
Abstract
Atherosclerosis, a chronic lipid-driven inflammatory disease affecting large arteries, represents the primary cause of cardiovascular disease in the world. The local remodeling of the vessel intima during atherosclerosis involves the modulation of vascular cell phenotype, alteration of cell migration and proliferation, and propagation of local extracellular matrix remodeling. All of these responses represent targets of the integrin family of cell adhesion receptors. As such, alterations in integrin signaling affect multiple aspects of atherosclerosis, from the earliest induction of inflammation to the development of advanced fibrotic plaques. Integrin signaling has been shown to regulate endothelial phenotype, facilitate leukocyte homing, affect leukocyte function, and drive smooth muscle fibroproliferative remodeling. In addition, integrin signaling in platelets contributes to the thrombotic complications that typically drive the clinical manifestation of cardiovascular disease. In this review, we examine the current literature on integrin regulation of atherosclerotic plaque development and the suitability of integrins as potential therapeutic targets to limit cardiovascular disease and its complications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Atherosclerotic cardiovascular disease
Atherosclerosis, a chronic inflammatory disease of the large arteries, involves the progressive accumulation of lipids, necrotic cell debris, and extracellular matrix proteins in the vessel intima that ultimately results in vessel occlusion or thrombotic complications. Initially, atherosclerotic plaques start as regions of local dysfunction in the endothelial lining of the blood vessel. Deposition of cholesterol [primarily packaged in low-density lipoproteins (LDL)] at these sites drives the accumulation of monocyte-derived macrophages in an attempt to clear the lipid deposits. This lipid-driven inflammatory response drives the formation of a microenvironment rich in oxidized LDL (oxLDL), cytokines, growth factors, and wound-associated extracellular matrix proteins (fibronectin, osteopontin) [1–4] (Fig. 1). These stimuli induce vascular smooth muscle cells in the media to lose their contractile properties, attain a synthetic, fibroproliferative phenotype, and migrate into the vessel intima [5]. While smooth muscle-driven fibrosis can produce stenotic lesions that restrict flow to target organs, smooth muscle remodeling is thought to confer protection against plaque rupture and thrombosis through the formation of a collagen-rich fibrous cap [6, 7].

Model for atherosclerotic plaque development. Progression of atherosclerotic plaques from alterations in early endothelial function to the progressive accumulation of macrophages and smooth muscle cells are shown. Intraplaque angiogenesis in large atherosclerotic lesions provide additional access sites for leukocyte targeting
Current treatments for atherosclerotic cardiovascular disease rely primarily on therapeutics that target systemic risk factors, such as hypercholesterolemia (e.g., statin family of HMG CoA reductase inhibitors). However, a large number of individuals do not show significant clinical benefit with lipid-lowering therapies, with two-thirds of all patients who receive statin therapy continuing to experience cardiovascular events within a 5-year timeframe [8]. While the disease is unquestionably lipid-driven, other systemic factors or components of the atherosclerotic microenvironment may perpetuate atherosclerotic disease even in the presence of normal plasma lipid levels. Of particular interest, the arterial microenvironment appears to regulate both the localization of where these plaques occur and the sensitivity of cells within the plaque to various atherogenic stimuli. Atherosclerotic lesions develop in arterial regions exposed to turbulent patterns of blood flow, including arterial curvatures, branch points, and bifurcations [9], whereas vascular regions exposed to unidirectional, laminar flow show protection from plaque formation. In addition to local hemodynamics, changes in the composition of the arterial matrix regulate multiple aspects of plaque formation, including lipid deposition, inflammation, and smooth muscle incorporation. The different mechanical properties of these matrix proteins serve to regulate tissue architecture, and the differential activation of cell-matrix receptors tunes cell function to microenvironmental conditions [10, 11].
Integrin activation and signaling
The integrin family of receptors plays a significant role in cellular crosstalk with its microenvironment [12, 13]. As transmembrane receptors, integrins serve to integrate the internal actomyosin cytoskeleton with extracellular matrix proteins or Ig superfamily counter-receptors [e.g., intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1)] on adjacent cells [12, 13]. As such, integrins affect cell function through mechanically coupling the cell to the surrounding environment, translating the composition and structure of the microenvironment into intracellular biochemical signals, and facilitating certain cell–cell interactions [13]. The mammalian cadre of integrins includes 18 α and 8 β subunits that form 24 distinct heterodimers, which can be divided by ligand affinity and cell type-restricted expression patterns (Fig. 2a) [12]. Collagens and laminins comprise a bulk of the integrin-binding matrix proteins in adult tissues, and the collagen-binding integrins (α1β1, α2β1, α10β1, α11β1) and laminin-binding integrins (α3β1, α6β1, α7β1, α6β4) are typically associated with a quiescent cell phenotype. In contrast, provisional matrix proteins, such as fibronectin, fibrinogen, and vitronectin, show enhanced production and deposition during tissue remodeling responses, and the integrins that bind to the RGD sequence in these proteins (α5β1, α8β1, αvβ1, αvβ3, αvβ5) are classically associated with enhanced cell proliferation and migration [12, 14]. Leukocyte-specific integrins (αLβ2, αMβ2, αXβ2, αDβ2, α4β7) interact with Ig superfamily counter-receptors expressed on activated endothelial cells, whereas the platelet-specific integrin αIIbβ3 classically interacts with the RGD sequence in fibrin [12, 15, 16].

