
Abstract
Vascular tissue engineering of the middle layer of natural arteries requires contractile smooth muscle cells (SMC) which can be differentiated from adipose-derived mesenchymal stem cells (ASC) by treatment with transforming growth factor-β, sphingosylphosphorylcholine and bone morphogenetic protein-4 (TSB). Since mechanical stimulation may support or replace TSB-driven differentiation, we investigated its effect plus TSB-treatment on SMC orientation and contractile protein expression. Tubular fibrin scaffolds with incorporated ASC or pre-differentiated SMC were exposed to pulsatile perfusion for 10 days with or without TSB. Statically incubated scaffolds served as controls. Pulsatile incubation resulted in collagen-I expression and orientation of either cell type circumferentially around the lumen as shown by alpha smooth muscle actin (αSMA), calponin and smoothelin staining as early, intermediate and late marker proteins. Semi-quantitative Westernblot analyses revealed strongly increased αSMA and calponin expression by either pulsatile (12.48-fold; p < 0.01 and 38.15-fold; p = 0.07) or static incubation plus TSB pre-treatment (8.91-fold; p < 0.05 and 37.69-fold; p < 0.05). In contrast, contractility and smoothelin expression required both mechanical and TSB stimulation since it was 2.57-fold increased (p < 0.05) only by combining pulsatile perfusion and TSB. Moreover, pre-differentiation of ASC prior to pulsatile perfusion was not necessary since it could not further increase the expression level of any marker.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tissue engineering of small caliber vascular grafts is an ongoing issue to develop optimized replacement strategies for damaged or lacking arteries and veins. Although being used for decades, alloplastic materials for vascular grafting exert severe limitations like the risk for infections31 and thrombosis.28 The latter is not only caused by the thrombogenicity of the material itself but also by its limited elasticity resulting in a compliance mismatch in particular in the anastomosis regions which results in disturbed blood flow and turbulences promoting to atherosclerotic lesions.6 Another deficit is the inability of the synthetic graft to adapt to the blood pressure due to lacking vasoconstrictive properties. The key players for this are smooth muscle cells (SMC) that form the tunica media in natural arteries. We have recently shown that SMC can be differentiated effectively from adipogenic stem cells (ASC) which were obtained on an autologous basis from the patient’s own adipose tissue.12 This differentiation was induced biochemically by the combination of three myogenic stimuli, namely transforming growth factor (TGF) β1, sphingosylphosphorylcholine (SPC) and bone morphogenetic protein (BMP) 4. This combination (abbreviated TSB) led to a strong and sustained expression of proteins of the contractile apparatus such as alpha smooth muscle actin (αSMA), calponin and smoothelin. αSMA was considered as early marker expressed in both the synthetic highly proliferative phenotype and the contractile phenotype with limited proliferation capacity.24 Smoothelin, in contrast, marks the end stage of differentiation towards the contractile phenotype and calponin is an intermediate marker molecule.24 Though the differentiation was verified by a functional contractility assay, the approach used recently displays certain limitations. First, the usage of growths factors involves several difficulties. Besides the fact that they are extremely cost intensive, their (patho-) physiological background warrants considerations. TGFβ is strongly associated with cardiovascular diseases like atherosclerosis29 and intima hyperplasia,16 which displays a considerable risk in the context of vascular replacement. Also BMP4 induces ectopic bone formation in vivo as may be involved in vascular calcification.7 Moreover, an underestimated problem is the putative contamination of growth factor solutions with endotoxins26 which can only be prevented by elaborate cleaning processes.17 Second, in our previous study, cells have been treated with TSB on cell culture dishes as two-dimensional (2D) cultures. However, it has been shown that the scaffold 3D structure20 and stiffness22 strongly affects the differentiation status of SMC. Finally, taking into account the engineering of a tunica media, the orientation of the cells in a tubular scaffold seems to be pivotal for their physiological function. A natural artery underlies a permanent mechanical load due to the pulsatile blood pressure that accounts for proper differentiation and orientation of SMC. The latter has been shown in an approach using mature SMC which were exposed to cyclic mechanical load after incorporation into fibrin scaffolds.5 However, so far there is no study investigating the clues mechanical stimulation will provide for SMC differentiation from ASC.
In this study, we systematically investigated the effect of mechanical load applied by pulsatile perfusion on ASC incorporated in tubular fibrin scaffolds with respect to myogenic differentiation, contractility, extracellular matrix (ECM) production and morphology. Moreover, the combination of mechanical load and biochemical stimulation using the myogenic factors TSB and the effect of a biochemical pre-differentiation of ASC towards SMC were tested. We thereby evaluated whether cyclic mechanical load is sufficient to fully or partially replace the differentiation of ASC towards SMC by myogenic factors and reflect the consequences that our findings have for autologous vascular tissue engineering.
Materials and Methods
Generation of Cellularized Fibrin Tubes
ASC were isolated from abdominal subcutaneous adipose tissue of one out of 7 donors (53 ± 14 years, min/max = 31/70 years, 1 male/6 female) scheduled for visceral surgery after ethical approval by the institutional review board (Ethikkommission der Medizinischen Hochschule Hannover) and informed consent. Biochemical pre-differentiation was performed as recently described.10,13 Fibrin was isolated from human plasma obtained from donors after informed consent (Institute for Transfusion Medicine, Hannover Medical School) by cryo-precipitation as described previously.11 Per graft, 100 mg fibrinogen, 400 U aprotinin (Bayer, Leverkusen, Germany) and 400 µl 10 × M199 (Sigma Aldrich, Steinheim, Germany) were replenished by blood serum to 2 mL and pH was neutralized by 5 M NaOH. Per tube, 107 untreated ASC or pre-differentiated ASC (SMC) were suspended in this solution and mixed with 2 mL of a solution containing 10 U thrombin resolved in 40 mM CaCl2, and 10 U factor XIII (both from CSL Behring, Marburg, Germany). The mixture was filled into custom-made 10 cm-long tubular molds with a central placeholder (Department of Medical Device Construction, Hannover Medical School; Supplemental Fig. S1-A) and was allowed to polymerize for 30 min. Then the placeholder was removed and the fibrin scaffold was centrifuged at 26×g for 10 min, at 103×g for 10 min and at 316×g for 15 min in a horizontal custom-made rotation unit (Department of Medical Device Construction; Supplemental Fig. S1-B). This resulted in a compaction and significant stabilization of the fibrin matrix confirmed by a 1.83-fold increased burst pressure and a 3.86-fold higher tensile strength (Supplemental Fig. S2).
