the full elicitation of the anti-inflammatory responses from phagocytes.

Responses of phagocytes . When phagocytes (both professional phagocytes and nonprofessional phagocytes) engage and engulf apoptotic cells, they produce anti-inflammatory mediators such as TGF-β, interleukin-10 (IL-10), platelet activating factor (PAF), prostaglandin E2, as well as the membrane protein ABCA1 that induces anti-inflammatory effects by a yet to be defined mechanism. Apoptotic cell recognition also suppresses the release of proinflammatory cytokines such as TNFα, interleukin-1β, interleukin-12, and interleukin-8. The apoptotic cell recognition process also regulates the numbers of neutrophils and hematopoietic precursor cells via the cytokine interleukin-23 (IL-23) and the chemokine CXCL12
Transforming Growth Factor-β (TGF-β) is a classic example of an anti-inflammatory signal released as a consequence of apoptotic cell engulfment. It is transcriptionally upregulated by apoptotic cell recognition in a p38 MAPK, JNK, and ERK-dependent process [77]. The apoptotic cell recognition-dependent transcription, translation, and release of TGF-β after PtdSer recognition is not unique to professional phagocytes such as macrophages but also observed in bronchial epithelial cell lines and vascular smooth muscle cells [78, 79]. Similar to TGF-β , the transcriptional upregulation of IL-10 in phagocytes requires p38 MAPK, and the pharmacological inhibition of p38 completely abrogates the IL-10 response to apoptotic cells [80]. Besides TGF-β and IL-10, other anti-inflammatory mediators are produced via different means. Several eicosanoids are made in response to apoptotic cell engulfment, including prostaglandins E2 and I2 (PGE2, PGI2) [74, 75]. The increased production of these is due to upregulation of important enzymes in their synthetic pathways, including COX-2, PGES, and PGIS. However, this response was found to be dependent on TGF-β autocrine and paracrine signaling to the phagocyte, indicating that TGF-β is a master regulator of the cell’s anti-inflammatory response after the engulfment of corpses [75].
Phagocytes also respond to apoptotic cells by upregulating the cholesterol transporter ABCA1 [81, 82]. This upregulation is transcriptional and induced by recognition of PtdSer [82]. Recent studies show that the upregulation of ABCA1 downstream of apoptotic cell recognition occurs via a novel membrane-initiated pathway [83]. This induction of ABCA1 transcription was rapid, and at least in part, involves the upstream phagocytic receptor BAI1, as well as the downstream signaling intermediates ELMO1 and Rac1. Importantly, this ABCA1 upregulation does not involve the classic LXR-dependent ABCA1 upregulation that is normally seen with increase in intra cytoplasmic oxysterol levels.
The upregulation of ABCA1 can have several effects important for the anti-inflammatory tone of the cells. First, the upregulation of cholesterol transporters unsurprisingly increases cholesterol efflux from the phagocyte. The presence of cholesterol in membranes affects the signaling of cell surface receptors as well as the function of transporters, and therefore the ability to efflux excess cholesterol is an important response to engulfed cells to prevent cholesterol loading [84]. The inability to maintain cholesterol homeostasis can lead to activation of Toll-like receptors (TLRs) and inflammatory signaling [85]. However, ABCA1 (and ABCG1) have other effects as the absence of ABCA1 causes an inflammatory cell death of the phagocyte mediated by sustained JNK activation [86]. Finally, ABCA1 itself can signal as an anti-inflammatory surface protein by signals through ABCA1 to Jak2 and STAT3 [87]. Whether ABCA1 is truly a significant anti-inflammatory molecule in phagocytes immediately after apoptotic cell engulfment remains to be seen, but it is clear that ABCA1 can play important functions in the response to apoptotic cells.
The increase in anti-inflammatory receptors, intracellular signals, and secreted mediators by the phagocyte are important for the dampening of immune responses/local inflammation within a tissue context. In addition to just acting in the engulfing phagocyte, the secreted immunosuppressive cytokines can signal back on the phagocyte as well as to neighboring cells to suppress their proinflammatory responses.
2.4.2 Suppression of Proinflammatory Signals
The suppression of proinflammatory signals is subtly different from the active initiation of an anti-inflammatory program, although the two are connected (Fig. 2.3). When peripheral blood mononuclear cells (PBMCs) were preincubated with apoptotic cells before being stimulated with LPS, they produced less of the inflammatory cytokines TNFα, IL-1β, and IL-12 [6]. The suppression of TNFα, IL-8, GM-CSF, and proinflammatory eicosanoids like leukotriene C4 is mediated by the autocrine signaling from released TGFβ, Platelet Activating Factor (PAF), and PGE2 [74]. Although some of the immunosuppressive effects of apoptotic cells can be traced back to the release of anti-inflammatory cytokines by phagocytes, this is not the only mechanism as LPS-induced production of IL-12 gets suppressed by treatment with apoptotic cells independent of TGFβ or IL-10 [88]. The suppression of IL-12 expression was found to be through GC-binding protein (GC-BP), a zinc finger nuclear factor that binds to the IL-12 promoter after apoptotic cell recognition [88]. Others have shown that LXR is crucial for the apoptotic cell-dependent suppression of IL-12 expression; however, LXR also controls a positive feedback loop whereby apoptotic cells signal through LXR to upregulate apoptotic cell receptors [89]. Sorting out the precise signaling pathways downstream of apoptotic cell recognition, the specific transcription factors activated, which genes they activate, and how they link to upregulation of anti-inflammatory cytokines and suppression of proinflammatory signals is a key challenge in the field.