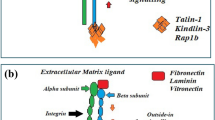
Vascular integrin signaling. a The 24 mammalian integrin dimers are shown. Shading indicates their ligand binding specificity for collagen (blue), laminin (purple), or RGD motif-containing proteins (red), or their leukocyte-specific expression pattern (green). b Structure of integrin signaling complexes with their signaling, force transduction, and actin coupling layers
While integrins do not possess any intrinsic enzymatic activity, integrin heterodimers mediate their mechanical and signaling functions through the recruitment of a vast array of proteins that assemble into large and functionally diverse complexes [17, 18]. Integrin ligation promotes the formation of nascent adhesions composed of loosely clustered integrin dimers and selects signaling and actin-binding proteins (Fig. 2b). These nascent adhesions converge to form higher order complexes at the end of contractile actin fibers. Based on the size, location, and composition, these adhesion sites can be termed focal complexes (smaller, linked to small actin bundles) or focal adhesions (larger, linked to actin stress fibers) (Fig. 2b). The application of force to integrin adhesions, either through stretching of the extracellular matrix or enhanced intracellular actomyosin contractility, results in a rapid structural reinforcement of adhesion sites, with enhanced actin filament bundling, increased recruitment of cytoskeletal adaptor proteins that link integrins to actin filaments (e.g., talin1, vinculin), and increased density of integrins within the adhesion site [13]. Both focal complexes and focal adhesions require myosin contractility to maintain their structure and rapidly disassemble in the presence of myosin inhibitors [19]. Additionally, application of force activates a variety of integrin-targeted signaling proteins that convert the physical force into intracellular biochemical signals, a process termed mechanotransduction [13, 20]. Super resolution microscopy identified an organization of these adhesome proteins into a membrane-proximal layer rich in signaling proteins [focal adhesion kinase (FAK), integrin-linked kinase (ILK)], a force transduction layer containing proteins involved in force-dependent reinforcement of adhesions (talin1, vinculin), and an actin regulation layer containing a variety of actin-binding proteins (zyxin, α-actinin) [21]. Together, this protein complex provides a 40-nm gap between the integrin tail and the actin filaments. These integrin clusters form a signaling hub at these focal adhesion sites, which can synergize with other extracellular stimuli such as growth factors to regulate cell function [17, 18].
Recent advances in proteomics have provided insight into the mechanisms of integrin signaling [18]. Proteomic analysis of isolated integrin complexes identified multiple cytoskeletal adaptors, Ser/Thr and Tyr kinases, signaling adaptor proteins, and guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs) for the Rho family GTPases. However, these studies have also uncovered unexpected recruitment of proteins involved in RNA translation, cellular metabolism, and membrane trafficking. Multiple studies have shown that both the integrin dimer involved and the force applied to the integrin play significant roles in determining which proteins are recruited [22–25]. These studies suggest that recruitment of certain proteins may be conserved among multiple integrins, and these tend to be proteins involved in cytoskeletal linkage, such as talin1, vinculin, filamin, kindlin, and VASP [18]. In contrast, recruitment of specific signaling proteins appears to be more integrin-specific. Ligation of α5β1 recruits higher levels of the ILK-PINCH-PARVIN complex and stimulates RhoA/ROCK-dependent myosin contractility [26]. However, ligation of αv integrin dimers (αvβ3, αvβ5) recruits Src and the Rho GEF (GEF-H1) resulting in high levels of RhoA activity but minimal myosin phosphorylation and tractional force [23]. Ligation of both α5β1 and αv integrin dimers is required for maximal myosin phosphorylation and cellular responses to increased substrate stiffness, suggesting that both integrins play functionally distinct but complementary roles in this response [23]. One weakness of the current proteomic approaches has been the concentration on a limited subset of integrins, as all of the previously mentioned studies focus only on α4β1, α5β1, αvβ3, and αvβ5 signaling.
In nonadherent cell types, such as platelets and leukocytes, integrins mostly exist in a bent conformation that sequesters the ligand binding pocket close to the cell surface and away from potential ligands [27, 28]. This structure is maintained through a salt bridge that forms between membrane-proximal regions of the α and β cytoplasmic tail [27, 28]. Integrin activation, a transition from a low affinity bent conformation to a high affinity extended confirmation, is induced by “inside-out” signaling pathways, signals inside the cell that disrupt this salt bridge and alter the integrin conformation on the outside of the cell. This alteration in integrin function critically regulates rapid induction of adhesion in circulating platelets and leukocytes to induce thrombosis and leukocyte homing, respectively [15, 16]. Also, rapid induction of integrin activation in adherent cells promotes a robust induction of integrin-specific signaling that can regulate cell function [29–31]. In the subsequent sections, we will examine the current literature related to how integrin signaling contributes to the various processes involved in atherosclerotic plaque formation and the potential for integrin-based therapeutics to modulate these responses.
Integrins in endothelial cell function
Like all epithelia, the vessel’s endothelial cell layer resides on a basement membrane composed of collagen IV, laminin, nidogen, perlecan, and other proteoglycans [32]. Macrovascular endothelial cells express multiple basement membrane-binding integrins (Table 1), including α2β1, α3β1, α6β1, and α6β4, and signaling through these integrins is widely associated with a quiescent endothelial cell phenotype [14, 33, 34]. During the early stages of atherosclerosis, provisional matrix proteins, such as the RGD-containing proteins fibronectin, fibrinogen, and thrombospondin-1, show significantly enhanced deposition within the endothelial cell matrix [31, 35, 36]. These provisional matrix proteins bind to a different subset of endothelial integrins (Table 1), including α5β1, αvβ3, and αvβ5, with signaling through these integrins associated with actin remodeling responses and endothelial activation [14, 33, 34]. Deposition of provisional matrix proteins into the subendothelial basement membrane may involve changes in endothelial permeability, resulting in the leak of provisional matrix proteins in the plasma into the vessel wall, or changes in endothelial gene expression to enhance deposition of endothelial cell matrices. Deletion of plasma fibronectin reduces fibronectin deposition during early atherogenesis [37], and atherosclerosis-prone sites show enhanced deposition of fibrinogen, which is not produced by vascular cells [31]. In partial carotid ligation models of disturbed flow, fibronectin deposition can only be reduced by global fibronectin knockout and not by deletion in the cells of the vascular wall (both endothelial cells and smooth muscle cells) [38], suggesting that leak of plasma fibronectin plays a primary role in fibronectin deposition in this model. However, atherosclerosis-prone sites also show enhanced endothelial fibronectin expression [36, 39] and deposition of fibronectin splice variants only expressed in cellular but not plasma fibronectin [40].
