
Abstract
In a DNA microarray dataset, gene expression data often has a huge number of features(which are referred to as genes) versus a small size of samples. With the development of DNA microarray technology, the number of dimensions increases even faster than before, which could lead to the problem of the curse of dimensionality. To get good classification performance, it is necessary to preprocess the gene expression data. Support vector machine recursive feature elimination (SVM-RFE) is a classical method for gene selection. However, SVM-RFE suffers from high computational complexity. To remedy it, this paper enhances SVM-RFE for gene selection by incorporating feature clustering, called feature clustering SVM-RFE (FCSVM-RFE). The proposed method first performs gene selection roughly and then ranks the selected genes. First, a clustering algorithm is used to cluster genes into gene groups, in each which genes have similar expression profile. Then, a representative gene is found to represent a gene group. By doing so, we can obtain a representative gene set. Then, SVM-RFE is applied to rank these representative genes. FCSVM-RFE can reduce the computational complexity and the redundancy among genes. Experiments on seven public gene expression datasets show that FCSVM-RFE can achieve a better classification performance and lower computational complexity when compared with the state-the-art-of methods, such as SVM-RFE.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
DNA microarray technology can monitor the expression level of a large number of genes, which helps us to detect the biological nature of gene expression data. Meanwhile, with the rapid development of DNA microarray technology, gene expression data grows explosively. The number of gene expression measurements remains in the hundreds, compared to tens of thousands of genes involved. However, several studies have shown that most genes measured in a DNA microarray experiment are not relevant in the accurate classification of different classes of the problem [15]. Thus, the importance of selecting relevant genes before designing a classifier can be over-emphasized [4]. The selected genes could provide vital clues for understanding the disease mechanism.
Gene expression data reduction involves two aspects: relevant and redundant. The relevancy between genes and the label information is measured with respect to the class labels, which is related to the importance of a gene for the classification task [24]. Highly correlated genes tend to deteriorate the generalization performance and become redundant for classification tasks [44]. Usually, an optimal performance could be achieved by a set of maximal relevance and minimal redundancy genes.
Feature(gene) selection as a means of dimension reduction in machine learning and pattern recognition has attracted many researchers. Feature selection aims to select the most representative feature subset with a high resolution by eliminating redundant and unimportant features [19]. The aim of feature selection is to select the subset of features which has feature metric maximization. Generally speaking, feature selection has three advantages. First, feature selection can reduce the dimension of the data. Second, feature selection can enhance the generalization performance of the classifier in case of feature redundancy. In other words, the classifier modelled by the feature subset can improve the classification accuracy. Third, feature selection can deepen the understanding of the data when data visualization is possible.
Considering whether the evaluation criterion involves classification models, gene selection methods can be divided into three categories: the filter gene selection [10, 15, 22, 27], the wrapper gene selection [9, 11, 16, 26, 34, 39] and the embedded gene selection [37]. Filter methods are independent of classifiers and can select a gene subset from an original dataset using specific evaluation criteria which are mostly based on statistical methods. Relief [22] and MRMR (minimal redundancy-maximal relevance) [10] are two typical filter gene selection algorithms. ReliefF was proposed as an extension of Relief in [25] by Kononeill et al. Zhang et al. proposed a hybrid method which combines ReliefF and MRMR in [45]. Specifically, the candidate gene set is first identified by ReliefF. Then, the redundancy is minimized with the help of MRMR, which facilitates to select an effectual gene subset from the candidate set. These algorithms are simple but efficient, and are widely used in gene selection. Generally, filter methods have a low computational complexity, but may result in an unsatisfactory classification accuracy. Wrapper methods can select a gene subset by employing the performance of the classifier to evaluate the importance of gene subsets. Compared with filter methods, wrapper methods can usually achieve a higher classification accuracy and have a higher computational complexity [18]. Embedded methods combine the advantages of filter and wrapper techniques by using a pre-determined classifier model to perform gene selection.
Among the three kinds of gene selection methods, wrapper methods have considerably attracted the attention of researchers. Support vector machine recursive feature elimination (SVM-RFE) is a typical wrapper feature selection method, which adopts the manner of a sequential backward elimination [16]. SVM-RFE can rank genes by taking weights generated by SVM as the ranking criterion of genes. However, weights generated by SVM does not account for the redundancy among the genes [42]. In addition, SVM-RFE has a high computational complexity when dealing with the high-dimension data. In [30], SVM-RFE and MRMR are combined to select genes. This new method incorporates a mutual-information-based MRMR filter into SVM-RFE to minimize the redundancy among selected genes, which can improve the accuracy of classification and yield a smaller gene set compared with both MRMR and SVM-RFE. However, the computational complexity of the new method is still high.