Cell viability after the compaction process was assessed in preliminary tests defining the optimal cell numbers in fibrin tubes. For this, Live/Dead staining was performed using a viability/cytotoxicity kit (Life-technologies, Darmstadt, Germany) following manufacturer’s instructions staining live cells green (calcein) and dead cells red (ethidium-homodimer). For the staining, sections of the fibrin tubes seeded with 0.5, 1 and 1.5 × 107 cells/tube were incubated with calcein/ethidium-homodimer solution 1, 5 and 8 days after seeding and compaction and assessed qualitatively by fluorescence microscopy. To quantify cell viability, segments with 1 × 107 cells/tube (which were used for the following experiments) were analyzed by counting the number of live and dead cells per microscopic view-field (10×—magnification) of 4 sections. Viability was calculated as the ratio of live cells to the total cell number (live cells + dead cells).
Bioreactor Set Up
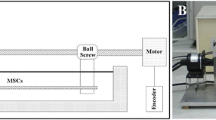
A custom made bioreactor-perfusion-system (Department of Medical Device Construction) was used for parallel pulsatile perfusion of four fibrin tubes. The main components of this system (Fig. 1) are an open reservoir (R), a non-occlusive pulsatile peristaltic pump (P1; modified heart–lung-machine-pump, Stoeckert-Shiley, Munich, Germany), a compliance chamber mimicking the “Windkessel”-effect of the aorta (C), four parallel aligned bioreactors containing the fibrin tubes (B1–B4), a pressure transducer (T) connected to a monitor-system (“CardioCap®” Datex, Helsinki, Finland) and a screwing clamp for flow- and pressure-modification (S). Due to minimal diffusion of culture medium from the luminal to the abluminal side through the tube-walls (≈ 2.5 mL tube−1 h−1), an open recirculation system was installed using a non-pulsatile peristaltic pump (P2; “Ismatec MCP Standard”, Cole Parmer, Wertheim, Germany).

Schematic illustration of the pulsatile bioreactor perfusion system. 4 fibrin segments were placed in parallel aligned bioreactors (B1–B4) and pulsatile perfusion was accomplished using a pulsatile peristaltic pump (P1) and a compliance chamber (C). The pressure was adjusted with a screwing clamp (S) and continuously monitored with a pressure transducer (T). An open reservoir and an open recirculation-system including a non-pulsatile pump (P2) ensured gas exchange within the incubator.
Pulsatile and Static Incubation
For pulsatile perfusion, fibrin scaffolds containing ASC or pre-differentiated ASC (SMC) were cut to a length of 5.5 cm, fixed on plastic hose nozzles using zip ties and inserted into the bioreactors. The tubes were stretched to a length of 6 cm to obtain a slight longitudinal tension and to prevent kinking of the scaffold during cyclic distention. The bioreactors and the reservoir of the perfusion system were filled with DMEM containing 1% FCS, 1% penicillin/streptomycin, 1% amphotericin B, 1% gentamycin, 1% glutaMAX, 7.5 U heparin sodium salt, 50 µg/mL ascorbic acid (to allow fully hydroxylated collagen production) and 100 U mL−1 aprotinin (to prevent fibrin degradation) complemented with or without TSB. In Table 1 the combinations of cells and perfusion medium used are given. The pulsatile incubation was performed with a flow rate of 64 mL min−1 at an impulse frequency of 60 bpm (Fig. 2a) for 10 days in a cell culture incubator at 37 °C and 5% CO2. Medium was changed every 3–4 days. As static controls, 4.5 cm long fibrin scaffolds were incubated in perfusion medium with or without TSB in vented cell culture tubes (Techno Plastic Products AG, Trasadingen, Switzerland) for 10 days at 37 °C and 5% CO2.

Pressure monitoring of the bioreactor perfusion system. Pressure profile was continuously measured using the CardioCap® monitoring system and documented every 24 h in each run. (a) pressure–time-curve in a 4 s time interval as displayed by the monitoring system. S systolic pressure, D diastolic pressure, M mean pressure. (b) Pressure course over the perfusion period of 10 days. Shown are mean ± SD of three independent experiments.
Determination of Wall Strain of the Fibrin Scaffolds
To determine the strain achieved by the pulsatile perfusion conditions, a video clip of the scaffold under pulsatile perfusion (n = 12) was taken and the outer diameter of the tubes at systolic and diastolic pressure was measured using the software “MB ruler”, version 5.3 (Windows Tools; Supplemental Fig. S3). The strain was calculated as follows: Strain (%) = (Diameter Systolic/Diameter Diastolic)*100-100. According to the formula for the circumference C = 2*radius*π and assuming a circular cross-section, the change in diameter is directly proportional to the change in circumference.
Characterization of Cell Orientation and ECM Production by Immunohistochemistry
After 10 days of pulsatile or static incubation, fibrin scaffolds were taken out of the bioreactors and cryo sections of 6 µm thickness were stained for the myo-specific marker proteins αSMA, calponin and smoothelin as well as for collagen-I as marker for ECM production. Moreover, α actin filaments were stained by TRITC-labeled phalloidin. For this, sections were fixed with 4% paraformaldehyde, permeabilized using 0.1% Triton-X-100 and blocked with PBS containing 10% FCS, 0.1% bovine serum albumin and 0.05% Tween-20 (all from Sigma-Aldrich). Immunostaining was performed using the following antibodies: Mouse αSMA; 1:100; DM001, Acris Antibodies, Herford, Germany); rabbit calponin (1:100; ab46794; Abcam), rabbit smoothelin (1:100; sc-28562; Santa Cruz Biotechnology, Heidelberg, Germany) and rabbit collagen-I (Thermo Scientific, Bremen, Germany). As secondary antibodies Alexa Fluor 555-coupled anti-rabbit IgG and Alexa Fluor 488-coupled anti-mouse IgG (Thermo Fisher Scientific, Bremen, Germany) were used. TRITC-labeled phalloidin (Sigma Aldrich) was diluted to 250 µg/mL and used parallel to the first antibodies. After incubation cells were washed with PBS and mounted with medium containing 4′6-diamidino-2-phenylindole (Roth, Karlsruhe, Germany) to counterstain cell nuclei. Staining was assessed by fluorescence microscopy (Nikon Eclipse TE300, Düsseldorf, Germany).
To test for ectopic calcification as a result of BMP4 treatment, cryo sections of each group were stained with Alizarin Red (Honeywell Fluka, Munich, Germany) according to histological standard procedures.