Integrin signaling in endothelial cell activation
In quiescent vessels, the endothelial layer regulates vascular tone, provides an anti-thrombotic surface, and forms a tight barrier to restrict the passage of blood components into surrounding tissue [41]. Activation of the endothelial cell layer during inflammatory responses involves a phenotypic transition that increases vascular permeability and enhances the expression of leukocyte counter-receptors (e.g., ICAM-1, VCAM-1), proinflammatory cytokines (tumor necrosis factor α (TNFα), interleukin-1B (IL-1β)), and procoagulant molecules (tissue factor (TF)) [41]. Molecularly, endothelial activation represents the first discernable sign of local atherosclerotic susceptibility and is maintained during subsequent stages of atherosclerotic progression [42]. Several stimuli promote endothelial activation, including mechanical stress, oxidized LDL (oxLDL), proinflammatory cytokines (e.g., TNFα, IL-1β), and the bacterial endotoxin LPS [41]. Much of the reprogramming in endothelial gene expression can be attributed to the proinflammatory transcription factor nuclear factor-κB (NF-κB) [43, 44]. The NF-κB family of homodimeric and heterodimeric transcription factors is maintained in an inactive state in the cytoplasm but translocates to the nucleus upon activation in response to a variety of endothelial cell activators [43]. Enhanced expression and nuclear localization of the NF-κB p65 subunit (hereafter NF-κB) can be observed at atherosclerosis-prone sites prior to the initiation of plaque formation [43, 44], and preventing NF-κB-driven VCAM-1 expression significantly reduces atherosclerotic plaque formation [43, 45].
Endothelial cells sense the frictional shear stress generated by blood flow resulting in integrin activation and ligation-dependent signaling [9, 20, 46]. Since different integrins activate disparate signaling responses, the endothelial response to flow differs based on the composition of the subendothelial matrix. Shear-induced ligation of provisional matrix-binding integrins results in activation of the Rac effector p21-activated kinase (PAK) which promotes activation of proinflammatory signaling (NF-κB, JNK), proinflammatory gene expression (ICAM-1, VCAM-1), and endothelial permeability [31, 47–49]. Fibronectin appears to play a major role in this response, as deleting fibronectin expression in the mouse plasma reduces PAK and NF-κB activation at sites of disturbed flow in vivo, corresponding to reduced plaque formation [37]. While shear stress activates multiple provisional matrix-binding integrins (α5β1, αvβ3 integrins), several groups have shown that inhibiting αvβ3 is sufficient to prevent proinflammatory signaling (PAK, NF-κB) on fibronectin [34, 50]. Furthermore, mice treated with αv integrin inhibitors and mice deficient in endothelial αv expression show reduced proinflammatory responses at both sites of endogenous disturbed flow and induced disturbed flow using the partial carotid ligation model [34]. In contrast to αvβ3, differing reports exist on the signaling through α5β1 in the endothelial response to flow. While Chen et al. showed no effect of α5β1 inhibition or α5 knockdown on the endothelial response to flow [34], Sun et al. demonstrated that α5 blocking antibodies and siRNA reduced oscillatory flow-induced ICAM-1 and VCAM-1 expression associated with reduced inflammasome activation and mature IL-1β production [51]. In addition, Yun et al. demonstrated that endothelial expression of chimeric integrins with the α5 extracellular domain and the α2 intracellular domain show reduced NF-κB activation. A potential confounding factor in these studies is the role of α5β1 in fibronectin deposition, suggesting that inhibiting α5 could limit provisional matrix deposition and subsequent signaling through αvβ3. Consistent with this hypothesis, α5/α2 chimeric mice show diminished fibronectin deposition in vivo [52]. In addition, the α5/α2 chimeric integrin enhances flow-induced PKA signaling in endothelial cells, which can inhibit αvβ3 activation [52].
Like mechanical forces, oxLDL also regulates endothelial integrin signaling and shows matrix-specific effects on endothelial cell function. Fibronectin deposition enhances oxLDL-induced NF-κB activation and subsequent ICAM-1 and VCAM-1 expression [52, 53]. This matrix-specific response occurs due to oxLDL’s ability to induce α5β1 activation and α5β1-dependent signaling. Unlike flow, α5β1 blocking antibodies and siRNA significantly blunt the inflammatory response to oxLDL [53], whereas αvβ3 inhibitors do not. Atherosclerosis-prone ApoE−/− mice fed a high fat, Western diet show reduced proinflammatory gene expression, monocyte recruitment, and plaque formation when treated with the α5β1 inhibitor ATN-161 [53]. Similarly, Sun et al. demonstrated reduced inflammation and early plaque formation in Western diet-fed LDLR−/− mice with reduced α5 expression (α5−/+) or treated with ATN-161 [51], demonstrating the contribution of α5 to plaque development. While shear stress-induced NF-κB activation involves PAK signaling [48], oxLDL-induced α5β1 signaling activates a FAK/ERK/p90 ribosomal S6-kinase (p90Rsk) pathway to induce NF-κB [54]. Consistent with this, FAK activation is enhanced in the endothelium overlying both mouse and human atherosclerotic plaques; mice expressing kinase-dead FAK in the endothelial cell layer show significantly reduced inflammation and early atherogenesis in response to high fat diet feeding [54].
