
Abstract
Microarray data analysis has been widely used for extracting relevant biological information from thousands of genes simultaneously expressed in a specific cell. Although many genes are expressed in a sample tissue, most of these are irrelevant or insignificant for clinical diagnosis or disease classification because of missing values and noises. Thus, finding a small, closely related gene set to accurately classify disease cells is an important research problem. At the same time, scalable gene selection methods are required for microarray data analysis due to rapidly increasing volume of microarray data. In this paper, we propose a scalable parallel gene selection method using the M a p R e u d c e programming model. The proposed method utilizes the kNN classifier algorithm for evaluating classification accuracy and uses four real and three synthetic datasets for experiments. Experimental results show that the proposed method can offer good scalability on large data with increasing number of nodes and it can also provide higher classification accuracy rather than using whole gene set for classification.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Microarray technology has recently become a popular technique for bioinformatics, especially in clinical diagnosis, disease classification and finding gene regulations. The technique observes expression values of thousands of genes simultaneously and analyzes expression levels for clinical diagnosis and discovers correlations among genes. For example, microarray gene expressions data are widely used for identifying candidate genes in various cancer studies [10]. These data usually contain thousands of genes (sometimes more than 10000 genes) and small number of samples (usually <100 samples). Although many genes are expressed in a microarray chip, most of these are irrelevant or useless for a particular analysis because some of the genes are differentially modulated in tissues under different conditions and an amount of noise in a microarray experiment is usually expected.
Therefore, an important step towards effective classification is to identify discriminatory genes and thus to reduce the number of genes used for classification purpose. The process of discriminatory gene identification is generally referred to as gene selection. Gene selection methods extract a small subset of discriminatory or the most relevant genes that can effectively classify test samples. Thus, it is possible to generate classification model from training data set at low cost while minimizing classification errors.
There is a variety of gene selection methods proposed in last few years [10, 12, 13]. They address various biological properties of microarray data and utilize those to extract relevant genes from large number of genes. Some of the methods use statistical analysis such as sampling technique [11, 12, 19] while others utilize machine learning algorithms such as genetic algorithm [6–8] and SVM classification model [21].
Meanwhile, due to recent developments of microarray chip technology, such experiments can handle more than 10,000 genes simultaneously in one chip and can generate large amount of microarray data at low cost. Thus, high performance computing for gene selection has become increasingly important in microarray data analysis. The MapReduce programming paradigm and its implementation Hadoop have a substantial base for biological data analysis including microarray data.
In this paper, we propose MapReduce based parallel gene selection method. Our method utilizes sampling technique to reduce irrelevant genes by using BW ratio [12] and uses kNN algorithm for comparison of classification accuracy. The method is implemented in MapReduce environment for achieving scalability with an increasing amount of microarray data. Major contributions of our work are as follows:
-
1.
We propose new gene selection method (MRGS) for microarray data by using the sampling technique.
-
2.
We devise MRkNN to execute multiple kNN in parallel using MapReduce programming model.
-
3.
Finally, the effectiveness of our method is verified through extensive experiments using several real and synthetic data-sets.
The rest of our paper is organized as follows: in Section 2, we discuss preliminary knowledge and existing works related to the scope of our work. Section 3 presents the proposed MapReduce gene selection algorithm. Section 4 explains experimental results and discusses several performance issues in our method. In Section 5, we conclude the paper with directions of future works.
2 Related works
In recent years, microarray technology has been widely used in biological researches. To analyze microarray gene expressions data, it is very important to select proper number of genes that are relevant for a data analysis. For this reason, several gene selection methods have been proposed in the last few years [10, 12, 13].