2.5 Defective Apoptotic Cell Clearance and Inflammatory Disease
2.5.1 Autoimmunity
Considering the anti-inflammatory effects of apoptotic cell clearance, it is no surprise that defects in the engulfment of apoptotic cells often result in systemic inflammation. The cause of the inflammation likely depends on the cause of the engulfment defect, but the presence of uncleared apoptotic cells which then undergo secondary necrosis provides a sufficient inflammatory stimulus to cause autoimmunity [3, 5, 90]. Apoptotic cells in the absence of clearance naturally progress to secondarily necrotic cells and lose their membrane integrity, resulting in the release of inflammatory intracellular contents [12, 91]. Some cells during apoptosis may even release proinflammatory cytokines over time [92]. This stresses the importance of efficient clearance early in the apoptotic program as a way to diffuse a ticking time bomb of inflammation and prevent homeostatic cell turnover from inducing a detrimental response. Before much of the apoptotic cell receptors or intracellular signals important for clearance were established, it was observed that excess apoptotic cells injected into mice were immunogenic, inducing the production of antibodies indicative of autoimmunity such as antinuclear and anticardiolipin antibodies [11]. Similarly, blocking the uptake of endogenous apoptotic cells in mice by masking PtdSer to prevent its recognition by phagocytes results in autoimmunity [93].
Now that some of the opsonins of apoptotic cells as well as engulfment receptors have been discovered, autoimmunity has been observed as a frequent result of their deletion in mice. Deficiency of the apoptotic cell bridging molecules C1q or MFG-E8 both result in marked autoimmunity as measured by autoantibodies and the development of glomerulonephritis [94, 95]. Consequently, disrupting the ability of phagocytes to bind MFG-E8 by deleting αv integrin also results in autoimmunity [96]. Receptors for apoptotic cells using other bridging molecules are not able to compensate for this loss to maintain immune tolerance, and in fact deleting a component of seemingly independent engulfment pathways all result in autoimmunity. For example, preventing signaling by Mer, a receptor for the apoptotic cell bridging molecule Gas6, also results in lupus-like autoimmunity in mice [55, 97]. If direct recognition of PtdSer on apoptotic cells by TIM4 is eliminated by genetic deletion, again autoimmunity results [98, 99].
In all of these mouse models in which autoimmunity occurs, there are also lingering uncleared apoptotic cells in vivo, making it difficult to uncouple these two features. However, DNaseII-deficient macrophages are able to phagocytose apoptotic cells but lack the ability to degrade the engulfed nuclei, resulting in an abundance of DNA-containing bodies throughout the embryo [100, 101]. Although global deletion of DNaseII in mice is embryonic lethal, the induced deletion of DNaseII in adult mice causes the development of autoimmune polyarthritis similar to rheumatoid arthritis [102]. The macrophages that are still able to efficiently engulf apoptotic cells are unable to process the corpses and subsequently have an inflammatory phenotype, including high TNFα expression [102]. These findings suggest that although cells undergoing secondary necrosis can be sufficiently immunogenic, the anti-inflammatory signaling in the phagocytes is also important to prevent autoimmunity.
In patients with systemic lupus erythematosus (SLE) , a prototypical autoimmune disease characterized by chronic systemic inflammation, there is an increase in uncleared apoptotic cells in lymph node germinal centers, suggesting failed clearance [103]. The engulfment capacity of peripheral blood mononuclear cells (PBMCs) isolated from SLE patients is markedly decreased compared to PBMCs from normal healthy donors [104]. Whether impaired clearance is a cause or an effect of autoimmunity in humans is yet to be definitively established.
2.5.2 Airway Inflammation
The lung is an interesting model organ to examine how apoptotic cells interact with multiple cell types, including professional phagocytes such as macrophages and nonprofessional phagocytes such as neighboring epithelial cells. The phagocytic responsibilities of these populations could vary based on the different circumstances. For example, during acute injury when there is increased apoptosis of epithelial cells as well as the infiltrating neutrophils, the professional phagocytes are likely the most capable cell type to handle the increased load. This is seen when excessive apoptotic cells are instilled into the lungs of mice. Although multiple cell types are able to clear them, only CD103+ dendritic cells are able to traffic the engulfed apoptotic cells to the lung-draining lymph node to present antigen [105]. In an acute lung injury model in mice, MFG-E8 deficiency exacerbated the effects, suggesting that engulfment is protective in the lung [106].
Mice in which Rac1 is deleted specifically in the epithelial cells are more susceptible to house dust mite (HDM) or ovalbumin-induced airway inflammation [79]. Interestingly, airway epithelial cells rather than the macrophages from these mice have impaired engulfment of apoptotic cells, suggesting that engulfment by the epithelial cells helps to maintain tolerance to allergens. In this context, the cytokines secreted by the engulfing epithelial cells, especially IL-10, appear to be important for the prevention of disease [79]. Although Rac1 is linked to many functions outside of apoptotic cell engulfment, the in vivo and in vitro evidence implicates Rac1 as important in the initiation of airway inflammation. In support of the importance of epithelial cells as engulfers of apoptotic cells, it has been shown that epithelial cells from cystic fibrosis patients, who have increased numbers of free apoptotic cells in their sputum, are defective in their ability to engulf apoptotic cells due to increased levels of RhoA, a GTPase with an inhibitory role in engulfment [107].