Integrins and endothelial dysfunction
Like endothelial activation, atherosclerotic plaque formation has long been associated with an impairment in endothelial nitric oxide (NO) production and vasodilation, termed endothelial dysfunction [55, 56]. Endothelial nitric oxide synthase (eNOS), an enzyme that converts the amino acid l-arginine to NO, is the primary source of endothelial NO production and a key contributor to vasodilation. During endothelial dysfunction, reduced endothelial NO production results from decreased eNOS activity and/or expression, increased eNOS uncoupling, and enhanced NO scavenging by reactive oxygen species [55, 56]. While NO’s vasodilatory properties do not likely contribute to its relationship to atherosclerosis, NO also inhibits NF-κB activation and prevents platelet and leukocyte integrin activation [55]. Shear stress promotes eNOS activation through induction of intracellular calcium and through eNOS phosphorylation [57]. While integrin signaling does not appear to regulate the initial calcium influx, integrin signaling may regulate the kinases involved in flow-induced eNOS phosphorylation, including Akt and PKA [57–59]. Yang et al. demonstrated that β1-blocking antibodies reduce Akt and eNOS activation early following onset of flow in bovine endothelial cells [60]. However, flow-induced Akt activation does not change based on matrix composition, and inhibiting either α5β1 or αvβ3 signaling does not affect flow-induced Akt activation or eNOS phosphorylation on the site classically phosphorylated by Akt (Ser1179) [34, 61]. In contrast, Yurdagul et al. showed enhanced flow-induced nitric oxide production in endothelial cells on basement membrane proteins compared to fibronectin, identifying enhanced PKA signaling as a primary mediator of this effect [62, 63]. While these studies found that flow-induced Ser1179 phosphorylation requires PKA, phosphorylation of another site known to contribute to eNOS activation, Ser635, does not require PKA activity and paradoxically shows enhanced flow-induced phosphorylation in the presence of PKA inhibitors [62]. Interestingly, PKA inhibition enhances αvβ3 activation in endothelial cells on collagen [61], and inhibiting αvβ3 reduces flow-induced eNOS Ser635 phosphorylation [34].
Integrins and plaque angiogenesis
Angiogenesis, the formation of new blood vessel from preexisting vessels, plays a dual role in atherosclerosis, both contributing to plaque progression and alleviating hypoxic injury to ischemic tissue affected by a stenotic plaque [64, 65]. Expansion of the vasa vasorum correlates with plaque progression in mouse models of atherosclerosis [66, 67], and intraplaque angiogenesis correlates with lipid deposition, inflammation, and hemorrhage [68–71]. Like peripheral tissue, hypoxia in the developing atherosclerotic plaque is thought to induce the expression of several proangiogenic growth factors, such as vascular endothelial growth factor and fibroblast growth factor, through the transcription factor hypoxia-inducible factor-1 [72, 73]. Additionally, oxLDL stimulates angiogenic growth factor production in plaque macrophages, consistent with the role of macrophages in regulating angiogenesis in other systems [74, 75]. Expansion of the vasa vasorum tends to occur in vessels with a media thickness >0.5 mm and is therefore more prominent in the arteries of large animals (i.e., pigs, humans) [64, 76]. However, vasa vasorum expansion also occurs in mouse models of atherosclerosis [77, 78]. In response to vessel injury, the adventitia serves as the primary site for acute inflammation [79]. Decreasing angiogenesis blunts stenotic vascular remodeling in injured vessels [80–82]. However, in mouse models of diet-induced atherosclerosis without vessel injury, only 28% of advanced atherosclerotic plaques (>250 µm thickness) and only 3% of intermediate plaques (100–250 µm thick) show angiogenic vessels in the intima [77].
Integrins, particularly the provisional matrix-binding integrins, play well-characterized roles in mediating endothelial proliferation and migration during angiogenic vascular remodeling in a variety of systems [14, 83]. However, the role of specific integrins and the processes regulating angiogenesis in the atherosclerotic plaque remain poorly characterized. Endostatin, a cleavage product of collagen XVIII, limits angiogenesis through inhibitory interactions with the endothelial integrins α5β1 and αvβ3 [84]. Endostatin treatment limited both plaque angiogenesis and neointimal area in hypercholesterolemic mice [77], and the endostatin-derivative Endostar reduces vasa vasorum density in hypercholesterolemic mini pigs associated with reduced intimal-medial thickness [85]. However, it is not clear from these studies that the reduction in angiogenesis caused the reduction in plaque area, as endostatin may affect other integrin-dependent functions. Both α5 and αvβ3 show enhanced expression in angiogenic vessels [86, 87], and this upregulation has been targeted by several PET tracers linked to RGD peptides used to visualize angiogenesis in vivo [88]. While αvβ3 inhibitors significantly blunt angiogenesis in multiple models [14, 83], αv- and β3-deficient mice do not show significant reductions in vasculogenesis or angiogenesis [89–91]. Similarly, endothelial α5 deletion or deletion of both α5 and αv does not impair vasculogenesis or angiogenesis in mice [92, 93]. Therefore, inhibiting endothelial α5 and αv integrin signaling may limit endothelial activation without significantly affecting physiological angiogenesis in the ischemic tissue.
Monocyte/macrophage integrins in plaque formation
Leukocytes express a distinct subset of integrins that mediate interactions with cell adhesion molecules on the endothelial cell surface. These include the β2 integrin subunit (CD18) and its four α subunits binding partners (αL (CD11a, LFA-1), αM (CD11b, MAC-1), αX (CD11c), and αD (CD11d)) and the β7 integrin subunit and its binding partner αE (CD103) (Fig. 2) [12]. In addition to these five integrin dimers, the α4 subunit (VLA-4) interacts with β7 only in leukocytes, whereas the α4β1 integrin dimer is found on both leukocyte and nonleukocyte cell populations [12]. While the β7 integrin heterodimers are primarily associated with lymphocyte targeting to intestinal tissue [94], multiple studies suggest a role for α4β1 and the various β2 integrins in atherosclerotic plaque formation. These integrins play vital roles in leukocyte targeting to the atherosclerotic plaque, and may also regulate leukocyte function following transendothelial migration into the plaque microenvironment. However, leukocytes also express several integrins involved in classic cell–matrix interactions that may play more dominant roles in cell motility and function in the tissue microenvironment. While several cell types, including dendritic cells, T cells, and B cells, uniquely and significantly contribute to plaque progression [95], monocyte/macrophages play a dominant role in disease progression and much of the data regarding integrins in atherosclerosis involves monocyte/macrophage biology. Therefore, integrin regulation of monocyte/macrophage function will serve as the focus of this section.