Among statistical methods, SVST [11] has introduced the concept of sample pruning to remove less relevant and outlier samples and has applied SVM to find biologically relevant genes. In order to improve accuracy of the classification technique, the method removes less relevant samples which are not located on support vectors. However, it may suffer from low training data since it drops around 50 % of samples while the number of samples is very small compared to the number of genes in a microarray experiment. RFGS [10] generates several hundreds of decision trees constructed from randomly selected gene subsets and considers root attributes for determining relevant gene sets. The method requires several executions since each execution produces different decision trees for random gene subsets. Moreover, in comparison with other approaches, it shows relatively lower accuracy in selecting biologically relevant genes.
On the contrary, other methods have adopted evolutionary approaches such as genetic algorithm because of their high learning capability. [12, 18] are representative methods of these approaches. GADP [12] has exploited probabilistic measures for crossover and mutation to improve the degree of fitness. The methods have common drawbacks that they do not emphasize on biological relevance of extracted information at each generation and also require many generations to achieve optimal result. Another interesting technique for gene selection is supervised clustering [14]. It generally begins with coarse clusters and incrementally refines clustering results by utilizing cluster features values. It also considers mutual relationship between genes rather than individual gene properties.
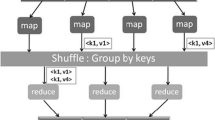
Furthermore, high performance computing has become extremely important for analyzing large amount of biological data. MapReduce is an easy-to-use and general purpose parallel programming model that is suitable for large data analysis on a commodity hardware cluster. Computation on MapReduce is divided into two major phases called map and reduce. The power of MapReduce is that map and reduce tasks are executed in parallel over a large number of processors with minimal effort by the application developer.
Figure 1 shows a schematic diagram to explain MapReduce [1] framework. It is being deployed increasingly in many biological data analysis projects. Several recent literatures [2–9] have proposed parallel classifications and learning methods for analyzing large datasets including biological data. Crossbow [4] is an open-source genotyping tool implemented on hadoop. It accelerates alignment and SNP calling tasks more than 100 times the capabilities of conventional computer systems. CloudBurst [5] efficiently maps next generation sequence data and achieves almost linear speed up with increasing number of processors. Until now, only a few works [6] have addressed the possibility of parallel processing in microarray data analysis. This scenario motivates us to develop a parallel gene selection method using MapReduce programming model.

Graphical representation of MapReduce programming model
3 Proposed method
In this section, we describe MapReduce based parallel gene selection method in detail. First, we explain overall outline and also the principles hidden in each step of our method. Then, we depict each parallelizable step with MapReduce framework.
3.1 Outline of our method
Figure 2 shows the overall procedures of our gene selection method. Generally, microarray data is presented as an N×M matrix, where N is the number of genes and M is the number of experimental samples involved. The transpose matrix presentation is particularly suitable since the number of genes is much larger than that of samples. Table 1 shows a sample microarray data which will be used for examples given in the paper.

Workflow of the proposed method
Typically, microarray data has many irrelevant genes that do not affect analysis results and have no correlation with other genes. Thus, it is not necessary to consider all genes for such analyses. Therefore, we eliminate unnecessary genes at the very beginning of our method. At first, we reduce the number of genes by measuring BW (Between-groups to Within-groups sum of square) ratio (1) [12] values. The BW ratio indicates degree of variances among gene expression values. If there are minor fluctuations among gene expressions, BW ratio value would be small. Smaller BW ratio value refers that corresponding gene might be irrelevant for a particular microarray analysis.
The potential genes extracted by BW ratio measure are defined in definition 1.
Definition 1
Potential Gene (g p ) Let \(G=\{g_{1},\,\, g_{2},\dots ,\) \(\left . g_{N-1}, g_{N}\right \}\), be a set of genes and \(S=\{s_{1}, \,\,s_{2},\dots ,\) \(\left .s_{M-1},s_{M}\right \}\), be a set of samples in a microarray dataset. If B W(g j )≥B W t h and g j ∈ G , then g j is a potential gene, g p . BW ratio of a gene can be calculated from (1). We also define a potential gene set \(G_{p} = \left \{\forall g_{p} \in G \right \}\).