In other diseases of the lung, such as emphysema and chronic obstructive pulmonary disease (COPD) , apoptotic cell clearance is beneficial. The porcine pancreatic elastase model of emphysema in mice is driven partially by apoptosis in the lung. Cotreatment of these mice with Annexin V to block apoptotic cell uptake worsened the emphysema [108]. Furthermore, smoking has been linked to impaired engulfment of apoptotic cells [109]. The alveolar macrophages from patients with COPD exhibit defective engulfment capacity in vitro, corresponding to the increase in free apoptotic cells in the lungs of these patients [110].
2.5.3 Inflammatory Colitis
An example of an inflammatory disease that may be affected by the clearance of apoptotic cells is inflammatory bowel disease. In mouse models of colitis, mice treated with dextran sulfate sodium (DSS) develop reversible acute intestinal inflammation associated with massive apoptosis. When DSS is given to mice lacking MFG-E8, the colitis is more severe than in treated wild-type mice [111]. Mirroring the effect of MFG-E8, mice with conditional knockout of αv integrin in hematopoietic cells develop spontaneous systemic inflammation, including severe colitis [96]. A causative link between impaired engulfment and the extent of colitis in mice has not been made, and in fact it has been suggested that the effects of MFG-E8 are through alternate anti-inflammatory mechanisms [112]. Recent studies suggest that the levels of the phagocytic receptor BAI1 are altered during DSS-induced colitis and also in human patients with ulcerative colitis [113]. Mice deficient in BAI1 have a much more pronounced disease with large numbers of uncleared apoptotic cells. Perhaps most telling of the relevance of apoptotic cell clearance in this disease model, mice with transgenic overexpression of BAI1 show attenuated disease, with reduction in proinflammatory cytokines and overall reduced inflammation. Interestingly, BAI1 expression in intestinal epithelial cells (rather than the myeloid cells) was critical for the beneficial effect, once again highlighting the key role played by nonprofessional phagocytes during inflammation [113].
2.6 Phagocytes, Anti-inflammatory Responses, and Cancer Context
2.6.1 Phagocytic Signaling Pathways in a Malignant Tissue
It is well known that apoptotic cells are found in many types of solid tumors along with mononuclear phagocytes, particularly, tumor-associated macrophages (TAMs) . The current evidence based on in vitro studies, and profiling of the TAMs suggests that the collection of engulfment proteins expressed in these TAMs is not vastly different from conventional tissue-resident macrophages. Therefore, the types of signaling pathways activated during apoptotic cell clearance in the tumor are likely not vastly different from the normal uptake mechanisms. But the real challenge is determining the interplay between the apoptotic tumor cells and the TAMs (see Chap. 3 for more discussion). Some of these challenges include: the type of responses initiated within the phagocytes upon contact with apoptotic and live tumor cells within the tumor tissue; the nature of death within the tumor; hypoxia versus normoxia (depending on the extent of vascularization of the tumor tissue) conditions when the phagocytes engulf; the effect of factors secreted by the healthy and dying tumor cells on the TAMs; the rates of proliferation within the tumor; and finally, the cross talk between the normal cells, tumor cells, and the TAMs and how they may influence the response of the engulfing phagocytes. Currently, there are significant efforts to induce immune responses to tumors via immunogenic cell death (see Chap. 7).
Interestingly, a number of engulfment genes that are linked to apoptotic cell clearance are also linked to tumor development in different tissues. These have been identified either via large-scale screening approaches, individual candidate gene approaches, or via altered expression profiles. We have chosen four examples below to highlight the link between engulfment genes and cancer.
2.6.2 BAI1
The gene encoding the phagocytic receptor BAI1 was initially identified as a p53-inducible gene whose expression was severely reduced or lost in glioblastomas (GBM) [114, 115]. Moreover, it was shown that the thrombospondin repeats of BAI1 (which also binds PtdSer) could function to inhibit angiogenesis [115]. Given the high degree of vascularization of GBM, it was initially hypothesized that the loss of BAI1 might influence the state of vascularization within the tumors [116–119]. However, given the recent studies directly linking BAI1 to apoptotic cell clearance, this needs to be revisited. GBMs are also known to have a number of apoptotic and necrotic cells, with characteristic “palisade necrosis” seen within the advanced stages of GBM (often lacking BAI1 expression). Whether this is due to defective clearance or defective angiogenesis or both remains to be determined.
2.6.3 ELMO1
The engulfment adapter protein ELMO1 has been linked to different types of cancers and is likely involved in apoptotic cell clearance and its role in tumor cell migration [120–124]. While macrophages express one of the highest levels of ELMO1, ELMO1 is widely expressed and mutations in ELMO1 that confer activation have been linked to head and neck tumors. Since ELMO1 associates with Dock180 to act as a guanine nucleotide exchange factor to activate Rac1 during cell migration [121], ELMO1 has been linked to metastasis of tumor cells in different contexts.
2.6.4 Pannexin Channels
In addition to the known role of Panx1 in the release of find-me signals from apoptotic cells, recently a mutant variant form of pannexin has been identified in highly metastatic human breast cancer cells [125]. This variant has been linked to extrusion of tumor cells from small vessels and metastasis. Although the precise mechanism by which the short fragment of Panx1 mediates this response of tumor cells is unclear, because Panx1 is a hexameric channel, it is possible that there is interference with the “holo channel.” Work in the context of apoptosis already suggests that Panx1 can influence cell shape and the cytoskeleton [126], and the short fragment of Panx1 might influence or augment the natural function of Panx1.