To quantify the wall strength of the scaffolds after pulsatile and static incubation, the thickness of 10 cross sections per condition was measured using light microscopy images and the software Image J (NIH, Bethesda, USA).
Quantitative Western Blot Analysis of SMC Markers
10 mm rings of fibrin scaffolds after pulsatile or static incubation were minced, washed in PBS and incubated in 100 µL 2x sodium dodecylsulfate (SDS) sample buffer containing 4% SDS (Roth), 10% β-mercaptoethanol (Sigma-Aldrich) and 1% dithiothreitol (Sigma-Aldrich) on ice for 5 min prior to centrifugation. Supernatants were heated to 95 °C and separated by SDS-PAGE, blotted onto PVDF membranes and stained by the following antibodies: αSMA (1:20,000; DM001, Acris Antibodies), calponin (1:20,000; ab46794, Abcam), smoothelin (1:500; ab8969; Abcam or 1:500; sc-28562; Santa Cruz Biotechnology) and GAPDH (1:10,000; 2118 s; Cell Signaling) and appropriate HRP-linked secondary antibodies (Anti-Mouse IgG, A9044 or Anti-Rabbit IgG, A9169; both Sigma Aldrich). SMC-specific bands were visualized using the ECL system (GE Healthcare, Chalfont St Giles, UK) and quantified densitometrically using the Quantity One software (Bio-Rad GmbH, Munich, Germany) prior to normalization to GAPDH expression of each cell lysate.
Contraction Assay
After 10 days of pulsatile or static incubation, fibrin scaffolds were taken out of the bioreactors and 5 mm rings of the scaffolds were cut open with a scalpel resulting in rectangular fibrin strips of approximately 15 mm length. The strips were incubated in 12-well-plates freely floating in perfusion media for 24 h at 37 °C and 5% CO2. Bending of the free-floating strips was assessed qualitatively by photo documentation after 0 and 24 h as an indicator for spontaneous contraction of functional SMC.
Statistics
Statistical analyses were performed using Graphpad Prism 6.04 (Graphpad Software, San Diego, California). Normal distribution of the data was tested using the d’Agostino & Pearson omnibus normality test. Results are presented as mean ± SD or SEM as indicated in the figure legends. Multiple comparisons between groups were performed by Two Way ANOVA followed by Sidak’s posttest. Differences were considered significant at p < 0.05. Significance levels were given as follows: *p < 0.05; **p < 0.01 or as indicated.
Results
Establishment of a Pulsatile Bioreactor Perfusion System
In this study we applied cyclic mechanical strain and pressure on fibrin scaffolds by a custom-made pulsatile perfusion system (Fig. 1). The system allowed the cyclic perfusion of 4 fibrin scaffolds in parallel under a maximal pressure of 19 ± 1 mmHg (‘systolic’) over 6 ± 1 mmHg (‘diastolic’; Fig. 2a) which was constant for 10 days (Fig. 2b). The pulsatile perfusion resulted in a wall strain of 6.85 ± 1.21% (n = 12; Supplemental Fig. S3), which did not differ significantly between the scaffolds perfused neither in parallel nor between the different runs.
Impact of Pulsatile Perfusion on Cell Morphology
To evaluate the effect of biomechanical forces on ASC or SMC, cells were embedded into the fibrin scaffolds prior to polymerization and fibrin compaction by centrifugation. In preliminary experiments a suitable cell number was evaluated by live-dead staining of the cells after the compaction process indicating a high cell survival (Supplemental Fig. S4). In particular the scaffolds seeded with 107 cells per scaffold showed a sufficient viability, which thus were used for all further experiments. Quantification of cell viability in these scaffolds on day 1 was calculated to be 95.78% ± 1.07%. We thus concluded that the compaction process did not impair cell viability.
The impact of pulsatile perfusion on cell morphology and the spatial arrangement of the cells within the scaffold were shown by phalloidin staining of cryo-sections obtained from scaffolds cultivated under pulsatile and static conditions (Fig. 3). In all groups, pulsatile perfusion reduced the thickness of the scaffold wall remarkably by 35.71% ± 11.2% (n = 10, p < 0.0001 by Student’s t test) as determined by light microscopy of cross sections of the tube wall. The cells under these conditions showed the same alignment parallel to the scaffold lumen as observed in natural arteries whereas under static conditions the cells showed a random distribution throughout the scaffold. Under pulsatile flow the majority of cells appeared to adapt an outstretched shape reminiscent of the spindle-like morphology of contractile SMC which seemed to be increasingly pronounced in ASC + TSB and SMC + TSB (see asterisks in Fig. 3). In contrast, the static controls showed round cells without any orientation.

Impact of pulsatile perfusion on cell morphology and alignment. Fibrin segments containing either adipose-derived mesenchymal stem cells (ASC) or pre-differentiated smooth muscle cells (SMC) were statically incubated or exposed to pulsatile perfusion for 10 days with or without the myogenic factors TGFβ1, SPC and BMP4 (TSB). Shown are cross sections of the tube walls after phalloidin staining of the cytoskeleton (red). The orientation of the cross section within the scaffolds is demonstrated in picture A: The luminal surface is at the bottom, the abluminal surface (not shown in the static probes) at the top of each picture. Representative cells presenting an elongated morphology are exemplarily marked with asterisks (*). Nuclei were stained blue by DAPI. Scale bar = 100 µm, TGFβ1 = Transforming growth factor β1, BMP4 = Bone morphogenetic protein 4, SPC = Sphingosylphosphorylcholine, DAPI = 4′,6-diamidino-2-phenylindole.
To test whether the treatment with myogenic factors including BMP4 induced the formation of ectopic calcium deposition, we stained cryo sections of each group with Alizarin Red. A specific staining indicating a starting calcification could not be observed (Supplemental Fig. S5).
Impact of Pulsatile Perfusion and Myogenic Factor Treatment on Myogenic Marker Protein Expression
In the next set of experiments, the embedded cells were stained for specific myogenic marker molecules. As we had shown previously,10 treatment of ASC with TSB resulted in strong and sustained expression of αSMA, calponin and smoothelin (Supplemental Fig. S6). We here stained cryo sections of fibrin scaffolds containing ASC or pre-differentiated ASC (SMC) incubated under static or pulsatile conditions for these molecules. As supposed, sections with embedded SMC in the presence of TSB showed a strong staining for the early marker αSMA under static and under pulsatile conditions, again confirming the spatial pattern observed before. In contrast, ASC, regardless of the treatment with the myogenic factors TSB, showed only a faint expression under static incubation whereas under pulsatile incubation the expression was comparable to that of SMC + TSB (Fig. 4a).