Here, m is the number of training samples, k is the number of classes and c represents corresponding class label. \(\bar {x}_{j}\) denotes overall expressions mean value of gene g j over training samples and \(\bar {x}_{c,j}\) denotes class based mean value of gene g j expressions belong to same class c k in training samples. Larger BW indicates significance of a gene for a particular analysis.
Let us consider, the expression values of g 2 for training samples in Table 1 (g 21=2, g 22=2, g 23=6, g 24=42). Thus, overall mean value, \(\bar {x}_{2}\)=(2 + 2 + 6 + 42)/4=13 and class mean values are \(\bar {x}_{1,2}\)=(6 + 42)/2=24 and \(\bar {x}_{2,2}\)=(2 + 2)/2=2 respectively. Finally, B W(g 2) is ((24−13)2+(2−13)2)/ ((2−2)2+(2−2)2)+(5−24)2+(42−24)2)=0.385. If 0.385 ≥ B W t h , g 2 would be a potential gene.
After extracting all potential genes, we generate a predefined number of potential gene subsets (S k ) of equal size. While generating the subsets, genes having higher BW value occur more frequently because higher BW value implies greater significance of a gene. Next, we calculate the classification accuracy of each subset by using the kNN algorithm. kNN classification algorithm requires to measure distance between each training and test sample. Based on the distance values, the algorithm selects k nearest training samples. Then, the class of a test sample is predicted by considering majority of k training samples class labels. Distance values are measured from the expression values of member genes of a subset. Finally, classification accuracy of a subset S k is determined by correct prediction ratio of test samples and denoted as A c c(S k ).
Definition 2
Candidate Genes (g c ) We define g i as a candidate gene, \(g_{c}=\left \{ g_{c} \in G_{p} \& g_{c} \in \exists S_{k}\right \}\), where S k shows higher accuracy than user given accuracy threshold, i.e., A c c(S k ) ≥ A c c t h . Such an S k is also considered as a candiate set, G c .
Finally, we consider top-k frequently occurred genes in candidate gene sets. This is the predictor set for a microarray data. We validate the predictor set’s classification accuracy over training and test samples using kNN. Moreover, we corroborate biological relevance of the top genes with publicly available domain knowledge.
3.2 Parallel gene selection method
In this section, we describe how to implement each step of the parallel gene selection method in MapReduce framework. For better understanding, we define frequently used symbols in Table 2.
-
Step 1: Generating potential genes (g p ) and subsets (S k )
This step consists of one MR job. The map tasks input each gene’s expression values in parallel and calculates the BW ratio value of a gene from training samples expressions. We utilize BW ratio (Between groups sum square to within groups sum square ratio) [12, 16] (1) to measure relevancy of genes based on domain knowledge. Higher BW ratio indicates that g i has significant variations in training samples expressions and thus contains more information to classify unknown test samples. If B W(i) is relatively larger than B W t h , g i is a significant potential gene and we ensure the gene occurs more frequently in potential gene subsets (S k ).
In the M a p R e d u c e framework, each map task inputs \(\langle g_{i}, (g_{i1}, g_{i2}, {\dots } g_{iM}) \rangle \) and emits \(\langle S_{1}, g_{i} \rangle , \langle S_{2}, g_{i}\rangle , \dots \) 〈S k ,g i 〉 to reduce task according to the subset id. Then, each reduce task collects 〈S k ,g i 〉 pairs with same subset id and emits \(\langle S_{k}, (g_{i_{1}}, g_{i_{2}}, {\dots } g_{i_{K}}) \rangle \).