Integrins in monocyte homing to the plaque
The inflammatory cascade contributes to immune surveillance of the vasculature and mediates leukocyte targeting to the atherosclerotic plaque [15]. Initial attachment of circulating leukocytes to the activated endothelium is mediated by selectins expressed on the endothelial cell surface. The weak bonds formed between endothelial selectins and the leukocyte counter-receptor P-selectin glycoprotein ligand-1 (PSGL-1) easily break in response to shear forces resulting in leukocyte rolling, which sufficiently slows leukocyte velocity to facilitate more stable interactions with the endothelium [15]. Both P-selectin and E-selectin show enhanced expression during early atherogenesis [96, 97], and deletion of either P-selectin or E-selectin reduces plaque formation in mouse models, although the effect of P-selectin deletion is considerably more prominent [98, 99]. Partial activation of leukocyte integrins resulting in an intermediate affinity state, such as in response to E-selectin ligation, allows these integrins to facilitate rolling and primes the integrins for full activation in response to arrest chemokines [100, 101]. The transition from rolling to firm cell adhesion requires integrin transition to a high affinity state, typically in response to arrest chemokines on the endothelial cell surface [102]. Multiple arrest chemokines play important roles in atherosclerosis, including monocyte chemotactic protein-1 (MCP-1, CCL2), fractalkine (CX3CL1), and regulated on activation, normal T cell expression and secretion (RANTES, CCL5) [103–108]. In addition to selectins and chemokines, cell–cell interactions between Eph receptors and their ephrin ligands, guidance molecules implicated in tissue patterning during development, also regulate leukocyte integrin activation [109]. Interactions between EphA2/ephrinA1, EphA4/ephrinA1, and EphA4/ephrinB2 have all been shown to stimulate monocyte adhesion [110–113], and expression levels of EphA2, ephrinA1, and ephrinB2 are enhanced in endothelial cells overlying atherosclerotic plaques [110, 114, 115]. Once arrested, integrins also play a major role in facilitating leukocyte migration along the endothelial surface and subsequent transendothelial migration into the neointimal matrix [15].
While difficulty visualizing leukocyte homing to atherosclerotic plaques in vivo confounds the study of specific integrins in this response, several groups have utilized genetic knockout models and blocking antibodies to assess the role of specific integrins in leukocyte accumulation within the plaque. Leukocyte integrins show redundancy in multiple integrin-ligand interactions, but specific leukocyte integrins have been shown to play prominent roles in plaque formation (Table 2). Mice deficient in either ICAM-1 or VCAM-1 show reduced early plaque formation [45, 116, 117]; however, only VCAM-1 deficiency limits atherosclerosis in the context of prolonged hypercholesterolemia [45]. The integrins αLβ2, αMβ2, and αXβ2 interact with ICAM-1/2 on the endothelial cell surface, whereas α4β1 serves as the primary leukocyte VCAM-1 receptor [118]. However, αXβ2 has been shown to bind VCAM-1 cooperatively with α4β1 to promote leukocyte adhesion [119]. Consistent with a prominent role for VCAM-1 in plaque formation, α4β1 blocking antibodies significantly reduce myeloid cell recruitment and neointimal growth in atherosclerosis-prone ApoE knockout mice [120, 121]. However, α4 deletion results in embryonic lethality [122], and these results have yet to be repeated with hematopoietic-specific α4 deletion. In addition to VCAM-1, α4β1 can interact with extracellular matrix ligands, such as osteopontin and the connecting segment-1 (CS1) domain of fibronectin [123]. While several groups suggest that CS1-positive fibronectin is present on the luminal atherosclerotic endothelium, blocking VCAM-1 prevents nearly 80% of monocyte adhesion to atherosclerotic endothelium, whereas blocking CS-1 only reduces monocyte adhesion by ~20% [120]. Deletion of β2 integrins similarly reduces early plaque formation [116, 124], suggesting that some β2 integrins also contribute to monocyte targeting to the plaque. Hypercholesterolemia enhances αL expression in human patients, and blocking antibodies to αLβ2 significantly reduce early macrophage recruitment in hypercholesterolemic rats, consistent with a role for ICAM-1 in early plaque formation [117, 125, 126]. While hematopoietic deletion of αM does not affect macrophage accumulation or plaque formation [127], αX integrin-deficient mice showed significantly reduced monocyte/macrophage accumulation in atherosclerotic plaques and decreased plaque formation [128]. The integrin αXβ2, expressed on myeloid cells and NK cells, binds a wide variety of ligands, including ICAM-1, VCAM-1, complement proteins, and fibrinogen [119, 129–131]. Hypercholesterolemic mice show enhanced levels of αX (CD11c)-positive monocytes [128, 132], and human peripheral blood monocytes show elevated αX expression following a high fat meal [133]. Interestingly, hypercholesterolemia results in the appearance of foamy monocytes in the circulation, and these monocytes show the greatest αX upregulation [132]. Signaling through αXβ2 in these cells promotes α4β1 activation, suggesting functional cooperativity between β1 and β2 integrins in leukocytes [132].
Several signaling pathways have been implicated in inside-out activation of leukocyte integrins, but the roles these pathways play in the context of atherosclerosis is not well defined [28]. The classic mechanism of integrin activation involves the activation of talin1, inducing the interaction of the FERM domain in the talin1 head region to interact with an NPxY motif in the integrin β subunit tail [28]. Rap1 and its effector protein RIAM interact with talin1 to stimulate its transition to an active confirmation [134]. Chemokine-induced G protein-coupled receptor (GPCR) activation typically stimulate Rap1 through a phospholipase Cγ pathway activating the Rap1 guanine nucleotide exchange factor CalDAG-GEFI [134]. Deletion of CalDAG-GEFI from bone marrow-derived cells significantly blunts atherosclerotic plaque formation in LDLR KO mice [135], underscoring the importance of this pathway to leukocyte integrin activation. In addition to talin1, binding between the kindlin family of proteins, particularly the hematopoietic isoform kindlin-3, and the β tail has also been shown to regulate integrin affinity [28]. Mutations in kindlin-3 result in severe defects in integrin activation resulting in leukocyte adhesion deficiency [136, 137]. These pathways may have integrin-specific effects as well, with talin1 and kindlin-3 both required for stable adhesion to ICAM-1 but only kindlin-3 required for stable adhesion to VCAM-1 [138].