To reduce the computational complexity of SVM-RFE, this paper proposes a gene selection method called Feature Clustering-based Support Vector Machine Recursive Feature Elimination (FCSVM-RFE).
The proposed method first performs gene selection roughly and then ranks the selected genes. First, a clustering algorithm is used to cluster genes into gene groups. Clustering genes according to their expression profiles is an important step for interpreting data from microarray studies. Clustering can help predict gene functions, as co-expressed genes are more likely to have similar functions than non-co-expressed genes [13]. The clustering method employed here is the widely used K-means, which outperforms the other algorithms, such as CRC (Chinese Restaurant Clustering) and ISA (Iterative Signature Algorithm), especially on typical microarray brain expression datasets [33]. Then, a gene that can be used to represent a gene group is found. By doing so, we can obtain a feature subset. Then, SVM-RFE is applied to rank genes from the obtained feature subset. Experiments on seven public gene expression datasets show that FCSVM-RFE achieves a better performance and a lower computational complexity than other compared methods.
The remainder of this paper is organized as follows. Section 2 briefly reviews SVM, Relief and MRMR, respectively. FCSVM-RFE is presented in Section 3. Section 4 gives extensive experimental results and analyzes the proposed model. Conclusions are provided in Section 5.
2 Related works on gene selection
2.1 Support vector machine
Support vector machine (SVM) proposed in [40] is a learning algorithm based on statistical learning theory. SVM implements the principle of structure risk minimization which is to minimize the empirical error and the complexity of the learner at the same time, and achieves good generalization performance in classification and regression tasks. The goal of SVM for classification is to construct the optimal hyperplane with the largest margin. In general, the larger the margin is, the lower the generalization error of the classifier has.
Given a set of training samples \(\{(\mathbf {x}_{i},y_{i})\}^{N}_{i=1}\), where \(\mathbf {x}_{i} \in \mathbb {R}^{D}\), y i ∈{(+1,−1)} is the label of x i ,D and N are the dimension and the number of samples, respectively, SVM solves the following primal optimization problem:
where w and b are the weight and threshold of the hyperplane w ⋅x + b = 0, respectively, C > 0 is a regulation parameter and ζ i is the slack variable. Tuning C can make a balance between the minimization of misclassification and the maximization of the margin of the hyperplane. By introducing Lagrange multipliers, the dual problem of (1) can be described as follows:
where α i is the Lagrange multiplier. The weight vector can be expressed using Lagrange multipliers and training samples:
The discriminant function of SVM has the form:
where s g n(⋅) is the sign function.
The training of linear SVM has a computational complexity of O(N D).
2.2 Relief
Relief is a feature weighting algorithm, which is limited to classification problems with two classes [22]. The main idea behind Relief is to estimate attributes according to how well their values distinguish among the instances that are near to each other. Relief first randomly selects a sample x. Then Relief finds one nearest neighbor from the same class, called nearest hits x H and one nearest neighbor from different classes, called nearest misses x M, respectively. Relief updates the quality estimation w for all features depending on their values for x i , hits x H and misses x M. The rationale of the formula for updating the weights of feature p is that a good attribute should have the same value for instances from the same class (subtracting the difference d i f f(p,x,x H) and should differentiate between instances from different classes (adding the difference d i f f(p,x,x M). The process is repeated for m times, where m is the number of iterations. The detail algorithm for Relief is shown in Algorithm 1 [10].
Relief is simple and efficient. However, Relief concerns the label information instead of the redundancy between the selected features.
For N training samples with D attributes, Relief has a computational complexity of O(m N D) where m can be taken as the iteration times [22].

2.3 MRMR (minimal redundancy-maximal relevance)
MRMR aims at selecting a maximally relevant and minimally redundant set of genes for discriminating tissue classes [10]. Here, we introduce the mutual-information-based MRMR criterion to find a set of genes with maximally relevance and minimally redundance.
If the expressions of genes are randomly or uniformly distributed in different classes, the mutual information among these classes is zero. If genes are strongly differentially expressed for different classes, then the mutual information should be large. Thus, MRMR uses the mutual information as a measure of relevance of genes [32].
Assume that there is a set of training samples \(\{(\mathbf {x}_{i},y_{i})\}^{N}_{i=1}\), where \(\mathbf {x}_{i} \in \mathbb {R}^{D}\), and y i ∈{(+1,−1)} is the label of x i . Let X be the sample matrix where x i is the i-th row. Without loss in generality, we use g i to represent the i-th column of X, or the i-th gene. Let S be the selected feature subset, where S ⊂{1,2,⋯ ,D}. The mutual information between the class label vector y = [y 1,⋯ ,y N ]T and g i can quantify the relevancy of gene i for the classification task. The relevancy R S of genes is defined as:
where I(y,g i ) is the mutual information between the class label vector y and gene i, and is defined as:
The redundancy of a gene subset is determined by the mutual information among the genes. The redundancy of gene with the other genes in the subset S is given by:
In MRMR, the gene ranking is performed by optimizing the ratio of the relevancy of a gene to the redundancy of the genes in the set. The maximally relevant and minimally redundant gene i ∗ in the set S is found by:
Exact solution to MRMR requires O(|S|2 D) computational complexity.