For example, \(BW_{g_{2}}\)=2.0, \(BW_{g_{4}}\)=1.0, \(BW_{g_{6}}\)=0.8 and \(BW_{g_{7}}\)=1.5 are obtained from Table 1 data. Let us consider, B W t h =0.5. Thus, possible potential gene subsets are S 1=\(\left \{g_{2}, g_{4}, g_{7}\right \}\), S 2=\(\left \{g_{2}, g_{4}, g_{7}\right \}\),…, S k =\(\left \{g_{2}, g_{4}, g_{6}\right \}\) according to Definition 1. B W(g 2)=2.0 indicates that g 2 is the most significant gene. Therefore, g 2 occurs most frequently in potential gene sets. g 7 is the next frequently occurred gene. The genes in a potential gene set are generated at 1st map task according to \(BW_{g_{i}}\) values.
Algorithm 1 describes map and reduce tasks in this step and also Fig. 3 shows the corresponding diagram of map and reduce tasks.

M a p R e d u c e jobs for generating potential gene subsets
-
Step 2: Measuring classification accuracy of each subset.
After generating potential gene subsets, we measure classification accuracy of the gene subsets. For the classification, we utilize kNN method using Euclidian distance function. kNN is a widely accepted method for classifying small number of classes and requires less time for classification compared to other classification methods such as SVM, Bayesian networks and so on. Standard kNN algorithm has nested iterations which are not suitable for MapReduce framework. For applying kNN, it is necessary to obtain distance values between training samples and a test sample. The second phase of the proposed method executes classification tasks. First, each map task inputs \(\langle S_{j}, (g_{i_{1}}, g_{i_{2}}, \dots ,g_{i_{k}})\rangle \) and calculates distance value between each training sample and given test sample considering genes in subset S j only. The Euclidian distance function is shown in (2).
The d i s t j implies distance value between a training sample j and a test sample l. After completing map tasks, reduce task takes \(\left \langle S_{i}, \left (dist_{1}, dist_{2} \ldots , dist_{j}\right ) \right \rangle \) inputs and predicts test sample class label based on majority of k nearest training samples class labels. R e d u c e task determines \(\left \langle S_{i},\left (ts_{1},T/F\right ) \right \rangle ,\left \langle S_{i},\left (ts_{2},T/F\right ) \right \rangle , \\ {\dots } \left \langle S_{i},\left (ts_{k},T/F\right ) \right \rangle \) (T/F means whether test sample is correctly classified or not) and emits A c c(S i ) to the following map tasks.
For parallel processing of this step, we distribute the computation of each subset to several map and reduce tasks. We devise a way to execute multiple instances of kNN algorithm in parallel using MapReduce programming model (MRkNN). The proposed algorithm has no iteration and relies on very small operations suitable for inherent architecture of MapReduce framework.
At this step, input files contain 〈S j , (\(g_{i_{1}}\), \(g_{i_{2}}\),\(\dots \) \(g_{i_{k}}) \rangle \) pairs. We assume that microarray data can be accessed by every map and reduce tasks. First, each map task calculates distance values of training samples and emits \(\left \langle S_{j}, dist_{j,s} \right \rangle \) pairs. Distance value of subset S j is calculated by \(\sqrt {{\sum }_{i=1}^{k} \left (g_{ij} - ts_{ji}\right )^{2}}\). After completion of all map tasks, reduce tasks sort \(\left \langle S_{j}, dist_{ji} \right \rangle \) pairs by d i s t j i value and produce top-k smallest \(\left \langle S_{j}, dist_{j_{i}} \right \rangle \) pairs. Then, the following map tasks collect class labels of top-k samples and predict the test sample class label based on majority class labels of k-nearest training samples. The reduce task checks it with correct class label and generates prediction accuracy value of each subset. If A c c(S j ) ≥ A c c t h , S j is a candidate set and reduce task emits < S j ,A c c(S j )>.
The MRkNN pseudocode is presented at Algorithm 2 and also Fig. 4 shows the diagram of map and reduce tasks in this step.