Impact of pulsatile perfusion on myogenic marker protein expression. Fibrin segments containing either adipose-derived mesenchymal stem cells (ASC) or pre-differentiated smooth muscle cells (SMC) were statically incubated or exposed to pulsatile perfusion for 10 days with or without the myogenic factors TGFβ1, SPC and BMP4 (TSB). Shown are cross sections of the tube walls stained for α smooth muscle actin (a, green), calponin (b, red) and smoothelin (c, red). For the orientation of the cross section within the scaffold see Fig. 3a and the respective legend. Nuclei were stained blue by DAPI. Scale bar = 100 µm, TGFβ1 = Transforming growth factor β1, BMP4 = Bone morphogenetic protein 4, SPC = Sphingosylphosphorylcholine, DAPI = 4′,6-diamidino-2-phenylindole.
The expression of the intermediate marker calponin followed virtually the same pattern. SMC + TSB under static incubation showed a clear expression of calponin which was even stronger under pulsatile incubation. Again, both ASC groups showed only a faint calponin expression under static conditions whereas pulsatile incubation induced its expression to the same extent than in SMC + TSB (Fig. 4b).
The late marker smoothelin, in contrast, was only weakly expressed under static incubation in either group. Cells under pulsatile incubation however, showed a clearly increased expression which was more pronounced in the presence of myogenic factors as shown for both, SMC + TSB and ASC + TSB (Fig. 4c).
Semi Quantitative Assessment of the Effect of Pulsatile Perfusion and Myogenic Factor Treatment on Myogenic Marker Protein Expression
To further investigate the impact of pulsatile perfusion and myogenic factor treatment on a semi quantitative basis, Western blot experiments were performed. In Fig. 5a specific bands at 42 kDa for αSMA, 34 kDa for calponin and at 110 kDa for smoothelin are shown. GAPDH was stained on each blot as internal loading control. Densitometric analysis of Western blots of at least 4 fibrin scaffolds for each marker protein confirmed the first impression the immunostaining gave (Fig. 5b). αSMA expression was rather low in ASC incubated under static conditions even in the presence of myogenic factors (ASC + TSB). Pre-differentiated SMC + TSB expressed significantly 7.23-fold (p < 0.05) more αSMA under the same conditions. Pulsatile incubation increased the expression of αSMA only significantly in ASC without treatment with myogenic factors (12.47-fold, p < 0.01). In the presence of myogenic factors, in ASC + TSB still a tendency towards increased αSMA expression (3.43-fold) was observed, whereas in SMC + TSB a slight tendency towards reduced αSMA levels were found. Again, as for the immunostaining, the calponin expression followed the same tendency among the groups. A strong though only marginally significant expression of calponin in ASC under pulsatile perfusion was found which was 38.3-fold higher than under static incubation (p = 0.075). In contrast to αSMA, this increase was also observed in the presence of myogenic factors, as the moderate expression in ASC + TSB under static incubation was 5.87-fold increased (p < 0.05) when the scaffolds were under pulsatile perfusion. Pre-differentiated SMC + TSB expressed high calponin levels also under static incubation which were unchanged by the pulsatile perfusion. Finally, the smoothelin expression was equally low in all three groups under static conditions, but was increased 2.70-fold (ASC + TSB) and 2.75-fold (SMC + TSB, both p < 0.05) by pulsatile perfusion only in those groups which had a parallel myogenic factor treatment. SMC + TSB did not express higher levels of smoothelin, thus indicating that a pre-differentiation is not advantageous for this tissue engineering application, but biomechanical stimulation and myogenic factor treatment both are necessary.

Semi quantitative assessment of myogenic marker proteins after pulsatile incubation. Fibrin segments, which contained either adipose-derived mesenchymal stem cells (ASC) or pre-differentiated smooth muscle cells (SMC) were statically incubated or exposed to pulsatile perfusion for 10 days with or without the myogenic factors TGFβ1, SPC and BMP4 (TSB). Extracts of the segments were submitted to Western blot analysis of α smooth muscle actin (αSMA), calponin and smoothelin. (a) Typical specific bands for each marker protein. (b) Semi quantitative analysis of specific bands for each marker protein by densitometry. Shown are mean ± SEM of 5 (ASC + TSB and SMC + TSB) or 4 (ASC) independently-run segments. *p < 0.05; **p < 0.01 and §p = 0.075 vs. ASC or as indicted by Two-way ANOVA and Sidak’s posttest. TGFβ1 = Transforming growth factor β1, BMP4 = Bone morphogenetic protein 4, SPC = Sphingosylphosphorylcholine.
Cell Contraction Capacity
To assess whether the expression of contractile marker proteins was also reflected by a changed contractility, a modified contraction assay was performed where spontaneous circular bending of the free-floating fibrin strips indicated a sustained contraction of the cells in the luminal part of the scaffold. Both ASC + TSB and SMC + TSB were positive for a contraction of the respective cells as indicated by bending of the strips after 24 h which was clearly stronger after pulsatile perfusion (Fig. 6). In the ASC-group, a slight bending was only observed in the pulsatile perfused probes, while the static controls did not show any cell contraction. Thus, again the combination of pulsatile flow and TSB was mostly supportive for an effective spontaneous contraction.

Qualitative contraction assay. 0.5 cm rings of each group where opened and placed in 12 well dishes floating freely in the corresponding perfusion media. The form of the strips as indicated by white dotted lines was assessed qualitatively immediately after placement and after 24 h in the cell culture incubator at 37 °C and 5% CO2.
Collagen Deposition in Fibrin Scaffolds
Finally, to test for beginning remodeling processes, the deposition of the extracellular matrix molecule collagen-I was stained on cryo sections obtained from fibrin scaffolds incubated under static and pulsatile conditions. Under static conditions, none of the cell types expressed significant amounts of collagen-I (Fig. 7). In contrast, pulsatile perfusion strongly induced the collagen-I expression in ASC, which seemed to be even more pronounced when myogenic factors were present confirming again the combination of mechanical and biochemical stimulation. There was no difference between ASC + TSB and SMC + TSB, indicating, that also for the expression of collagen-I a pre-treatment with myogenic factors was not necessary. However, throughout all groups, collagen-I expression was restricted to the cellular bodies and not released into the extracellular space.