2.6.5 MER Tyrosine Kinase
The tyrosine kinase phagocytic receptor MER-TK has been linked to many different types of cancers [127, 128]. Besides its high expression in macrophages, and the induction of MER-TK expression by different stimuli and conditions, MER-TK is also expressed on certain epithelial cells [113]. Moreover, in gene expression profiling studies MER-TK expression has been shown to be both upregulated and downregulated [129]. Given the anti-inflammatory properties associated with MER-TK expression on macrophages [130], there could be complex scenarios depending on MER-TK expression in the macrophages versus tumor cells (see Chap. 6 for more details).
2.7 Concluding Thoughts
Our understanding of apoptotic cell clearance has expanded greatly in the past few decades, both mechanistically and in its physiological roles. There is also much work left to do to parse the complex signaling networks involved in clearance. There are inevitably more components of the engulfment machinery that remain to be identified, whether eat-me signals, engulfment receptors or coreceptors, or intracellular signaling pathways. Many of the players already known do not yet have clearly defined roles, which offer the opportunity to make new progress in this area. Furthermore, it is still unclear how different cell types utilize different engulfment pathways to clear cells and how these differences affect the response to apoptotic cells. A key aspect of tissue homeostasis is the loss/disposal of used/aged cells while replacing them with newer cells/regeneration. This is particularly important for cell types such as neutrophils that are produced in very large numbers, have a relatively short lifespan, and whose production has to be tightly coordinated. Since the loss of neutrophil numbers is a critical complication in many cancer therapies, defining the specific molecular details how the neutrophil death/engulfment/production cycle is coordinated in healthy and treated patients could be highly therapeutically relevant. Another of the beautiful challenges ahead in the context of cancer and cell death is determining under which contexts the apoptotic cell clearance is altered in tumors, whether the anti-inflammatory properties of apoptotic cell recognition are dampening an immune response or modifying the macrophages, how the tumor cells (via engulfment) might be made immunogenic, and how manipulation of the engulfment machinery can be of benefit. In addition, with recent evidence that apoptotic cell clearance can be augmented in vivo [113], the next level challenge is boosting cell clearance via small molecules and to target the myriad of inflammatory diseases.
References
Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. 2015;16:907–17.
Gregory CD, Pound JD. Cell death in the neighbourhood: direct microenvironmental effects of apoptosis in normal and neoplastic tissues. J Pathol. 2011;223:177–94.
Henson PM. Dampening inflammation. Nat Immunol. 2005;6:1179–81.
Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007;7:964–74.
Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–75.
Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–1.
Haslett C, Savill JS, Whyte MK, Stern M, Dransfield I, Meagher LC. Granulocyte apoptosis and the control of inflammation. Philos Trans R Soc Lond B Biol Sci. 1994;345:327–33.
Savill J, Dransfield I, Hogg N, Haslett C. Vitronectin receptor-mediated phagocytosis of cells undergoing apoptosis. Nature. 1990;343:170–3.
Savill J, Hogg N, Ren Y, Haslett C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J Clin Invest. 1992;90:1513–22.
Savill JS, Henson PM, Haslett C. Phagocytosis of aged human neutrophils by macrophages is mediated by a novel “charge-sensitive” recognition mechanism. J Clin Invest. 1989;84:1518–27.
Mevorach D, Zhou JL, Song X, Elkon KB. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med. 1998;188:387–92.
Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 2010;584:4491–9.
Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353–63.
Kroemer G, Senovilla L, Galluzzi L, Andre F, Zitvogel L. Natural and therapy-induced immunosurveillance in breast cancer. Nat Med. 2015;21:1128–38.
Galluzzi L, Bravo-Sanpedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, Baehrecke EH, Bazan NG, Bertrand MJ, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Campanella M, Candi E, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Dawson TM, Dawson VL, de Laurenzi V, de Maria R, Debatin KM, di Daniele N, Dixit VM, Dynlacht BD, el-Deiry WS, Fimia GM, Flavell RA, Fulda S, Garrido C, Gougeon ML, Green DR, Gronemeyer H, Hajnoczky G, Hardwick JM, Hengartner MO, Ichijo H, Joseph B, Jost PJ, Kaufmann T, Kepp O, Klionsky DJ, Knight RA, Kumar S, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, Lopez-Otin C, Lugli E, Madeo F, Malorni W, Marine JC, Martin SJ, Martinou JC, Medema JP, Meier P, Melino S, Mizushima N, Moll U, Munoz-Pinedo C, Nunez G, Oberst A, Panaretakis T, Penninger JM, Peter ME, Piacentini M, Pinton P, Prehn JH, Puthalakath H, Rabinovich GA, Ravichandran KS, Rizzuto R, Rodrigues CM, Rubinsztein DC, Rudel T, Shi Y, Simon HU, Stockwell BR, Szabadkai G, Tait SW, Tang HL, Tavernarakis N, Tsujimoto Y, vanden Berghe T, Vandenabeele P, Villunger A, Wagner EF, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58–73.
Arandjelovic S, Ravichandran KS. A MERry response after myocardial infarction. Circ Res. 2013;113:949–51.
Elliott MR, Ravichandran KS. ELMO1 signaling in apoptotic germ cell clearance and spermatogenesis. Ann N Y Acad Sci. 2010;1209:30–6.
Burstyn-Cohen T, Lew ED, Traves PG, Burrola PG, Hash JC, Lemke G. Genetic dissection of TAM receptor-ligand interaction in retinal pigment epithelial cell phagocytosis. Neuron. 2012;76:1123–32.
Prasad D, Rothlin CV, Burrola P, Burstyn-Cohen T, Lu Q, Garcia De Frutos P, Lemke G. TAM receptor function in the retinal pigment epithelium. Mol Cell Neurosci. 2006;33:96–108.
Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14:166–80.
Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. 2011;35:445–55.
Lauber K, Bohn E, Kröber SM, Xiao Y-J, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–30.
Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, Melville L, Melrose LA, Ogden CA, Nibbs R, Graham G, Combadiere C, Gregory CD. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 2008;112:5026–36.
Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–7.
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–6.
Leonardi-Essmann F, Emig M, Kitamura Y, Spanagel R, Gebicke-Haerter PJ. Fractalkine-upregulated milk-fat globule EGF factor-8 protein in cultured rat microglia. J Neuroimmunol. 2005;160:92–101.
Miksa M, Amin D, Wu R, Ravikumar TS, Wang P. Fractalkine-induced MFG-E8 leads to enhanced apoptotic cell clearance by macrophages. Mol Med. 2007;13:553–60.
Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–16.
Martin SJ, Reutelingsperger CP, Mcgahon AJ, Rader JA, van Schie RC, Laface DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–56.
Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39–51.
Segawa K, Nagata S. An apoptotic ‘eat me’ signal: phosphatidylserine exposure. Trends Cell Biol. 2015;25:639–50.
Borisenko GG, Matsura T, Liu S-X, Tyurin VA, Jianfei J, Serinkan FB, Kagan VE. Macrophage recognition of externalized phosphatidylserine and phagocytosis of apoptotic Jurkat cells—existence of a threshold. Arch Biochem Biophys. 2003;413:41–52.
Huynh M-LN, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50.
Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344:1164–8.
Suzuki J, Fujii T, Imao T, Ishihara K, Kuba H, Nagata S. Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J Biol Chem. 2013;288:13305–16.
Gardai SJ, Mcphillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg P-A, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–34.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61.
Ogden CA, Decathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, Henson PM. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. 2001;194:781–95.
Arur S, Uche UE, Rezaul K, Fong M, Scranton V, Cowan AE, Mohler W, Han DK. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev Cell. 2003;4:587–98.
Eda S, Yamanaka M, Beppu M. Carbohydrate-mediated phagocytic recognition of early apoptotic cells undergoing transient capping of CD43 glycoprotein. J Biol Chem. 2004;279:5967–74.
Gregory CD, Devitt A. The macrophage and the apoptotic cell: an innate immune interaction viewed simplistically? Immunology. 2004;113:1–14.
Moffatt OD, Devitt A, Bell ED, Simmons DL, Gregory CD. Macrophage recognition of ICAM-3 on apoptotic leukocytes. J Immunol. 1999;162:6800–10.
Yoon KW, Byun S, Kwon E, Hwang SY, Chu K, Hiraki M, Jo SH, Weins A, Hakroush S, Cebulla A, Sykes DB, Greka A, Mundel P, Fisher DE, Mandinova A, Lee SW. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science. 2015;349:1261669.
Zitvogel L, Kroemer G. CANCER. A p53-regulated immune checkpoint relevant to cancer. Science. 2015;349:476–7.
Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RA, Henson PM. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature. 2000;405:85–90.
Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–9.
Park D, Tosello-Trampont A-C, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–4.
Park S-Y, Jung M-Y, Kim H-J, Lee S-J, Kim S-Y, Lee B-H, Kwon T-H, Park R-W, Kim I-S. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15:192–201.
de Freitas A, Banerjee S, Xie N, Cui H, Davis KI, Friggeri A, Fu M, Abraham E, Liu G. Identification of TLT2 as an engulfment receptor for apoptotic cells. J Immunol. 2012;188:6381–8.
He M, Kubo H, Morimoto K, Fujino N, Suzuki T, Takahasi T, Yamada M, Yamaya M, Maekawa T, Yamamoto Y, Yamamoto H. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 2011;12:358–64.
Nishi C, Toda S, Segawa K, Nagata S. Tim4- and MerTK-mediated engulfment of apoptotic cells by mouse resident peritoneal macrophages. Mol Cell Biol. 2014;34:1512–20.
Abu-Thuraia A, Gauthier R, Chidiac R, Fukui Y, Screaton RA, Gratton JP, Cote JF. Axl phosphorylates Elmo scaffold proteins to promote Rac activation and cell invasion. Mol Cell Biol. 2015;35:76–87.
Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–7.
Nakano T, Ishimoto Y, Kishino J, Umeda M, Inoue K, Nagata K, Ohashi K, Mizuno K, Arita H. Cell adhesion to phosphatidylserine mediated by a product of growth arrest-specific gene 6. J Biol Chem. 1997;272:29411–4.
Scott RS, Mcmahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–11.
Gaipl US, Kuenkele S, Voll RE, Beyer TD, Kolowos W, Heyder P, Kalden JR, Herrmann M. Complement binding is an early feature of necrotic and a rather late event during apoptotic cell death. Cell Death Differ. 2001;8:327–34.
Hamon Y, Trompier D, Ma Z, Venegas V, Pophillat M, Mignotte V, Zhou Z, Chimini G. Cooperation between engulfment receptors: the case of ABCA1 and MEGF10. PLoS One. 2006;1, e120.
Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–69.
Kinchen JM, Cabello J, Klingele D, Wong K, Feichtinger R, Schnabel H, Schnabel R, Hengartner MO. Two pathways converge at CED-10 to mediate actin rearrangement and corpse removal in C. elegans. Nature. 2005;434:93–9.
He B, Lu N, Zhou Z. Cellular and nuclear degradation during apoptosis. Curr Opin Cell Biol. 2009;21:900–12.