-
Step 3: Generating top genes from G c and validation with biological data

M a p R e d u c e jobs for generating top-k significant genes
After completing execution of MRkNN, we obtain candidate gene sets (G c ), having high prediction accuracy. In this MR job, we measure the frequency of each gene in the candidate sets. Each map task inputs \(\left \langle G_{c}, \left (g_{id_{1}}, g_{id_{2}}, \ldots , g_{id_{K}}\right ) \right \rangle \) and emits \(\left \langle g_{i}, 1 \right \rangle \) for every gene in G c s. The following reduce task aggregates value for keys (g i ) and emits \(\left \langle g_{i}, freq_{i} \right \rangle \) into file system. The driver class sorts \(\left \langle g_{i}, freq_{i} \right \rangle \) by descending order of f r e q i . From the sorted list, we consider top genes as final predictor set. Our experimental results show that such a predictor set gives higher accuracy while classifying test samples. Then, we validate top genes with biological resources [26]. Majority of the genes are found meaningful regarding existing cancer literatures and gene data.
4 Experimental results
We have conducted experiments in a seven-physical node cluster, each node having four cores. There are four virtual machines in each physical node. Therefore, we have a total of 28 nodes considering each virtual machine a node. The memory size is 15 GB and the storage capacity is 800 GB. The operating system is Ubuntu 11.10. We use Apache Hadoop’s distribution of 1.1.0 for MapReduce library. One node is set as the master node. The remaining nodes are set as worker nodes. Each worker node has two slots of Map and Reduce. Thus, there are maximum 54 map tasks and 54 reduce tasks that can run concurrently. The HDFS block size is 32MB and each block has three replications. We apply our proposed method to four publicly available microarray datasets and three synthetic datasets. Along with predictor set generation capability, we examine various MapReduce metrics such as node scalability, data scalability and I/O costs. Since the sizes of publicly available datasets are small, we validate our method’s scalability by using synthetically generated data sets. We generate three synthetic microarray datasets from a prostate cancer dataset[21] and use those to exhibit data scalability in MapReduce environment. For node scalability, we change the number of active nodes in the cluster and observe execution time differences.
4.1 Datasets
Table 3 gives a brief description of the four real datasets used in our experiments. The colon dataset [15] contains 62 microarray samples of tumor and normal colon tissues. Among these, five samples are reported as outliers in the existing literature [19]. Therefore, we drop those samples from the experiments. Three other datasets, ALL/AML (Leukemia) [17], Lymphoma [20] and Prostate cancer [21] contain 72, 77 and 102 samples respectively. All of the datasets have two class labels: normal and affected.
We generate three synthetic datasets from the prostate cancer dataset. Though the generated datasets are not very large with regards to MapReduce model, our intention is to observe the MapReduce scalability metrics on relatively larger data. Table 4 shows a brief description of the synthetic datasets.
4.2 Predictor set accuracy
To validate the accuracy metric, we experiment with four real microarray datasets. The method works in three steps. In the first step, potentially informative genes are chosen based on domain knowledge. For instance, g 245 in the colon dataset produces high BW value. Therefore, it is included in G p . In the second step, MRkNN produces G c sets from S k sets. The A c c t h requirements further reduces the chance of picking irrelevant genes. In our experiments, we set candidate set accuracy threshold (A c c t h ), 90 %-100 % for three datasets and 80 % for Lymphoma dataset. The reason is three out of four datasets show very high prediction accuracy. We then sort genes in G c according to descending order of frequency and select top genes which occur most frequently. Resultant top genes show strong prediction capability. Fig. 5 shows the differences in prediction capability for varied count of top genes. According to our experiments, optimum accuracy can be achieved by using top 20-30 genes.

Candidate set and top genes classification accuracy
Moreover, our method shows satisfactory accuracy compared to existing methods. Table 5 shows accuracy comparisons with optimal gene count on three real datasets. We can see that SVST [11] and GADP [12] produce a bit better accuracy than that of proposed method. GADP gives priority on selecting minimum number of genes; however, it is widely accepted that tiny predictor set may cost generalization capability. Though SVST and GADP give slightly better accuracy, the proposed method is more scalable than these methods. In Section 4.4, we compare scalability of SVST and GADP through existing parallel algorithms of their core methods.