Assessment of extracellular matrix production by collagen-I staining. Fibrin segments containing either adipose-derived mesenchymal stem cells (ASC) or pre-differentiated smooth muscle cells (SMC) were statically incubated or exposed to pulsatile perfusion for 10 days with or without the myogenic factors TGFβ1, SPC and BMP4. Shown are cross sections of the tube walls after collagen-I staining (red). For the orientation of the cross section within the scaffold see Fig. 3a and the respective legend. Nuclei were stained blue by DAPI. Scale bar = 100 µm, TGFβ1 = Transforming growth factor β1, BMP4 = Bone morphogenetic protein 4, SPC = Sphingosylphosphorylcholine, DAPI = 4′,6-diamidino-2-phenylindole.
Discussion
ASC have been utilized for vascular tissue engineering purposes due to their capacity to differentiate towards the myogenic lineage for several years. A number of studies suggest that besides chemical agents also mechanical stimulation can promote their myogenic differentiation.3 In this study, we evaluated the impact of mechanical cues applied by a pulsatile perfusion system alone or in combination with myogenic factor stimulation on myogenic characteristics of ASC or SMC incorporated into small diameter fibrin-based vascular grafts. The major findings are: (i) that tubular small diameter fibrin grafts are a suitable 3D matrix to culture ASC or SMC under pulsatile conditions in vitro, (ii) that pulsatile perfusion results in a circumferential alignment and increased collagen-I production independently of the use of TSB; (iii) either biochemical or mechanical stimulation alone leads to an increased expression of early- and intermediate-stage SMC-markers, but (iv) only the combination of both induces the expression of smoothelin as a marker of the late contractile SMC phenotype, whereby biochemical pre-differentiation does not further improve myogenic differentiation.
Fibrin Scaffolds and Bioreactor Technology
In this study fibrin was used as a novel biomaterial that comprises several benefits. Fibrin can be isolated very easily and efficiently from the patient’s own plasma by cryo precipitation which is applicable even in cachectic and pediatric patients and completely prevents graft rejections or inflammatory responses. The biocompatibility of fibrin is well known since fibrin glue is used successfully for adjuvant hemostasis in cardiovascular surgery for many years.25 Moreover, it contains multiple cell adhesion sites (see below) enabling its proper reseeding and remodeling. However, weak biomechanical properties so far have prevented its use as vascular graft. To manufacture tubular scaffolds suitable for a pulsatile perfusion, the fibrin matrices were manufactured by a rotation technique1 that results in effective fibrin compaction and thereby in significantly improved mechanical strength of the matrices.
The imitation of physiologic mechanical stress native blood vessels are exposed to in the human body is crucial for successful bioreactor-guided myogenic differentiation in vascular tissue engineering. With a mean strain of 6.85% ± 1.21% at 60 bpm, the mechanical stress generated by the system is accurately within the physiological range of 5–10% strain at 0.5–2 Hz in adults,9 although the mean pressure was distinctively lower compared to the physiologic blood pressure of the systemic circulation. This is most likely due to the relatively higher compliance of the fibrin matrices since fibrin in general displays a biomaterial with a high flexibility and instability although the compaction technique significantly increased burst pressure and tensile strength of the scaffolds. The secant modulus of scaffolds produced by the same rotation technique by Aper et al. was 0.29 ± 0.003 kPa/mm.1 Slight variations in the stiffness of the scaffolds and the mechanical forces acting on the scaffolds in the different runs cannot be ruled out. However, since the fibrin matrices in both the statically and the pulsatile incubated groups were fabricated and centrifuged identically, the mechanical stiffness should be comparable in all groups. Moreover, the longitudinal distension in the dynamic groups also is supposed to be identical since all scaffolds were stretched similarly during the implementation into the bioreactor. In this study, the effect of the pulsatile perfusion (pressure, wall strain) was quantified and controlled; nevertheless, the mechanic effects on the scaffolds are more complex since they include not only pressure and cyclic distention but also longitudinal distension and axial tension. The latter two could not be quantified in this study and thus were not included in our investigations systematically what can be considered as a certain limitation. Pulsatile perfusion resulted in direct exposure of the fibrin tubes to intraluminal pressure, which is suggested to play a role in the maintenance of the contractile SMC-phenotype in vitro.2 In contrast, in studies using fibrin scaffolds with lower stability, either extremely long culture times prior to mechanical stimulation were required8 or the grafts were wrapped around silicon tubes to allow mechanical stimulation,5,27 which resulted merely in plain stretching of the tube wall without any intraluminal pressure and most likely biased the mechanical conditioning. Considering all these studies, we conclude that (i) obtaining physiologic stretch amplitudes (5–10%) is more important for SMC differentiation than generating physiologic intraluminal pressure and that (ii) direct exposure of the scaffolds to intraluminal pressure marks a step towards a more physiologic conditioning in vitro. Since we cannot rule out that the low intraluminal pressure interferes with SMC differentiation, further experiments are necessary to improve the mechanical strength of the fibrin matrices and to enable direct perfusion under physiologic pressure values of 120 over 80 mmHg.
Cell Alignment, Morphology and Extracellular Matrix Production
Previous studies using uniaxial or equiaxial stretch in 2D models showed that mesenchymal stem cells and SMC align perpendicular to the direction of stretch.14,21 However, when exposed to circular dilatation by pulsatile stretch in a 3D scaffold as performed in this study, SMC in the small-diameter fibrin tubes align circumferentially around the lumen as shown by cytoskeleton staining with phalloidin. This finding confirms the observation by Gui et al.,5 where circular stretching of fibrin scaffolds seeded with mature SMC likewise resulted in circumferential alignment of the cells. As mentioned above, in that set-up the scaffolds were stretched on silicon tubes without intraluminal pressure, indicating that predominantly the circular mechanical load and not the intraluminal pressure induced the spatial arrangement of the cells. The achieved circumferential alignment of the cells resembles the arrangement of SMC in the tunica media of native blood vessels, thereby enabling the modulation of the vascular resistance by contraction or relaxation. The divergence between 2D and 3D cultures implies that the pulsatile stretch in tubular perfusion systems and in native vessels is more complex than a simple variation of the vessel diameter. It may also include a longitudinal stretch since the fibrin matrices were stretched during fixation in the bioreactor to avoid kinking and also natural vessels underlie a permanent longitudinal tension. These considerations underline the importance of 3D culture systems in vascular tissue engineering to achieve a physiological orientation of vascular SMC, which is critical for blood vessel function.