3 FCSVM-RFE (feature clustering based support vector machine recursive feature elimination)
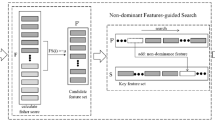
Because SVM-RFE does not account for the redundancy among the genes and has a high computational complexity, this paper presents an enhanced feature selection algorithm based on SVM-RFE, called feature clustering based support vector machine recursive feature elimination (FCSVM-RFE). The framework of FCSVM-RFE is shown in Fig. 1. There are three stages in the proposed method, gene clustering, gene representation and gene ranking. We first roughly cluster the gene expressions into gene groups so as to eliminate the redundancy of the gene data, and then find representative genes for these gene groups. Finally, we rank the representative gene set using SVM-RFE.

Framwork of FCSVM-RFE
3.1 Gene clustering
In gene clustering, genes having similar expression profiles would be clustered into a gene group. Genes which belong to the same gene group could contain partially redundant information for the classification task, whereas the information held by different gene groups is different. It is necessary for us to consider a good clustering algorithm. There is not a best clustering algorithm according to the No Free Lunch Theorem. However, it has shown that the K-means clustering algorithm outperforms CRC and ISA when clustering gene expressions [33]. It is well known that K-means is a single cluster membership method that has been in use for several decades [17]. Thus, K-means is used here for its simplicity and efficiency in practice.
In K-means, each gene belongs to only one cluster or one group. Essentially, K-means distributes K centers throughout the data. A gene would be assigned to the group whose center is the nearest to it. At the same time, the centers are removed to minimize the distance between them and their assigned genes. This process is repeated until the center are stable. A number of distance measures can be used to define the distance between genes and centers. The Euclidean distance is one of the most commonly used and simplest measures.
Assume that we have a set of training samples \(\{(\mathbf {x}_{i},y_{i})\}^{N}_{i=1}\), where \(\mathbf {x}_{i} \in \mathbb {R}^{D}\), and y i ∈{(+1,−1)} is the label of x i ,D and N are the dimension and the number of samples, respectively. In gene clustering, it does not require the label information. Let the sample matrix be \(\textbf {X} = [\mathbf {x}^{T}_{1},\cdots ,\mathbf {x}^{T}_{N}] \in \mathbb {R}^{N \times D}\) and the gene g i be the i-th column of X. K-means is summarised in Algorithm 2 [17].
The key parameter in K-means is the number of clusters. If the number of clusters is too large, the most information contained in the feature subset would be redundant. On the other hand, if the number of clusters is too small, some useful information contained in genes would be lost. In both cases, the classifier performance would be decreased. So a proper number of cluster centers is crucial to improve the classifier performance. In experiments, a 10-fold cross validation [3] is adopted to determine the number of clusters.

3.2 Gene representative
Although we compress genes and obtain K gene groups using gene clustering, the operation of gene selection is not performed. Each gene group consists of some genes having a similar profile and a potentially similar function. Thus, we can use a gene to represent the gene group. It is necessary to select the representative gene which should carry most useful information compared with other genes in the cluster. In doing so, we reduce the number of genes from D to K.
Generally, the center of the cluster could be used as a representative point. However, the center may be not a real gene, just a synthetic gene. Although center can not be taken as the representative genes directly, we can select the gene from a gene group, which has the minimal distance from the corresponding center [20].
Let G j be the j-th gene group. The representative gene \(\overline {\mathbf {g}}_{j}\) for the j-th gene group can be determined by:
where ∥⋅∥2 denotes the Euclidian distance, \(\mathbf {m}_{j}=\frac {1}{|G_{j}|}{\sum }_{\mathbf {g}_{i}\in G_{j}} \mathbf {g}_{i}\) is the center of the jth cluster. By doing so, we can retain the original information carried by gene expressions which is more convincing for gene classification.
3.3 Gene ranking
Since K representative genes are roughly selected, we can not guarantee all K genes are useful. Thus, we need to rank these genes according to their importance with respect to classification tasks. SVM-RFE is used to rank the K representative genes.
The main idea behind SVM-RFE is that each gene is related to a score which determines the importance of a gene.