Kinchen JM, Doukoumetzidis K, Almendinger J, Stergiou L, Tosello-Trampont A, Sifri CD, Hengartner MO, Ravichandran KS. A pathway for phagosome maturation during engulfment of apoptotic cells. Nat Cell Biol. 2008;10:556–66.
Kinchen JM, Hengartner MO. Tales of cannibalism, suicide, and murder: programmed cell death in C. elegans. Curr Top Dev Biol. 2005;65:1–45.
Kinchen JM, Ravichandran KS. Journey to the grave: signaling events regulating removal of apoptotic cells. J Cell Sci. 2007;120:2143–9.
Zhou Z, Yu X. Phagosome maturation during the removal of apoptotic cells: receptors lead the way. Trends Cell Biol. 2008;18:474–85.
Park S-Y, Kang K-B, Thapa N, Kim S-Y, Lee S-J, Kim I-S. Requirement of adaptor protein GULP during stabilin-2-mediated cell corpse engulfment. J Biol Chem. 2008;283:10593–600.
Su HP, Nakada-Tsukui K, Tosello-Trampont A-C, Li Y, Bu G, Henson PM, Ravichandran KS. Interaction of CED-6/GULP, an adapter protein involved in engulfment of apoptotic cells with CED-1 and CD91/low density lipoprotein receptor-related protein (LRP). J Biol Chem. 2002;277:11772–9.
Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, Tosello-Trampont A-C, Macara IG, Madhani H, Fink GR, Ravichandran KS. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol. 2002;4:574–82.
Debakker CD, Haney LB, Kinchen JM, Grimsley C, Lu M, Klingele D, Hsu P-K, Chou B-K, Cheng L-C, Blangy A, Sondek J, Hengartner MO, Wu Y-C, Ravichandran KS. Phagocytosis of apoptotic cells is regulated by a UNC-73/TRIO-MIG-2/RhoG signaling module and armadillo repeats of CED-12/ELMO. Curr Biol. 2004;14:2208–16.
Gumienny TL, Brugnera E, Tosello-Trampont AC, Kinchen JM, Haney LB, Nishiwaki K, Walk SF, Nemergut ME, Macara IG, Francis R, Schedl T, Qin Y, van Aelst L, Hengartner MO, Ravichandran KS. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell. 2001;107:27–41.
Tosello-Trampont A-C, Kinchen JM, Brugnera E, Haney LB, Hengartner MO, Ravichandran KS. Identification of two signaling submodules within the CrkII/ELMO/Dock180 pathway regulating engulfment of apoptotic cells. Cell Death Differ. 2007;14:963–72.
Park D, Hochreiter-Hufford A, Ravichandran KS. The phosphatidylserine receptor TIM-4 does not mediate direct signaling. Curr Biol. 2009;19:346–51.
Toda S, Hanayama R, Nagata S. Two-step engulfment of apoptotic cells. Mol Cell Biol. 2012;32:118–25.
Uderhardt S, Herrmann M, Oskolkova OV, Aschermann S, Bicker W, Ipseiz N, Sarter K, Frey B, Rothe T, Voll R, Nimmerjahn F, Bochkov VN, Schett G, Krönke G. 12/15-lipoxygenase orchestrates the clearance of apoptotic cells and maintains immunologic tolerance. Immunity. 2012;36:834–46.
Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–8.
Freire-De-Lima CG, Xiao YQ, Gardai SJ, Bratton DL, Schiemann WP, Henson PM. Apoptotic cells, through transforming growth factor-beta, coordinately induce anti-inflammatory and suppress pro-inflammatory eicosanoid and NO synthesis in murine macrophages. J Biol Chem. 2006;281:38376–84.
Ramos GC, Fernandes D, Charão CT, Souza DG, Teixeira MM, Assreuy J. Apoptotic mimicry: phosphatidylserine liposomes reduce inflammation through activation of peroxisome proliferator-activated receptors (PPARs) in vivo. Br J Pharmacol. 2007;151:844–50.
Xiao YQ, Freire-De-Lima CG, Schiemann WP, Bratton DL, Vandivier RW, Henson PM. Transcriptional and translational regulation of TGF-beta production in response to apoptotic cells. J Immunol. 2008;181:3575–85.
Fries DM, Lightfoot R, Koval M, Ischiropoulos H. Autologous apoptotic cell engulfment stimulates chemokine secretion by vascular smooth muscle cells. Am J Pathol. 2005;167:345–53.
Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, Borish L, Ravichandran KS. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature. 2013;493:547–51.
Chung EY, Liu J, Homma Y, Zhang Y, Brendolan A, Saggese M, Han J, Silverstein R, Selleri L, Ma X. Interleukin-10 expression in macrophages during phagocytosis of apoptotic cells is mediated by homeodomain proteins Pbx1 and Prep-1. Immunity. 2007;27:952–64.
Gerbod-Giannone M-C, Li Y, Holleboom A, Han S, Hsu L-C, Tabas I, Tall AR. TNFalpha induces ABCA1 through NF-kappaB in macrophages and in phagocytes ingesting apoptotic cells. Proc Natl Acad Sci U S A. 2006;103:3112–7.
Kiss RS, Elliott MR, Ma Z, Marcel YL, Ravichandran KS. Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr Biol. 2006;16:2252–8.
Fond AM, Lee CS, Schulman IG, Kiss RS, Ravichandran KS. Apoptotic cells trigger a membrane-initiated pathway to increase ABCA1. J Clin Invest. 2015;125:2748–58.