Both ASC and SMC in the mechanically stimulated segments had an outstretched spindle-like morphology with a length of approximately 100 µm, which is characteristic for the contractile phenotype of smooth muscle cells23,24 and which was not displayed by the cells in the static controls. Moreover, collagen-I production as indicator for proper extracellular matrix production which is characteristic for mature SMC, was significantly induced by mechanical stimulation. However, myogenic factors seemed to slightly increase this expression. Under any condition, a deposition into the extracellular space was not observed. This limitation is most likely due to the relative short culture time of only 10 days since a pronounced collagen production culture has been described after 30 days5 and up to 2 months30 under comparable conditions. Although longer stimulation times are supposed to result in proper extracellular matrix localization and in further increased stability, we here prefer not to incubate the scaffolds longer than 10 days since our approach was guided by our overarching goal of clinical translation for what culture times of up to 2 months are practically not suitable. Fibrin matrices seeded with the patient’s own ASC present a potential autologous method for vessel replacement (e.g. as coronary bypass graft or dialysis shunt) and thus should be generated by an efficient and highly reliable protocol which would be compromised by extended culture times. We suppose that after implantation the natural regeneration process also includes a proper ECM deposition. This, however, has to be confirmed in animal experiments which will be faced soon. In summary, biomechanical stimulation alone induced a properly aligned, outstretched and ECM-secreting SMC phenotype.
Myogenic Differentiation of ASC
We here showed by qualitative (immunohistochemistry) and semi-quantitative approaches (Western blot) that for the expression of the early- and intermediate-stage SMC markers αSMA and Calponin in ASC two treatment options were sufficient: Either a pre-differentiation followed by static incubation with myogenic factors or mechanical stimulation alone. The latter is consistent with observations of Nieponice et al., where bone marrow-derived mesenchymal stem cells were seeded on planar fibrin matrices and exposed to static or cyclic 10% longitudinal stretch at a frequency of 1 Hz for 6 days. αSMA and calponin expression was induced without treatment with myogenic factors by both static and cyclic stretch but not in non-stretched samples as assessed qualitatively by immunohistochemistry.19 In contrast, in our study smoothelin expression indicating specifically the late contractile SMC phenotype was not induced sufficiently solely by mechanical stimulation but required the combination with myogenic factors. There is one study determining late marker proteins under comparable conditions. Lin et al. perfused polyurethane-based scaffolds seeded with mouse embryonic multipotent mesenchymal stem cells at 1 Hz at 40 or 8 mL min−1 and found a significant expression not only of αSMA but also of the late myogenic marker protein myosin heavy chain (MHC) although a clear circumferential alignment of the cells was not observed.15 Myosin heavy chain and smoothelin both have been considered as appropriate markers for the contractile phenotype24 and thus should indicate a comparable differentiation status. There are many possible explanations for this discrepancy. First, embryonic mouse cells may be more susceptible to biomechanical cues than adult ASC. Second, the strain applied was probably within a different order of magnitude. Third, MHC -although characterized as late myogenic marker- may be induced differently by biomechanical stimulation and thus cannot be compared directly with smoothelin. Finally and most important, the scaffold material may influence also the expression level of myogenic molecules.
Effect of Fibrin on the Differentiation Status of SMC
As shown previously, treatment with TSB strongly induced the expression of αSMA, calponin, MHC and smoothelin in ASC cultured statically on tissue culture plastic.10 In the current study, ASC incorporated in fibrin scaffolds, however, expressed only low levels of αSMA, calponin and smoothelin indicating an inhibitory effect of the scaffold itself. Indeed, this phenomenon has been described previously by O’Cearbhaill et al. who observed a significantly lower expression of αSMA and calponin in mesenchymal stem cells embedded in fibrin compared to tissue culture plastic-cultivated cells.20 The molecular basis for this effect may involve insufficient cellular binding sites on the fibrin scaffold. Fibrin contains binding sites for thrombocytes via integrin α2β3, for macrophages via αMβ5 and for fibroblasts via αVβ318 that is also present in SMC, though it is only expressed in the proliferative phenotype.4 Thus, matrix-cell interactions that are in general involved in myogenic differentiation32 are strongly reduced in fibrin scaffolds, particularly in SMC of the contractile phenotype. On the other hand, it was shown that the stiffness of a scaffold positively influences the differentiation status of SMC in response to TGFβ.22 Taking into account that compaction of the fibrin scaffolds increases the stiffness of the matrix as we published previously,1 it could be assumed that the embedded cells underlie stimuli promoting myogenic differentiation as well.
Thus, myogenic differentiation of ASC in scaffolds is influenced by multiple factors and further studies are needed to unravel the underlying mechanisms for this phenomenon.
Contractility of Differentiated SMC
The results of the contraction assay follow virtually the same pattern as observed for the myogenic marker expression: The combination of biochemical and biomechanical stimulation induced SMC-functionality most effectively while both conditions alone were still sufficient to generate functioning SMC, though in a less effective manner. Taking into account the capacity of contraction as an indicator for the desired contractile SMC phenotype plus the ability of extracellular matrix secretion, these results again underline the importance of combined biochemical and biomechanical stimulation for complete myogenic differentiation of ASC.
Finally, the addition of mechanical stress on biochemically pre-differentiated SMC did not further improve the expression of either marker protein or contractile properties indicating that a pre-differentiation prior to ongoing chemical and additional mechanical stimuli is not beneficial. In the light of the enormous cost of myogenic factor treatment and -as indicated above- the risks that walk along with the usage of TGFβ and BMP4, our findings suggest that pre-differentiation of ASC towards SMC as also performed in Ref. 30 can be abandoned consequently.
Conclusion
We here showed that for fibrin- and ASC-based autologous tissue engineering of the tunica media it is indispensable to apply a combination of mechanical and biochemical cues to achieve SMC of the desired contractile phenotype aligned circumferentially around the vessel lumen and producing extracellular matrix while a myogenic pre-differentiation is not necessary. Thus, mechanical stimulation alone cannot completely replace TSB-guided differentiation. On the other hand, by abandoning growth factor driven pre-differentiation definitively the costs and most probably the risks related to long-term TSB treatment can be minimized considerably. To our knowledge, this is the first study comparing directly the effects exerted by either treatment showing that a proper cell morphology, alignment and extracellular matrix deposition can be achieved by biomechanical stimulation alone whereas co-treatment with myogenic factors was necessary to induce the switch from early/intermediate-staged SMC towards late-staged SMC. Though 10 days of perfusion were not enough for extracellular collagen-I deposition we anticipate that under in vivo conditions a remodeling process will cure most probably this deficit. In the light of the overarching goal of clinical translation short and effective process times are highly desirable. For this, in vivo-tests in appropriate animal models will be faced in the near future which will also reveal whether the unavoidable use of growth factors will trigger the risk of vascular calcification.