After gene clustering and gene representative, we obtain a new training sample set \(\{(\mathbf {x}^{\prime }_{i},y_{i})\}^{N}_{i=1}\), where \(\mathbf {x}_{i} \in \mathbb {R}^{K}\), and y i ∈{(+1,−1)}. In each iteration, a linear SVM is trained with the selected feature subset to generate the weight vector w = [w 1,⋯ ,w K ]T. The score for gene i is defined as follows:
The higher the score c i is , the greater the importance of the feature is. The detail algorithm for SVM-RFE is shown in Algorithm 3 [16].

3.4 Computation complexity
Now, we discuss the computational complexity of FCSVM-RFE. Since FCSVM-RFE consists of three stages: gene clustering, gene representation and gene ranking, the computational complexity of FCSVM-RFE depends on the them.
K-means is used to cluster genes and has the computational complexity of O(N D K T), where N is the number of samples, D is the total number of genes, K is the required number of clusters and T is the number of iterations [31, 43]. The computational complexity of gene representation is O(D N). For SVM-RFE, its computational complexity largely depends on the number of features. The training of linear SVM has a computational complexity of O(N D). If only one feature is removed from the feature list in each iteration, SVM-RFE has a computational complexity of O(N D 2) [38].
Thus, we have O(N D K T) + O(D N) + O(N K 2) for FCSVM-RFE. In gene expression data, we usually have D ≫ N. In addition, D ≫ K and T ≪ D in our algorithm. Thus, the computational complexity of FCSVM-RFE should be O(N D K T) which is much lower than that of SVM-RFE.
Table 1 lists the computational complexity of five methods. We can see that FCSVM-RFE has the same order of magnitude as Relief with respect to D.
4 Experimental design and results
To validate the efficiency of FCSVM-RFE, we perform experiments on seven public gene microarray datasets available which are summarized in Table 2. Seven gene expression datasets are Leukemia [2], CNS Tumor [2], ColonTumor [2], DLBCL [1], BreastCancer [2], Lung Cancer [2], and Prostate [2].
All genes in these datasets are expressed as numerical values at different measurement levels. All samples are normalized to zero mean and unit variance, based on gene expressions of a particular sample.
In this paper, we use accuracy and recalls to evaluate the performance of compared methods. The accuracy is defined as
The recall of the positive class is calculated by
where TP is the number of correctly classified positive samples, and FN is the number of wrongly classified positive samples. Similarly, the recall of the negative class is defined as
where TF and FP are the number of correctly and wrongly classified negative samples, respectively.
In our experiments, all source codes are implemented with Matlab R2015a and experiments are conducted on a Pentium PC with 3.3GHz processor and 6GB main memory.
4.1 Experiments on the Leukemia dataset
In the Leukemia dataset, there are two sets for training and test, respectively. The training set of Leukemia consists of 38 bone marrow samples (27 ALL and 11 AML), over 7129 probes from 6817 human genes. The other 34 samples for test is provided, with 20 ALL and 14 AML.
4.1.1 Parameter setting
For comparison, we consider five gene selection methods, including SVM-RFE, Relief, MRMR, MRMR+SVM-RFE, and the proposed FCSVM-RFE. We use the linear SVM as the subsequent classifier. In both SVM and SVM-RFE, there is the regularization parameter C which is determined from the set {1, 10, 100} by using the 10-fold cross-validation, and finally let C = 10.
For MRMR and MRMR+SVM-RFE, we need to determine how many genes are remained. Xin and Tuck showed that a maximum of 400 genes are identified in all experiments [46]. To validate this conclusion, we perform feature selection with SVM-RFE on the Leukemia dataset, and the accuracy on the test data is shown in Fig. 2. We can see that when the number of selected genes is larger than 400, the accuracy on the test set is not significantly increased. Thus, the gene subset has at most 400 genes for three methods, SVM-RFE, MRMR and MRMR+SVM-RFE.

Accuracy vs. gene number on the Leukemia dataset with SVM-RFE
For Relief and FCSVM-REF, let the iteration times be the sample size N. In other words, m = T = N in Table 1. For Relief, the threshold of relevancy δ = 0.001, which is the experience value. For FCSVM-REF, the number of clusters must also be determined in advance. The 10-fold cross validation is adopted to get the optimal number of cluster centers K which is selected from the set {10, 20, 30, 50, 80, 100, 150}. Figure 3 shows the experimental result on the Leukemia dataset. We can see from the accuracy curve that the accuracy is highest when the number of clusters is 80. Thus, let K = 80 in the following experiments. To save evaluation time, we also adopt C = 10, K = 80, m = T = N, δ = 0.001 and selected gene number is 400 in other six gene microarray datasets.