Integrins in macrophage migration
Leukocyte migration within the plaque likely affects multiple aspects of plaque biology, including fibrous cap stability, adaptive immunity, and plaque regression. To facilitate cell migration, macrophages express several extracellular matrix-binding integrins, including the collagen receptor α1β1, the laminin receptors α3β1 and α6β1, and the provisional matrix-binding integrins α4β1, α5β1, α9β1, αvβ3, and αvβ5 [139–143]. Ligation of α1β1 on collagen limits macrophage egression from inflamed tissue [139], and α1-deficient mice show reduced plaque macrophage and T cell content [144]. Osteopontin, known to be expressed in atherosclerotic plaques, interacts with the monocyte integrins α4β1, α9β1, and αvβ3, and promotes monocyte migration and survival [145]. However, the role of the provisional matrix-binding integrins in macrophage migration has not been examined in the context of atherosclerosis. In addition to classic matrix-binding integrins, several integrins implicated in leukocyte homing also show affinity for extracellular matrix proteins. Of particular interest, the promiscuous integrin αDβ2 binds to the provisional matrix proteins fibronectin and vitronectin and shows prominent upregulation during macrophage foam cell formation [146]. High levels of αDβ2 expression hinders macrophage migration, suggesting that upregulation of αDβ2 may serve as a migratory stop signal to limit macrophage egression from atherosclerotic plaques [147].
Integrin signaling in macrophage function
Ligation of specific macrophage integrins may affect various aspects of macrophage function in atherosclerosis, including the ability of macrophages to clear local lipid deposits, to phagocytose apoptotic cell debris, and to facilitate local proinflammatory gene expression (Table 3). Fibronectin in the atherosclerotic plaque can bind αMβ2 and αvβ3 in plaque macrophages [140, 148], and both αMβ2 and αvβ3 signaling reduces macrophage scavenger receptor expression (CD36, SRA) and foam cell formation [140, 149]. Similarly, αMβ2 signaling can reduce macrophage lipid uptake, although αMβ2 expression is downregulated in plaque macrophages [150]. Furthermore, peritoneal macrophages from β2 knockout mice show enhanced native and acetylated LDL uptake as well as apoptotic thymocytes [124], suggesting integrin dimers may attenuate macrophage lipid accumulation and cellular debris. Atherosclerotic plaques show enhanced expression of the integrin α4β7, classically involved in homing to gut-associated lymphoid tissue, and macrophages from β7 knockout mice show reduced native and acetylated LDL uptake [151]. Macrophage αvβ3 signaling has been shown to drive sustained NF-κB signaling and proinflammatory cytokine (TNF-α, IL-1β, IL-8) production and to augment the macrophage proinflammatory response to TNFα and LPS [152]. While deletion of β3 integrins in myeloid cells paradoxically enhances TNFα production and atherosclerotic plaque formation in vivo [153], myeloid-specific deletion of the β3-binding partner αv results in severe ulcerative colitis that may exacerbate atherosclerosis in the β3 knockout mice [154]. In addition to lipid uptake and proinflammatory cytokine production, plaque macrophages also critically clear apoptotic cell debris in the atherosclerotic plaque through the process of efferocytosis [155]. Several receptors are implicated in the engulfment of apoptotic cells, including the vitronectin-binding integrins αvβ3 and αvβ5 [156]. These integrins recognize the matricellular extracellular matrix protein thrombospondin and the phosphatidylserine-binding protein MFG-E8 on the surface of dying cells to facilitate phagocytosis [156, 157]. Macrophage efferocytosis becomes deficient in the atherosclerotic plaque, and treatment with damage-associated molecular pathogen HMGB1, known to accumulate in atherosclerosis, prevents efferocytosis through αvβ3 inhibition [155, 158, 159]. However, the role of these integrins in efferocytosis in the context of atherosclerosis has not been explored.
Integrins in smooth muscle migration and proliferation
Vascular smooth muscle cells, known for their contractile function in blood flow regulation and vessel tonicity [160], play a critical role in atherosclerotic progression through their remarkable ability to transdifferentiate to a proliferative and migratory phenotype. Under quiescent conditions in the media, smooth muscle cells express contractile markers (smooth muscle actin (SMA), calponin, SM22-α) with minimal proliferation and migration. However, upon stimulation with various factors such as cytokines, growth factors, oxidized lipids, and extracellular matrix proteins [1–3, 161], vascular smooth muscle cells downregulate their contractile markers and become increasingly fibroproliferative and migratory. Moreover, vascular smooth muscle exhibits the capacity to undergo an inflammatory, macrophage-like phenotypic switch, characterized by enhanced inflammatory gene expression (VCAM-1, ICAM-1) and phagocytic properties [162, 163]. The multifarious contribution by smooth muscle during atherogenesis is evident, as early and intermediate plaques accumulate endoplasmic reticulum and lipid droplet-rich vascular smooth muscle cells, while smooth muscle-derived fibrous tissue and fibrous cap formation is observed in advanced lesions [6]. Furthermore, recent lineage-tracing studies show up to 80% of all cells in atherosclerotic lesions are derived from smooth muscle cell origin [164], suggesting a previously underestimated vascular smooth muscle contribution during atherogenesis.
Integrin signaling in smooth muscle phenotypic modulation
With the known roles of integrins in matrix remodeling, proliferation, and cell migration, the role of integrin signaling in smooth muscle biology in atherosclerosis has garnered considerable attention. Quiescent medial vascular smooth muscle is surrounded by a thin basement membrane rich in collagen IV and laminin [165], and basement membrane proteins tend to promote a quiescent smooth muscle phenotype (Table 4). Smooth muscle cells express the collagen-binding integrins α1β1 and α2β1 [166]; however, α1β1 binds more strongly to collagen IV and α2β1 shows a higher affinity for collagen I [167]. Consistent with a role for basement membrane proteins in limiting endothelial activation, α1β1 shows reduced expression during smooth muscle phenotypic modulation [168], and α1 knockout mice show enhanced smooth muscle incorporation into atherosclerotic plaques [144]. Like α1β1, the laminin-binding integrins α6β1 and α7β1 show enhanced expression in quiescent smooth muscle cells [169, 170], and ligation of α7β1, such as through interactions with cartilage oligomeric matrix protein, maintains vascular smooth muscle quiescence [171] and reduces smooth muscle proliferation [172].