References
Aper, T., M. Wilhelmi, C. Gebhardt, K. Hoeffler, N. Benecke, A. Hilfiker, and A. Haverich. Novel method for the generation of tissue-engineered vascular grafts based on a highly compacted fibrin matrix. Acta Biomater 29:21–32, 2016.
Birukov, K. G., N. Bardy, S. Lehoux, R. Merval, V. P. Shirinsky, and A. Tedgui. Intraluminal pressure is essential for the maintenance of smooth muscle caldesmon and filamin content in aortic organ culture. Arterioscler Thromb Vasc Biol 18:922–927, 1998.
Dan, P., E. Velot, V. Decot, and P. Menu. The role of mechanical stimuli in the vascular differentiation of mesenchymal stem cells. J Cell Sci 128:2415–2422, 2015.
Finney, A. C., K. Y. Stokes, C. B. Pattillo, and A. W. Orr. Integrin signaling in atherosclerosis. Cell Mol Life Sci 74:2263–2282, 2017.
Gui, L., M. J. Boyle, Y. M. Kamin, A. H. Huang, B. C. Starcher, C. A. Miller, M. J. Vishnevetsky, and L. E. Niklason. Construction of tissue-engineered small-diameter vascular grafts in fibrin scaffolds in 30 days. Tissue Eng Part A 20:1499–1507, 2014.
Haruguchi, H., and S. Teraoka. Intimal hyperplasia and hemodynamic factors in arterial bypass and arteriovenous grafts: a review. J Artif Organs 6:227–235, 2003.
Hruska, K. A., S. Mathew, and G. Saab. Bone morphogenetic proteins in vascular calcification. Circ Res 97:105–114, 2005.
Isenberg, B. C., C. Williams, and R. T. Tranquillo. Endothelialization and flow conditioning of fibrin-based media-equivalents. Ann Biomed Eng 34:971–985, 2006.
Jufri, N. F., A. Mohamedali, A. Avolio, and M. S. Baker. Mechanical stretch: physiological and pathological implications for human vascular endothelial cells. Vasc Cell 7:8, 2015.
Lau, S. Strategies for the generation of fully autologous tissue-engineered fibrin-based vascular grafts resembling three-layered natural arteries. In: Department of Cardiothoracic, Transplant and Vascular Surgery. Hannover: Hannover Medical School, 2017, p. 143.
Lau, S., D. Eicke, M. Carvalho Oliveira, B. Wiegmann, C. Schrimpf, A. Haverich, R. Blasczyk, M. Wilhelmi, C. Figueiredo, and U. Boer. Low immunogenic endothelial cells maintain morphological and functional properties required for vascular tissue engineering. Tissue Eng. A 24:432–447, 2018.
Lau, S., M. Klingenberg, A. Mrugalla, F. Helms, D. Sedding, A. Haverich, M. Wilhelmi, and U. Boer. Biochemical myogenic differentiation of ASC is donor-dependent and requires sound characterization. Tissue Eng. A 2019
Lau, S., C. Schrimpf, M. Klingenberg, F. Helfritz, T. Aper, A. Haverich, M. Wilhelmi, and U. Böer. Evaluation of autologous tissue sources for the isolation of endothelial cells and adipose tissue-derived mesenchymal stem cells to pre-vascularize tissue-engineered vascular grafts. BioNanoMaterials 16:309–321, 2015.
Lee, W. C., T. M. Maul, D. A. Vorp, J. P. Rubin, and K. G. Marra. Effects of uniaxial cyclic strain on adipose-derived stem cell morphology, proliferation, and differentiation. Biomech Model Mechanobiol 6:265–273, 2007.
Lin, S., and K. Mequanint. Bioreactor-induced mesenchymal progenitor cell differentiation and elastic fiber assembly in engineered vascular tissues. Acta Biomater 59:200–209, 2017.
Low, E. L., A. H. Baker, and A. C. Bradshaw. TGFbeta, smooth muscle cells and coronary artery disease: a review. Cell Signal 2018.
Mack, L., B. Brill, N. Delis, and B. Groner. Endotoxin depletion of recombinant protein preparations through their preferential binding to histidine tags. Anal Biochem 466:83–88, 2014.
Mosesson, M. W., K. R. Siebenlist, and D. A. Meh. The structure and biological features of fibrinogen and fibrin. Ann N Y Acad Sci 936:11–30, 2001.
Nieponice, A., T. M. Maul, J. M. Cumer, L. Soletti, and D. A. Vorp. Mechanical stimulation induces morphological and phenotypic changes in bone marrow-derived progenitor cells within a three-dimensional fibrin matrix. J Biomed Mater Res A 81:523–530, 2007.
O’Cearbhaill, E. D., M. Murphy, F. Barry, P. E. McHugh, and V. Barron. Behavior of human mesenchymal stem cells in fibrin-based vascular tissue engineering constructs. Ann Biomed Eng 38:649–657, 2010.
Park, J. S., J. S. Chu, C. Cheng, F. Chen, D. Chen, and S. Li. Differential effects of equiaxial and uniaxial strain on mesenchymal stem cells. Biotechnol Bioeng 88:359–368, 2004.
Park, J. S., J. S. Chu, A. D. Tsou, R. Diop, Z. Tang, A. Wang, and S. Li. The effect of matrix stiffness on the differentiation of mesenchymal stem cells in response to TGF-beta. Biomaterials 32:3921–3930, 2011.
Patel, S., Y. Shi, R. Niculescu, E. H. Chung, J. L. Martin, and A. Zalewski. Characteristics of coronary smooth muscle cells and adventitial fibroblasts. Circulation 101:524–532, 2000.
Rensen, S. S. M., P. Doevendans, and G. van Eys. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Netherlands Heart Journal 15:100–108, 2007.
Rousou, J. A. Use of fibrin sealants in cardiovascular surgery: a systematic review. J Card Surg 28:238–247, 2013.
Schwarz, H., M. Schmittner, A. Duschl, and J. Horejs-Hoeck. Residual endotoxin contaminations in recombinant proteins are sufficient to activate human CD1c + dendritic cells. PloS ONE 9:e113840, 2014.
Syedain, Z. H., J. S. Weinberg, and R. T. Tranquillo. Cyclic distension of fibrin-based tissue constructs: evidence of adaptation during growth of engineered connective tissue. Proc Natl Acad Sci USA 105:6537–6542, 2008.