Accuracy vs. cluster number on the Leukemia dataset
4.1.2 Comparison of accuracy and computational time
Based on the above parameter settings, Fig. 4 shows the best recalls on the Leukemia dataset for the five feature selection methods. Here, the best performance of SVM-RFE, MRMR and MRMR+SVM-RFE is obtained using some top-ranked genes from the remaining 400 genes instead of using all remaining 400 genes. So does FCSVM-REF algorithm.

Classification performance on the Leukemia dataset dataset
As we can see, FCSVM-RFE algorithm and SVM-RFE algorithm outperform the other three methods and achieve the average recall of 100%. It may be confused that the combination method MRMR+SVM-RFE does not beat both of MRMR and SVM-RFE. The main reason is that MRMR+SVM-RFE is sensitive to the first selected gene which is M55150_at from the Leukmia dataset, and M55150_at is excluded from the good gene list. Additionally, FCSVM-RFE requires only 7 genes to achieve the 100% average recall, which is the smallest among five methods. The number of selected genes is 59, 258, 240 and 239 for SVM-RFE, Relief, MRMR and MRMR+SVM-RFE, respectively.
Table 3 lists the running time of five methoeds. Obviously, FCSVM-RFE is the fastset one among five methods, followed by Relief. FCSVM-RFE is faster than SVM-RFE in three orders of magnitude. The runing time of MRMR+SVM-RFE is even longer than SVM-RFE.
4.1.3 Selected genes
Table 4 presents top-ranked 10 genes obtained by FCSVM-RFE. The genes listed in Table 4 have been reported by many previous works [35, 36], which shows the effectiveness of FCSVM-RFE. In fact, previous experiments and clinical studies indicate that the 7791th (Zyxin, probe ID: X95735_at), the 945th (CD33, probe ID: M23197_at) and the 973th (MB-1, probe ID: U05259_rna1_at) genes are associated with leukemia. For example, the Zyxin gene has been shown to encode an LIM domain protein important in cell adhesion of fibroblasts and CD33 has been developed for targeted antibody therapy to kill leukemia AML cells [35, 36]. The distribution of training data with the two top-ranked genes is shown in Fig. 5. According to these two genes, we can exactly discriminate ALL and AML on the trainning set.

Distribution of training data on the Leukemia dataset
4.2 Experiments on the prostate dataset
The Prostate dataset has two classes, Tumor versus Normal: training set contains 52 prostate tumor samples and 50 non-tumor (labelled as “Normal”) prostate samples with around 12600 genes. An independent set of testing samples is also prepared, which is from a different experiment and has a nearly 10-fold difference in overall microarray intensity from the training data. We also have removed extra genes contained in the testing samples. In the above publication, the testing set is indicated to have 27 tumor and 8 normal samples. However, from our extraction, there are 25 tumor and 9 normal samples.
4.2.1 Comparison of accuracy and computational time
The experimental setting is the same as before. We report the results on the Prostate dataset in Table 5, where the bold values indicate the best performance among five algorithms. It is easy to see that FCSVM-RFE achieves the best average recall and has the fastest selection speed among five methods. Although both SVM-RFE and MRMR have 100% tumor recall, their normal recalls are decreases. MRMR+SVM-RFE has a bad classification performance on the Prostate dataset.
4.2.2 Selected genes
Table 6 presents the 11 selected genes obtained by FCSVM-RFE. These genes are consistent with the results reported in previous studies. For example, the 3249th gene (probe ID: 37639_at) has been selected by other papers [6, 21, 29, 35, 41]. This gene is known as a potential prostate cancer biomarker [8, 28] because it has been reported to encode hepsin, a serine protease that is significantly upregulated in human prostate cancer and it promotes cancer progression and metastasis of prostate. The 3329th gene (probe ID: 37720_at) has been correlated to different cancer types with consistent upregulation in tumor [23].
The distribution of training data with the two top-ranked genes is shown in Fig. 6. Obviously some samples are over-lapped, but the spearablity of the two top-ranked genes is acceptable. Actually, we use 11 top-ranked genes to classify Tumor and Normal.

Distribution of training data on the Prostate dataset
4.3 Experiments on the Lung Cancer dataset
The Lung Cancer dataset consists of 96 tissue samples of which 86 primary lung adenocarcinomas samples and 10 non-neoplastic lung samples are included. Each sample is described by 7129 genes.
The experimental setting is the same as before. Since the Lung Cancer dataset has no partition for training and test, we randomly take half of samples as training ones and the rest as test ones, which is repeated 10 times. We report the average results on 10 trials in Table 7, which shows the comparison of the best classification performance obtained by five methods. In Table 7, the bold values represent the best performance among five algorithms.
We can see from Table 7, Both FCSVM-RFE and MRMR+SVM-RFE have the best average performance, 100%. In fact, other algorithms also obtain comparable performance. However, these algorithms take more time to perform gene selection except for FCSVM-RFE and Relief.