4.3 Biological relevance
We validate biological significance of predictor genes with NCBI [26] knowledge base. NCBI-Gene resources are confidently recommended because of its completeness and periodical synchronization with other major repositories such as BIND, GO, HGNC and EMBL. Table 6 provides a summary of the top 24 genes of the colon dataset (CRIP1 and CRP1 are same gene with different features) extracted by the proposed method. The genes are evaluated by GO terms http://www.geneontology.org/, bibliographic results and RefSeq summaries. Among them, 71 % are mentioned in existing cancer studies. 11 genes (marked by a) have known effects on cancer diseases including rectal cancer and 6 others (marked by b) are identified as significant bio-markers for cancer detection. Hence the proposed method can extract good predictor set and we recommend further biological experiments on these genes.
4.4 Hadoop scalability
We measure effectiveness of the distributed and parallel processing model on two widely discussed metrics: node scalability and data scalability. A MapReduce implementation has some overhead to initialize execution environment. Therefore, relatively large datasets are desirable for observing significant performance upgrades through parallelization. The publicly available microarray datasets are fairly small in size. Thus, we generate three synthetic datasets shown in Table 4 along with real datasets. Lab size clusters typically consist of a small amount of memory and a small number of nodes. Therefore, increasing the number of nodes and memory significantly speed up MR job execution. Publicly available cloud services are good candidates for such systems unless security is a major issue.
Figure 6a shows Hadoop scalability over real datasets. As the number of worker nodes is increased, the execution time decreases. The decrement is not linear because of initial overhead imposed on each MR job. We can see the difference in execution time between two datasets. For example, for a 15-node configuration (1 master node and 14 worker nodes), the Colon and Leukemia datasets require 58 seconds and 104 seconds respectively. The Leukemia dataset is four times larger than colon dataset regarding matrix size.

Hadoop Scalability on Real and Synthetic Datasets
However, the difference in execution time is not very large. In fact, all of the MR jobs except 2nd MR job in MRkNN, take almost same amount of time to execute. The MR jobs in MRkNN generate large intermediate data which cause memory spills and large shuffle/sort phase. MapReduce model is suitable for the shared nothing architecture. We observe effectiveness of this property in our experiments. Another interesting observation is that the Lymphoma and Leukemia datasets require different amounts of time over varying configuration while Lymphoma data size is similar to that of Leukemia. The genes have relatively large BW ratio value in Lymphoma dataset. Thus, potential gene set (G p ) is much larger and the number of intermediate operations is significantly higher than that of the Leukemia dataset.
In Fig. 6(b), the execution times of synthetic datasets are shown over varying number of nodes. We maintain the proportion of normal and affected samples in the synthetic data also. Each newly added synthetic sample is generated by averaging three randomly selected samples of the same class. As the number of nodes is decreased, the slope gets sharper over increasing data size. Each virtual machine has one core and can only run two map and reduce tasks at a time. With smaller nodes, all of the data splits cannot run in parallel. The intermediate outputs are also larger and require more time for I/O.
Table 5 shows that SVST and GADP is suitable for producing highly accurate gene sets. SVST deploys SVM and BPNN for finding relevant genes. Both SVM and NN algorithms are computationally intensive and they require much iteration to converge. In MapReduce model too many iterations cause excessive I/O and reduce scalability. The existing parallel SVM algorithms [24, 25] indicate that they are less suitable for MapReduce model. Moreover, SVM is used to determine relevant samples only. BPNN is applied afterwards on relevant samples which also require too much iteration. Therefore, despite producing high accuracy, the method lacks scalability. Similarly, GADP introduces genetic algorithm with dynamic probability measures to select relevant genes from a dataset. It requires several iterations to produce expected result. Considering MapReduce model, each such iteration incurs costly I/O. Jin, Chao et al. [7, 8] discussed challenges of implementing efficient GAs in MapReduce programming model. Moreover, the method initially creates 50 potential gene subsets (S k ) and updates those iteratively based on fitness value while our proposed method executes more than 1000 S k s and can extend the instances far more with increasing number of nodes. Thus, MRGS acquires a good mixture of accuracy and scalability.