During neointimal recruitment, smooth muscle cells are exposed to a variety of provisional matrix proteins such as fibronectin, vitronectin, osteopontin, and tenascin-C [10]. Similarly, several provisional matrix-binding integrins, such as α5β1 and αvβ3, show enhanced expression in neointimal smooth muscle [173]. While other integrins likely play important functions, current literature strongly supports a role for signaling through αvβ3 in smooth muscle proliferation and migration. Deletion of plasma fibronectin significantly reduces smooth muscle content in atherosclerotic lesions [37], suggesting fibronectin-binding integrins may contribute to smooth muscle migration and proliferation during neointimal recruitment. Fibronectin binds both α5β1 and αvβ3, and while both α5β1 inhibitors (ATN-161) and αvβ3 inhibitors (S247) reduce atherosclerotic plaque formation, only αvβ3 inhibition reduced the incidence of fibrous cap formation [34, 53]. While αvβ3 expression can be observed in the medial layer [174, 175], smooth muscle cells upregulate αvβ3 in response to arterial injury in vivo and thrombin, transforming growth factor β (TGFβ), and platelet-derived growth factor-BB (PDGF-BB) in vitro [175–177]. Smooth muscle proliferation in response to osteopontin and tenascin-C in culture can be blunted by inhibiting αvβ3 but not β1 integrins [178–180], and inhibitors of αvβ3 prevent restenosis following vascular injury [181, 182]. While β3 knockout mice show enhanced atherosclerosis [183], this response is thought to occur due to the enhanced production of TNFα in myeloid cells in these mice, and β3 knockout mice show reduced neointimal formation following femoral artery injury [184], consistent with αvβ3-dependent smooth muscle recruitment and proliferation.
Integrin signaling promotes smooth muscle cell proliferation and migration through multiple mechanisms. Ligation of αvβ3 and αvβ5 integrins mediates FAK activity [185] and drives vascular smooth muscle migration through Akt and paxillin phosphorylation [186–188]. Conversely, the endogenous FAK inhibitor FAK-related nonkinase (FRNK) is highly expressed in vascular smooth muscle cells and reduces smooth muscle migration [189]. In addition to direct effects by integrin-linked signaling pathways, integrins crosstalk with growth factor signaling affects smooth muscle proliferation. Tenascin-C promotes αvβ3 interactions with PDGF receptor to further enhance proliferation [179, 180]. Following vascular injury in models of hyperglycemia, enhanced αvβ3 signaling upregulates Insulin Growth Factor-1 (IGF-1) [190]. In addition, IGF-1 reciprocally promotes vascular smooth muscle proliferation on αvβ3-binding matrix proteins, but not on laminin or collagen IV, although specific integrins involved were not identified [191].
Angiotensin II (AngII), a well-characterized vasoconstrictor in the renin–angiotensin system, contributes to smooth muscle proliferation through increased integrin activation and enhanced application of mechanical forces to integrin adhesions [192, 193]. AngII induces smooth muscle adhesion to collagen and fibronectin through the AT1 receptor [192–194], resulting in FAK-dependent ERK activation and enhanced cell proliferation [195, 196]. Cytoskeletal stiffening and contraction of smooth muscle cells treated with AngII results in elevated blood pressure [192, 193, 197], thereby enhancing mechanical load on the smooth muscle integrins. However, integrin signaling may also contribute to vasoconstriction, as treatment with the integrin inhibitor RGDS reduced vasoconstriction to AngII but not other vasoconstrictors [198]. While α1β1 has largely been implicated in maintaining the contractile phenotype in atherosclerosis, smooth muscle hypertrophy to chronic AngII stimulation is reduced in α1 knockout mice [199], and inhibiting α1β1 using siRNA knockdown or peptide inhibitors blunts AngII-induced ERK activation and proliferation [195, 200]. Consistent with this association, AngII promotes α1 integrin expression [199] along with the expression of multiple provisional matrix-binding integrins, such as α8, β1, and β3 [201, 202].
Not all integrin–matrix interactions conform to the concept that collagen/laminin-binding integrins suppress smooth muscle phenotypic modulation and provisional matrix-binding integrins promote phenotypic modulation. The provisional matrix-binding integrin α8β1 is associated with smooth muscle quiescence, as smooth muscle phenotypic modulation reduces α8 expression, while overexpression of α8 promotes the contractile phenotype [208, 209]. In contrast to these findings, Zargham et al. demonstrated enhanced α8 expression in the neointima and showed that α8 overexpression promotes smooth muscle migration, whereas deletion reduces migration [210, 211]. However, α8 knockout mice show enhanced injury-induced restenosis and diet-induced atherosclerotic plaque formation [212], consistent with a role for α8β1 signaling in maintaining smooth muscle quiescence. In contrast, upregulation of collagen VIII in the atherosclerotic plaque promotes smooth muscle proliferation and migration into the neointima presumably through α2β1, since α2β1 blockade reduces smooth muscle adhesion to collagen VIII by over 70% [213, 214]. Signaling through α2β1 in vascular smooth muscle cells promotes smooth muscle growth through upregulating PDGF-BB expression and transphosphorylation of the PDGF receptor [215, 216]. Collagen VIII knockout mice show significantly reduced fibrous cap thickness in atherosclerotic plaques [217], consistent with a role for collagen VIII and α2β1 in fibrous cap formation.