The probe IDs of the 2 top-ranked genes obtained by FCSVM-RFE are U60115_at and X62466_at, where U60115_at is FHL1- Juvenile dermatomyositis muscle profile (HuGeneFL), and X62466_at is CD52 - CD34+ cell analysis. The distribution of training data with the top-ranked 2 genes is shown in Fig. 7. We can see tht the separability between AD and NL is very high.

Distribution of training data on the Prostate dataset
4.4 More experiments on gene datasets
More datasets are used here, including Breast Cancer [2], Colon Tumor [2], Diffuse large B-cell lymphoma (DLBCL) [1] and CNS Tumor [2].
The Breast Cancer dataset has 97 samples belonging to two classes, or Relapse (positive class) and Non-relapse (negative class). Each sample has 24481 genes. The training set contains 78 patient samples, 34 of which are from patients who had developed distance metastases within 5 years (labelled as “relapse”), and the remaining 44 samples are from patients who remained healthy from the disease after their initial diagnosis for an interval of at least 5 years (labelled as “non-relapse”). Correspondingly, there are 12 relapse and 7 non-relapse samples in the test set.
The Colon Tumor dataset contains 62 samples collected from colon-cancer patients. Among them, 40 tumor biopsies are from tumors (labelled as “negative”) and 22 normal (labelled as “positive”) biopsies are from healthy parts of the colons of the same patients. Two thousand out of around 6500 genes were selected based on the confidence in the measured expression levels.
The diffuse large B-cell lymphoma (DLBCL) and follicular lymphomas, the most common lymphoid malignancy in adults, is curable in less than 50% of patients. There are 77 samples, 58 of them are from DLBCL group (labelled as “negative”) while 19 are FL group (labelled as “positive”). Each sample is described by 5469 genes.
The CNS dataset contains 60 patient samples, 21 are survivors (alive after treatment) which are labelled as positive class, and 39 are failures (succumbed to their disease) which are labelled as negative class. There are 7129 genes in the dataset. The training set consists of the first 10 survivors and 30 failures, the other 11 survivors and 9 failures are testing points.
The experimental setting on the Colon Tumor and DLBCL datasets is the same as that of the Lung Cancer dataset, and setting on the Breast Cancer and CNS Tumor datasets is the same as that of the Leukemia Dataset. We report the results on the four datasets in Table 8, which shows the comparison of five methods when obtaining the best classification performance. Inspection on Table 8 implies that FCSVM-RFE has a very fast gene selection speed. In addition, the classification performance of FCSVM-RFE is also comparable.
4.5 Statistical comparison over multiple datasets
In the previous section, we perform experiments on seven datasets, and compare the classification performance and running time of FCSVM-RFE with other methods. For the sake of comparison, statistical tests on multiple data sets for multiple algorithms are preferred for comparing different algorithms over multiple datasets [7]. In the following, we conduct two different statistical analyses, the win-loss-tie summary and the Friedman test.
First, the win-loss-tie times are summarized to compare FCSVM-RFE with the other four methods. Table 9 shows the win-loss-tie summary of FCSVM-RFE in terms of the average recall and the running time for gene selection, respectively. From Table 9, it is observed that FCSVM-RFE outperforms SVM-RFE in five out of seven datasets, both Relief and MRMR in six out of seven datasets, and MRMR+SVM-RFE in four out of seven datasets in terms of the average recall. Briefly speaking, FCSVM-RFE can achieve a better performance than other methods. On the performance of the running time, FCSVM-RFE is the fastest among these five methods.
Second, we conduct the Friedman test with the corresponding post-hoc tests, which is a non-parametric equivalence of the repeated-measures analysis of variance (ANOVA) under the null hypothesis that all the algorithms are equivalent and so their ranks should be equal [14]. According to [14], the Friedman test is carried out to test whether all the algorithms are equivalent. If the test result rejects the null hypothesis, i.e., these algorithms are equivalent, we can proceed to a post-hoc test. The power of the post-hoc test is much greater when all learners are compared with a control learner and not among themselves. We do not need to make pairwise comparisons when we in fact only test whether a newly proposed method is better than the existing ones.
FCSVM-RFE is taken as the control learner to be compared with. The Bonferroni-Dunn test [12] is used as post-hoc tests when all learners are compared to the control one. The performance of pairwise classifiers is significantly different if the corresponding average ranks differ by at least the critical difference
where j is the number of algorithms, T is the number of data sets, the critical values q α can be found in [14], and the subscript α is the threshold value. Generally, let α = 0.1 [5], and in Table 10 we find that the critical value q 0.10 = 2.241. Here, we have j = 5, T = 7, then C D = 1.8940.