Tatterton, M., S. P. Wilshaw, E. Ingham, and S. Homer-Vanniasinkam. The use of antithrombotic therapies in reducing synthetic small-diameter vascular graft thrombosis. Vasc Endovasc Surg 46:212–222, 2012.
Toma, I., and T. A. McCaffrey. Transforming growth factor-beta and atherosclerosis: interwoven atherogenic and atheroprotective aspects. Cell Tissue Res 347:155–175, 2012.
Wang, C., L. Cen, S. Yin, Q. Liu, W. Liu, Y. Cao, and L. Cui. A small diameter elastic blood vessel wall prepared under pulsatile conditions from polyglycolic acid mesh and smooth muscle cells differentiated from adipose-derived stem cells. Biomaterials 31:621–630, 2010.
Wilson, W. R., T. C. Bower, M. A. Creager, S. Amin-Hanjani, P. T. O’Gara, P. B. Lockhart, R. O. Darouiche, B. Ramlawi, C. P. Derdeyn, A. F. Bolger, M. E. Levison, K. A. Taubert, R. S. Baltimore, and L. M. Baddour. Vascular graft infections, mycotic aneurysms, and endovascular infections: a scientific statement from the American Heart Association. Circulation 134:e412–e460, 2016.
Zhang, D., M. B. Sun, J. Lee, A. A. Abdeen, and K. A. Kilian. Cell shape and the presentation of adhesion ligands guide smooth muscle myogenesis. J Biomed Mater Res A 104:1212–1220, 2016.
Acknowledgments
We thank S. Zippusch for her help with the fibrinogen isolation.
Funding
This work has been carried out as an integral part of the BIOFABRICATION FOR NIFE (2012) Initiative, which is financially supported by the Lower Saxonian Ministry for Science and Culture and the VolkswagenStiftung. (NIFE is the Lower Saxonian Center for Biomedical Engineering, Implant Research and Development—a joint translational research center of the Hannover Medical School, the Leibniz University Hannover, the Foundation University of Veterinary Medicine Hannover and the Laser Center Hannover).
Author Contributions
There were no contributions that do not justify authorship.
Conflict of interest
No conflict of interest exists.
Author information
Authors and Affiliations
Corresponding author
Additional information
Associate Editor Joel Stitzel oversaw the review of this article.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
10439_2019_2234_MOESM1_ESM.tif
Supplemental Fig. S1: A: Composition of the vessel mold. The vessel mold consisted of two half-shells of polyether ether ketone (PEEK), two Teflon®-cuffs with drill holes for the application of the fibrinogen/thrombin-mixture and a tubular placeholder (I). The mold was fixed using a hose clamp and adhesion tape and brought into an upright position for initial static polymerization (II). The casted tubes were 10 cm in length with a luminal diameter of 4.75 mm and a wall thickness of 1.63 mm. B: Manufacturing setup of the rotation unit. The rotation unit consisted of a computer-controlled water-cooled electric motor and a rotation chamber (I) containing a rotating metal tube. After static polymerization, the vessel mold including the fibrin segment was placed in the metal tube, which contained drill holes to allow the outflow of excessive fluid during rotation and was closed with a sealing cap (II). Supplementary material 1 (TIFF 3961 kb)
10439_2019_2234_MOESM2_ESM.tif
Supplemental Fig. S2: Effect of fibrin compaction on biomechanical strength. Burst pressure or tensile strength was measured of uncompacted or compacted fibrin scaffolds. Shown are means ± SD of 6 scaffolds. **p < 0.01 and #p < 0.0001 by student’s T test. Supplementary material 2 (TIFF 103 kb)
10439_2019_2234_MOESM3_ESM.tif
Supplemental Fig. S3: Quantification of cyclic strain. Fibrin segments (n = 12, 3 runs) were exposed to pulsatile perfusion under video recordings. Images of segments under maximal (systolic) and minimal (diastolic) pressure were subsequently analyzed using the measurement-software “MB Ruler”©. Wall strain was calculated using the formula Strain (%) = (Diameter Systolic/Diameter Diastolic)*100–100. Supplementary material 3 (TIFF 7318 kb)
10439_2019_2234_MOESM4_ESM.tif
Supplemental Fig. S4: Live/dead staining for analyzing Zell viability after the compaction process and determine the optimal zell count per tube. Fibrin scaffolds where seeded with 0.5, 1 and 1.5 × 107 cells/tube and live/dead staining was performed on plane sections of 0.5 cm Rings on day 1, 5 and 8 after the compaction process. Living cells are stained green (Calcein) and dead cells are stained red (Ethidium-Homodimer). Scale bar = 100 µm. Supplementary material 4 (TIFF 4965 kb)
10439_2019_2234_MOESM5_ESM.tif
Supplemental Fig. S5: Impact of treatment with myogenic factors on scaffold calcification. Fibrin segments containing either adipose-derived mesenchymal stem cells (ASC) or pre-differentiated smooth muscle cells (SMC) were statically incubated or exposed to pulsatile perfusion for 10 days with or without the myogenic factors TGFβ1, SPC and BMP4 (TSB). Shown are cryo sections stained by Alizarin Red. No calcium deposits in either group (A-C) were observed as shown in the positive control (D; see purple arrows pointing to calcium crystals in calcified decellularized equine carotid artery as vascular graft in sheep as published in Böer et al., Int J Art Org, 2013, 36(3) 184). Scale bar: 100 µm. Supplementary material 5 (TIFF 26182 kb)
10439_2019_2234_MOESM6_ESM.tif
Supplemental Fig. S6: Expression of myogenic marker proteins in myogenic factor pre-differentiated ASC. Adipose-derived mesenchymal stem cells (ASC) cultured with 1 % FCS or differentiated towards the myogenic phenotype with TGFβ1, BMP4 and SPC for 8 days (n = 3 in both groups) were analyzed by Western blot analysis for the expression of α smooth muscle actin (αSMA), calponin and smoothelin prior to embedding into the fibrin graft. FCS = fetal calf serum, TGFβ1 = Transforming growth factor β1, BMP4 = Bone morphogenetic protein 4, SPC = Sphingosylphosphorylcholine. Supplementary material 6 (TIFF 5721 kb)
Rights and permissions
About this article

Cite this article
Helms, F., Lau, S., Klingenberg, M. et al. Complete Myogenic Differentiation of Adipogenic Stem Cells Requires Both Biochemical and Mechanical Stimulation. Ann Biomed Eng 48, 913–926 (2020). https://doi.org/10.1007/s10439-019-02234-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10439-019-02234-z