Lee AG. How lipids affect the activities of integral membrane proteins. Biochim Biophys Acta. 2004;1666:62–87.
Sun Y, Ishibashi M, Seimon T, Lee M, Sharma SM, Fitzgerald KA, Samokhin AO, Wang Y, Sayers S, Aikawa M, Jerome WG, Ostrowski MC, Bromme D, Libby P, Tabas IA, Welch CL, Tall AR. Free cholesterol accumulation in macrophage membranes activates toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K. Circ Res. 2009;104:455–65.
Yvan-Charvet L, Pagler TA, Seimon TA, Thorp E, Welch CL, Witztum JL, Tabas I, Tall AR. ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ Res. 2010;106:1861–9.
Tang C, Liu Y, Kessler PS, Vaughan AM, Oram JF. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem. 2009;284:32336–43.
Kim S, Elkon KB, Ma X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21:643–53.
A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Díaz M, Gallardo G, de Galarreta CR, Salazar J, Lopez F, Edwards P, Parks J, Andujar M, Tontonoz P, Castrillo A. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–58.
Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–30.
Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189:1059–70.
Cullen SP, Henry CM, Kearney CJ, Logue SE, Feoktistova M, Tynan GA, Lavelle EC, Leverkus M, Martin SJ. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol Cell. 2013;49:1034–48.
Asano K, Miwa M, Miwa K, Hanayama R, Nagase H, Nagata S, Tanaka M. Masking of phosphatidylserine inhibits apoptotic cell engulfment and induces autoantibody production in mice. J Exp Med. 2004;200:459–67.
Botto M, Dell’agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–9.
Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–50.
Lacy-Hulbert A, Smith AM, Tissire H, Barry M, Crowley D, Bronson RT, Roes JT, Savill JS, Hynes RO. Ulcerative colitis and autoimmunity induced by loss of myeloid alphav integrins. Proc Natl Acad Sci U S A. 2007;104:15823–8.
Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RAS, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196:135–40.
Rodriguez-Manzanet R, Sanjuan MA, Wu HY, Quintana FJ, Xiao S, Anderson AC, Weiner HL, Green DR, Kuchroo VK. T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc Natl Acad Sci. 2010;107:8706–11.
Wong K, Valdez PA, Tan C, Yeh S, Hongo J-A, Ouyang W. Phosphatidylserine receptor Tim-4 is essential for the maintenance of the homeostatic state of resident peritoneal macrophages. Proc Natl Acad Sci U S A. 2010;107:8712–7.
Kawane K, Fukuyama H, Yoshida H, Nagase H, Ohsawa Y, Uchiyama Y, Okada K, Iida T, Nagata S. Impaired thymic development in mouse embryos deficient in apoptotic DNA degradation. Nat Immunol. 2003;4:138–44.
Krieser RJ, Maclea KS, Longnecker DS, Fields JL, Fiering S, Eastman A. Deoxyribonuclease IIalpha is required during the phagocytic phase of apoptosis and its loss causes perinatal lethality. Cell Death Differ. 2002;9:956–62.
Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, Yoshikawa H, Nagata S. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006;443:998–1002.
Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, Kirchner T, Kalden JR, Herrmann M. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:191–201.
Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41:1241–50.
Desch AN, Randolph GJ, Murphy K, Gautier EL, Kedl RM, Lahoud MH, Caminschi I, Shortman K, Henson PM, Jakubzick CV. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J Exp Med. 2011;208:1789–97.
Aziz M, Matsuda A, Yang W-L, Jacob A, Wang P. Milk fat globule-epidermal growth factor-factor 8 attenuates neutrophil infiltration in acute lung injury via modulation of CXCR2. J Immunol. 2012;189:393–402.
Vandivier RW, Richens TR, Horstmann SA, Decathelineau AM, Ghosh M, Reynolds SD, Xiao Y-Q, Riches DW, Plumb J, Vachon E, Downey GP, Henson PM. Dysfunctional cystic fibrosis transmembrane conductance regulator inhibits phagocytosis of apoptotic cells with proinflammatory consequences. Am J Physiol Lung Cell Mol Physiol. 2009;297:L677–86.
Yoshida S, Minematsu N, Chubachi S, Nakamura H, Miyazaki M, Tsuduki K, Takahashi S, Miyasho T, Iwabuchi T, Takamiya R, Tateno H, Mouded M, Shapiro SD, Asano K, Betsuyaku T. Annexin V decreases PS-mediated macrophage efferocytosis and deteriorates elastase-induced pulmonary emphysema in mice. Am J Physiol Lung Cell Mol Physiol. 2012;303:L852–60.
Minemetsu N, Blumental-Perry A, Shapiro SD. Cigarette smoke inhibits engulfment of apoptotic cells by macrophages through inhibition of actin rearrangement. Am J Respir Cell Mol Biol. 2011;44:474–82.
Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81:289–96.
Chogle A, Bu H-F, Wang X, Brown JB, Chou PM, Tan X-D. Milk fat globule-EGF factor 8 is a critical protein for healing of dextran sodium sulfate-induced acute colitis in mice. Mol Med. 2011;17:502–7.
Aziz MM, Ishihara S, Mishima Y, Oshima N, Moriyama I, Yuki T, Kadowaki Y, Rumi MAK, Amano Y, Kinoshita Y. MFG-E8 attenuates intestinal inflammation in murine experimental colitis by modulating osteopontin-dependent alphavbeta3 integrin signaling. J Immunol. 2009;182:7222–32.