Table 11 lists the mean rank of five feature selection algorithms including FCSVM-RFE, SVM-RFE, Relief, MRMR and MRMR+SVM-RFE. Table 12 shows the Friedman test results. On the performance of average recall, although FCSVM-RFE is the first, we could not find any significant differences between FCSVM-RFE and Relief, MRMR and MRMR+SVM-RFE since all rank differences are below the critical difference. However, we can see that FCSVM-RFE is significantly better than SVM-RFE according to the difference (2.1429) between them.
For the performance of running time, the differences between FCSVM-RFE and other algorithms including SVM-RFE, MRMR and MRMR+SVM-RFE are greater than the critical difference, so the differences are significant, which means the performance of running time of FCSVM-RFE is significantly better than SVM-RFE, MRMR and MRMR+SVM-RFE in this current experimental setting. The difference (1.1429) between FCSVM-RFE and Relief is below the critical difference. We could not detect any significant difference between FCSVM-RFE and Relief.
5 Conclusion
We propose FCSVM-RFE to enhance SVM-RFE for gene selection by incorporating the K-means clustering method, and apply it to cancer classification. FCSVM-RFE can reduce the computational complexity and the redundancy a mong genes. There are three stages in the proposed method, gene clustering, gene representation, and gene ranking. Gene clustering is implemented by applying K-means clustering. The goal of gene representation is to find the representative genes for gene clusters. SVM-RFE is used to rank the representative genes. Extensive experiments are performed on seven public gene expression datasets, including Leukemia, CNS Tumor, ColonTumor, DLBCL, Breast Cancer, Lung Cancer, and Prostate. All experimental results show that FCSVM-RFE can achieve better classification performance and much lower computational complexity when compared with the state-the-art-of methods. The Friedman test also shows that FCSVM-RFE is ranked the first on the performance of both average recall and running time.
In the framework of FCSVM-RFE, each stage can also be implemented by other methods. For example, we could use other clustering algorithms to perform gene clustering. Thus, we will continue to research this model further for better classification performance and faster gene selection.
References
The dataset is download from gene expression model selector. http://www.gems-system.org/
The dataset is download from kent ridge bio-medical dataset. http://datam.i2r.a-star.edu.sg/datasets/krbd/
Ambroise C, McLachlan GJ (2002) Selection bias in gene extraction on the basis of microarray gene-expression data. Proc Nat Acad Sci 99(10):6562–6566
Blum AL, Langley P (1997) Selection of relevant features and examples in machine learning. Artif Intell 97(1):245–271
Chen H, Tiho P, Yao X (2009) Predictive ensemble pruning by expectation propagation. IEEE Trans Knowl Data Eng 21(7):999–1013
Chu W, Ghahramani Z, Falciani F, Wild DL (2005) Biomarker discovery in microarray gene expression data with Gaussian processes. Bioinformatics 21(16):3385–3393
Demṡar J (2006) Statistical comparisons of classifiers over multiple data sets. J Mach Learn Res 7:1–30
Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S (2001) Delineation of prognostic biomarkers in prostate cancer. Nature 412(6849):822–826
Díaz-Uriarte R, De Andres SA (2006) Gene selection and classification of microarray data using random forest. BMC Bioinform 7(1):1
Ding C, Peng H (2005) Minimum redundancy feature selection from microarray gene expression data. J Bioinform Comput Biol 3(02):185–205
Duan KB, Rajapakse JC, Wang H, Azuaje F (2005) Multiple svm-rfe for gene selection in cancer classification with expression data. IEEE Trans NanoBiosci 4(3):228–234
Dunn OJ (1961) Multiple comparisons among means. J Am Stat Assoc 56(293):52–64
Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Nat Acad Sci 95(25):14,863–14,868
Friedman M (1937) The use of ranks to avoid the assumption of normality implicit in the analysis of variance. J Amer Stat Assoc 32(200):675–701
Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA et al (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286(5439):531–537
Guyon I, Weston J, Barnhill S, Vapnik V (2002) Gene selection for cancer classification using support vector machines. Mach Learn 46(1–3):389–422
Hartigan JA, Wong MA (1979) Algorithm as 136: a k-means clustering algorithm. J R Stat Soc Series C (Appl Stat) 28(1):100– 108
Inza I, Larrañaga P., Blanco R, Cerrolaza AJ (2004) Filter versus wrapper gene selection approaches in dna microarray domains. Artif Intell Med 31(2):91–103
Islam AT, Jeong BS, Bari AG, Lim CG, Jeon SH (2015) Mapreduce based parallel gene selection method. Appl Intell 42(2):147–156