5 Conclusions
In this paper, we first address the possibility of utilizing MapReduce programming model for gene selection technique. The proposed MRGS method is based on our own sampling technique and kNN algorithm. In order to execute multiple kNN algorithms in parallel, we develop the MRkNN algorithm in MapReduce framework. We experiment with four real cancer datasets and three synthetically generated datasets. During the experiments, we observe accuracy along with Hadoop scalability measures. Extensive experimental results verify the effectiveness of our method. Our next objective is to devise parallel gene association analysis (GAA) algorithm for microarray data using MapReduce framework.
References
Dean J, Ghemawat S (2008) Mapreduce: simplified data processing on large clusters. Commun ACM 51(1):107113
Akdogan A, Demiryurek U, Banaei-Kashani F, Shahabi C (2010) Voronoi-based geospatial query processing with mapreduce In: Cloud computing technology and science (CloudCom), IEEE 2nd international conference on, pages 9–16. IEEE
Ji C, Dong T, Li Y, Shen Y, Li K, Qiu W, Qu W, Guo M (2012) Inverted grid-based knn query processing with mapreduce. ChinaGrid Annual Conference (ChinaGrid), 7th, pages 25–32. IEEE
Langmead B, Schatz MC, Lin J, Pop M, Salzberg SL (2009) Searching for SNPs with cloud computing. Genome Biol 10(11):R134
Schatz MC (2009) CloudBurst: highly sensitive read mapping with MapReduce. Bioinformatics 25(11):1363–1369
Palomino R, Benites A, Liang LR Cloud parallel genetic algorithm for gene Microarray data analysis. Tools with artificial intelligence (ICTAI), 2011 23rd IEEE international conference on, pp 932–933
Chao J, Vecchiola C, Rajkumar B (2008) MRPGA: an extension of MapReduce for parallelizing genetic algorithms eScience, eScience’08. IEEE 4th international conference on, pp 214–221
Verma A, Xavier L, Goldberg DE, Campbell RH (2009) Scaling genetic algorithms using mapreduce In: Intelligent systems design and applications, 2009. ISDA’09. 9th international conference on. IEEE Press, pp 13–18
Xin D, Youcong N, Zhiqiang Y, Ruliang X, Datong X (2013) High performance parallel evolutionary algorithm model based on MapReduce framework. Int J Comput Appl Technol 46(3):290–295. Inderscience
Austin C, Yin-Wu T, Ching-Heng L (2010) Novel methods to identify biologically relevant genes for leukemia and prostate cancer from gene expression profiles. BMC Genomics: 11
Chen AH, Lin CH (2011) A novel support vector sampling technique to improve classification accuracy and to identify key genes of leukaemia and prostate cancers. Expert Syst Appl 38(4):3209–3219
Lee CP, Leu Y (2011) A novel hybrid feature selection method for microarray data analysis. Appl Soft Comput 11(1):208–213
Leu Y, Lee CP, Tsai HY (2010) A gene selection method for microarray data based on sampling. Comput Collective Intell Technol Appl: 68–74
Pradipta M, Chandra D (2012) Relevant and significant supervised gene clusters for Microarray cancer classification. NanoBioscience, IEEE Trans 11(2):161–168
Uri A, Naama B, Notterman DA, Kurt G, Suzanne Y, Mack D, Levine AJ (1999) Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci 96(12):6745–6750
Dudoit S, Fridlyand J, Speed TP (2002) Comparison of discrimination methods for the classification of tumors using gene expression data. J Am Stat Assoc 97(457):77–87