Lee CS, Penberthy KK, Wheeler KM, Juncadella IJ, Vandenabeele P, Lysiak JJ, RavichandranKS. Boosting Apoptotic Cell Clearance by Colonic Epithelial Cells Attenuates Inflammation In Vivo. Immunity. 2016 Apr 19;44(4):807–20. doi: 10.1016/j.immuni.2016.02.005.
Kaur B, Brat DJ, Devi NS, van Meir EG. Vasculostatin, a proteolytic fragment of brain angiogenesis inhibitor 1, is an antiangiogenic and antitumorigenic factor. Oncogene. 2005;24:3632–42.
Nishimori H, Shiratsuchi T, Urano T, Kimura Y, Kiyono K, Tatsumi K, Yoshida S, Ono M, Kuwano M, Nakamura Y, Tokino T. A novel brain-specific p53-target gene, BAI1, containing thrombospondin type 1 repeats inhibits experimental angiogenesis. Oncogene. 1997;15:2145–50.
Cork SM, VAN Meir EG. Emerging roles for the BAI1 protein family in the regulation of phagocytosis, synaptogenesis, neurovasculature, and tumor development. J Mol Med (Berl). 2011;89:743–52.
Kaur B, Brat DJ, Calkins CC, van Meir EG. Brain angiogenesis inhibitor 1 is differentially expressed in normal brain and glioblastoma independently of p53 expression. Am J Pathol. 2003;162:19–27.
Stephenson JR, Paavola KJ, Schaefer SA, Kaur B, van Meir EG, Hall RA. Brain-specific angiogenesis inhibitor-1 signaling, regulation, and enrichment in the postsynaptic density. J Biol Chem. 2013;288:22248–56.
Zhu D, Hunter SB, Vertino PM, van Meir EG. Overexpression of MBD2 in glioblastoma maintains epigenetic silencing and inhibits the antiangiogenic function of the tumor suppressor gene BAI1. Cancer Res. 2011;71:5859–70.
Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, Bandla S, Imamura Y, Schumacher SE, Shefler E, McKenna A, Carter SL, Cibulskis K, Sivachenko A, Saksena G, Voet D, Ramos AH, Auclair D, Thompson K, Sougnez C, Onofrio RC, Guiducci C, Beroukhim R, Zhou Z, Lin L, Lin J, Reddy R, Chang A, Landrenau R, Pennathur A, Ogino S, Luketich JD, Golub TR, Gabriel SB, Lander ES, Beer DG, Godfrey TE, Getz G, Bass AJ. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45:478–86.
Grimsley CM, Kinchen JM, Tosello-Trampont AC, Brugnera E, Haney LB, Lu M, Chen Q, Klingele D, Hengartner MO, Ravichandran KS. Dock180 and ELMO1 proteins cooperate to promote evolutionarily conserved Rac-dependent cell migration. J Biol Chem. 2004;279:6087–97.
Li H, Yang L, Fu H, Yan J, Wang Y, Guo H, Hao X, Xu X, Jin T, Zhang N. Association between Galphai2 and ELMO1/Dock180 connects chemokine signalling with Rac activation and metastasis. Nat Commun. 2013;4:1706.
Shi L, Zhang B, Sun X, Zhang X, Lv S, Li H, Wang X, Zhao C, Zhang H, Xie X, Wang Y, Zhang P. CC chemokine ligand 18(CCL18) promotes migration and invasion of lung cancer cells by binding to Nir1 through. Nir1-ELMO1/DOC180 signaling pathway. Mol Carcinog. 2016 Jan 12. doi: 10.1002/mc.22450.
Zhang B, Shi L, Lu S, Sun X, Liu Y, Li H, Wang X, Zhao C, Zhang H, Wang Y. Autocrine IL-8 promotes F-actin polymerization and mediate mesenchymal transition via ELMO1-NF-kappaB-Snail signaling in glioma. Cancer Biol Ther. 2015;16:898–911.
Furlow PW, Zhang S, Soong TD, Halberg N, Goodarzi H, Mangrum C, Wu YG, Elemento O, Tavazoie SF. Mechanosensitive pannexin-1 channels mediate microvascular metastatic cell survival. Nat Cell Biol. 2015;17:943–52.
Poon IK, Chiu YH, Armstrong AJ, Kinchen JM, Juncadella IJ, Bayliss DA, Ravichandran KS. Unexpected link between an antibiotic, pannexin channels and apoptosis. Nature. 2014;507:329–34.
Lemke G, Burstyn-Cohen T. TAM receptors and the clearance of apoptotic cells. Ann N Y Acad Sci. 2010;1209:23–9.
Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8:327–36.
Bosurgi L, Bernink JH, Delgado Cuevas V, Gagliani N, Joannas L, Schmid ET, Booth CJ, Ghosh S, Rothlin CV. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc Natl Acad Sci U S A. 2013;110:13091–6.
Rothlin CV, Carrera-Silva EA, Bosurgi L, Ghosh S. TAM receptor signaling in immune homeostasis. Annu Rev Immunol. 2015;33:355–91.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Fond, A.M., Ravichandran, K.S. (2016). Clearance of Dying Cells by Phagocytes: Mechanisms and Implications for Disease Pathogenesis. In: Gregory, C. (eds) Apoptosis in Cancer Pathogenesis and Anti-cancer Therapy. Advances in Experimental Medicine and Biology, vol 930. Springer, Cham. https://doi.org/10.1007/978-3-319-39406-0_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-39406-0_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-39404-6
Online ISBN: 978-3-319-39406-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)