Jäger J, Sengupta R, Ruzzo WL (2002) Improved gene selection for classification of microarrays. In: Proceedings of the eighth Pacific symposium on biocomputing. Lihue, pp 53–64
Karan D, Kelly DL, Rizzino A, Lin MF, Batra SK (2002) Expression profile of differentially-regulated genes during progression of androgen-independent growth in human prostate cancer cells. Carcinogenesis 23(6):967–976
Kira K, Rendell LA (1992) The feature selection problem: traditional methods and a new algorithm. In: AAAI, vol 2, pp 129–134
Kishino H, Waddell PJ (2000) Correspondence analysis of genes and tissue types and finding genetic links from microarray data. Genome Inform 11:83–95
Kohavi R, John GH (1997) Wrappers for feature subset selection. Artif Intell 97(1):273–324
Kononenko I (1994) Estimating attributes: analysis and extensions of relief. In: Machine learning: ECML-94. Springer, pp 171–182
Lee S, Park YT, d’Auriol BJ, et al. (2012) A novel feature selection method based on normalized mutual information. Appl Intell 37(1):100–120
Liu X, Krishnan A, Mondry A (2005) An entropy-based gene selection method for cancer classification using microarray data. BMC Bioinform 6(1):1
Magee JA, Araki T, Patil S, Ehrig T, True L, Humphrey PA, Catalona WJ, Watson MA, Milbrandt J (2001) Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Res 61(15):5692–5696
Mao Z, Cai W, Shao X (2013) Selecting significant genes by randomization test for cancer classification using gene expression data. J Biomed Inform 46(4):594–601
Mundra PA, Rajapakse JC (2010) Svm-rfe with mrmr filter for gene selection. IEEE Trans NanoBiosci 9(1):31–37
Nazeer KA, Sebastian M (2009) Improving the accuracy and efficiency of the k-means clustering algorithm. In: Proceedings of the world congress on engineering, vol 1, pp 1–3
Peng H, Long F, Ding C (2005) Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell 27(8):1226–1238
Richards AL, Holmans P, O’Donovan MC, Owen MJ, Jones L (2008) A comparison of four clustering methods for brain expression microarray data. BMC Bioinform 9(1):1
Ruiz R, Riquelme JC, Aguilar-Ruiz JS (2006) Incremental wrapper-based gene selection from microarray data for cancer classification. Pattern Recogn 39(12):2383–2392
Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, Tamayo P, Renshaw AA, D’Amico AV, Richie JP et al (2002) Gene expression correlates of clinical prostate cancer behavior. Cancer Cell 1(2):203–209
Sun S, Peng Q, Shakoor A (2014) A kernel-based multivariate feature selection method for microarray data classification. PloS One 9(7):e102,541
Szedmak S, Shawe-Taylor J, Saunders CJ, Hardoon DR et al (2004) Multiclass classification by l1 norm support vector machine. In: Pattern recognition and machine learning in computer vision workshop. Citeseer, pp 02–04
Tan M, Wang L, Tsang IW (2010) Learning sparse svm for feature selection on very high dimensional datasets. In: Proceedings of the 27th international conference on machine learning (ICML-10), pp 1047–1054
Tang Y, Zhang YQ, Huang Z (2007) Development of two-stage svm-rfe gene selection strategy for microarray expression data analysis. IEEE/ACM Trans Comput Biol Bioinform (TCBB) 4(3):365–381
Vapnik VN, Vapnik V (1998) Statistical learning theory, vol 1. Wiley, New York
Wang X, Gotoh O (2009) Accurate molecular classification of cancer using simple rules. BMC Med Genom 2(1):1
Xie ZX, Hu QH, Yu DR (2006) Improved feature selection algorithm based on svm and correlation. In: Advances in neural networks-ISNN 2006. Springer, pp 1373–1380
Yedla M, Pathakota SR, Srinivasa T (2010) Enhancing k-means clustering algorithm with improved initial center. Int J Comput Sci Inform Technol 1(2):121–125
Yu L, Liu H (2004) Efficient feature selection via analysis of relevance and redundancy. J Mach Learn Res 5:1205–1224
Zhang Y, Ding C, Li T (2008) Gene selection algorithm by combining relieff and mrmr. BMC Genom 9(2):1
Zhou X, Tuck DP (2007) Msvm-rfe: extensions of svm-rfe for multiclass gene selection on dna microarray data. Bioinformatics 23(9):1106–1114
Acknowledgment
This study was funded by the National Natural Science Foundation of China (grant numbers 61373093, 61672364, and 61672365), by the Natural Science Foundation of Jiangsu Province of China (grant number BK20140008), by the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (grant number 13KJA520001), and by the Soochow Scholar Project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Huang, X., Zhang, L., Wang, B. et al. Feature clustering based support vector machine recursive feature elimination for gene selection. Appl Intell 48, 594–607 (2018). https://doi.org/10.1007/s10489-017-0992-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10489-017-0992-2