Golub TR, Donna SK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA et al (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Sci 286(5439):531–537
Jirapech-Umpai T, Aitken S (2005) Feature selection and classification for microarray data analysis, evolutionary methods for identifying predictive genes. BMC bioinforma 6(1):148
Li L, Weinberg CR, Darden TA, Pedersen LG (2001) Gene selection for sample classification based on gene expression data: study of sensitivity to choice of parameters of the ga/knn method. Bioinforma 17(12):1131–1142
Shipp MA, Ross KN, Tamayo P, Weng AP, Kutok JL, Aguiar RCT, Gaasenbeek M, Angelo M, Reich M, Pinkus GS et al (2002) Feature selection and classification for microarray data analysis: evolutionary methods for identifying predictive genes. Nat Med 8(1):68–74
Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, Tamayo P, Renshaw AA, D’Amico AV, Richie JP (2002) Gene expression correlates of clinical prostate cancer behavior. Cancer cell 1(2):203–209
Cho J-H, Lee D, Park JH, Lee I-B (2004) Gene selection and classification from microarray data using kernel machine. FEBS letters 571(1):93–98. Elsevier
Armano G, Chira C, Hatami N (2011) A new gene selection method based on random subspace ensemble for microarray cancer classification. Pattern Recognit Bioinforma 571(1):191–201. Springer
Caruana G, Li M, Qi MA MapReduce based parallel SVM for large scale spam filtering Fuzzy systems and knowledge discovery (FSKD), 2011 8th international conference on vol 4, pp2659–2662, 2011, IEEE
Kiran M, Kumar A, Mukherjee S, Prakash RG (2013) Verification and validation of MapReduce program model for parallel support vector machine algorithm on Hadoop cluster, vol 10, pp 317– 325
National Center for Biotechnology Information - (Gene), http://www.ncbi.nlm.nih.gov/gene
The Gene Ontology, http://www.geneontology.org/
Yotov WV, Hamel H, Rivard G-E, Champagne MA, Russo PA, Leclerc J-M, Bernstein ML, Levy E (1999) Amplifications of DNA primase 1 (PRIM1) in human osteosarcoma. Genes, Chromosom Cancer 26(1):62–69. Wiley Online Library
Hu H, Zhang H, Ge W, Liu X, Loera S, Chu P, Chen H, Peng J, Zhou L, Yu S et al (2012) Secreted protein acidic and rich in cysteines-like 1 suppresses aggressiveness and predicts better survival in colorectal cancers. Clin Cancer Res 18(19):5438–5448. AACR
Hao J, Serohijos A WR, Newton G, Tassone G, Wang Z, Sgroi DC, Dokholyan NV, Basilion JP (2008) Identification and rational redesign of peptide ligands to CRIP1, a novel biomarker for cancers. PLoS Comput Biol 4(8):e1000138. Public Library of Science
Lin D-T, Lechleiter JD (2002) Mitochondrial targeted cyclophilin D protects cells from cell death by peptidyl prolyl isomerization. J Biol Chem 277(34):31134–31141. ASBMB
Zhuo J, Tan EH, Yan B, Tochhawng L, Jayapal M, Koh S, Tay HK, Maciver SK, Hooi SC, Salto-Tellez M et al (2012) Gelsolin induces coclorectal tumor cell invasion via modulation of the urokinase-type plasminogen activator cascade. PloS one 7(8):e43594. Public Library of Science
Acknowledgments
This work was supported by a grant from the Kyung Hee University in 2013 (KHU-20130441)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Islam, A.K.M.T., Jeong, BS., Bari, A.T.M.G. et al. MapReduce based parallel gene selection method. Appl Intell 42, 147–156 (2015). https://doi.org/10.1007/s10489-014-0561-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10489-014-0561-x