Integrin signaling in extracellular matrix remodeling
Vascular smooth muscle cells drive the development of the fibrous cap, largely composed of smooth muscle and collagen, to stabilize the plaque and prevent plaque rupture [6]. Therefore, alterations in the balance between matrix deposition and proteolysis and perturbations in the microenvironment that regulate smooth muscle apoptosis critically determine the stability of the fibrous cap. Smooth muscle collagen deposition requires both α2β1 integrins and the deposition of fibronectin [202]. Fibronectin fibrillogenesis, classically mediated by α5β1, can be blunted by inhibiting α5β1 in smooth muscle cells in culture, and α5β1 shows enhanced expression in neointimal smooth muscle cells [218, 219]. Loading smooth muscle cells with lipid, as observed in atherosclerotic plaques, significantly diminishes fibronectin and collagen deposition, associated with perturbations in α5β1-dependent fibrillar adhesion formation [220]. This suggests that lipid accumulation within the plaque or defective macrophage lipid clearance may reduce fibrous cap stability through effects on the smooth muscle integrins. Interestingly, αvβ3 integrin signaling prevents smooth muscle apoptosis in response to oxidized LDL [221], and αvβ3 integrins are largely absent from sites of plaque rupture [222]. While αvβ3 can also contribute to fibronectin deposition, the contribution of α5β1 and αvβ3 to neointimal matrix deposition has not been shown in vivo, likely due to difficulties in separating the roles of these integrins in smooth muscle recruitment and subsequent matrix deposition.
Platelet integrins in thrombotic complications
Platelets express a number of integrins, namely αIIbβ3 (the fibrinogen receptor), αvβ3, as well as three β1 integrins at lower expression levels (α2β1, α5β1, α6β1) (Table 5). A number of proteins present in the plasma or released from activated platelets bind platelet integrins and act as bridging proteins to receptors on other cells. These bridging proteins include fibrin and fibrinogen, fibronectin, von Willebrand Factor (vWF), vitronectin, and other ligands that contain the RGD integrin-binding domain. Platelet αIIbβ3 binds many of these, and its role, in particular, in the thrombotic complications of atherosclerosis is well documented. In more recent years, the importance of the crosstalk between platelets, leukocytes, and vascular endothelium during inflammatory processes of atherogenesis has become more appreciated. Furthermore, there is evidence for a contribution of platelet integrins in these responses.
Platelet integrin activation
During early atherogenesis, endothelial activation results in platelet activation and recruitment of platelets to the vessel wall. This early recruitment establishes a positive feedback loop in which the platelets release substances that cause further endothelial activation. Inhibiting early platelet adhesion decreased atherosclerotic plaque formation and is therefore thought to be a key step in the initiation of atherogenesis [223, 224]. While a number of receptor–ligand pairs are important, platelet integrins play a key role in firm adhesion. After rolling along the endothelium, platelets become activated leading to inside-out signaling which causes a conformational change in αIIbβ3. This activated form of αIIbβ3 facilitates the firm adhesion of platelets to the endothelium through a fibrinogen cross-bridge with endothelial αvβ3 [225]. Activated platelets facilitate recruitment of leukocytes to regions of injury or inflammation. Platelets bind circulating leukocytes to form platelet–leukocyte aggregates in the circulation, which are subsequently recruited to the vessel wall. Initial interactions between platelet P-selectin and leukocyte PSGL-1 lead to signaling through a Src family tyrosine kinase pathway, which enhances leukocyte β2 integrin expression [226]. Furthermore, ligation of platelet P-selectin activates αIIbβ3 through inside-out signaling, which stabilizes platelet–leukocyte aggregate formation. These aggregates occur either in circulation or via platelet binding to the endothelial surface, and facilitate leukocyte recruitment through leukocyte β2 integrin and platelet αIIbβ3 co-interactions with fibrinogen [227–229]. In contrast, adherent neutrophils within venules have been shown to interact with P-selectin- and αIIbβ3-expressing platelets, although a specific role for αIIbβ3 was not identified [230].
Integrin signaling in platelet function
As a result of outside-in signaling from the integrins, activated platelets release preformed substances from their granules and can make new proteins from pre-packaged mRNA. These can promote further platelet aggregation. In addition, this integrin-dependent degranulation places the platelets in a position to direct the inflammatory process of atherosclerosis by releasing soluble factors such as cytokines (e.g., IL-1b, CD40L) and chemokines (e.g., CXCL4, CCL5). These in turn activate the vascular endothelium leading to enhanced expression of adhesion molecules such as ICAM-1 and VCAM-1 and release of IL-6 and other inflammatory mediators that can promote the recruitment of monocytes to the plaque by processes described above. For example, deposition of CCL5 onto the atherosclerotic endothelium from platelet granules promotes monocyte arrest [231]. What exactly happens to the platelets that are attached to leukocytes as they undergo diapedesis has not been fully elucidated. However, there is evidence that platelets relocate to the rear of a monocyte and transfer to the endothelium as the monocyte begins to transmigrate [232].
The signaling initiated via platelet αIIbβ3 ligation can affect other parts of the plaque formation. For example, the destabilization of the plaque that occurs before rupture is also orchestrated in part by platelets. Activated platelets release MMP-2 and MMP-9 during aggregation [233, 234], and platelet αIIbβ3 signaling stimulates endothelial production and secretion of MMP-9 and urokinase-type plasminogen activator receptor (uPAR) [235], potentially through the generation of CD40L and its subsequent interaction with endothelial CD40 receptors. Ligation of αIIbβ3 promotes CD40L secretion from activated platelets [236], and CD40L has been shown to have an integral role in plaque development, inflammation, and atherothrombosis [237]. More recently, Simic et al. provided evidence in human platelets that α5β1 is also a receptor for soluble CD40L, and that engagement of α5β1 by sCD40L leads to platelet activation [238], suggesting that we have yet to understand the complete roles of platelet integrins in the atherogenic process.
Platelet integrins in thrombotic complications
Platelets are key players in thrombotic complications of atherosclerosis, as they immediately respond to exposure of the subendothelial matrix due to disruption of the endothelium in atherothrombosis. Initial contact, or tethering, is mediated by platelet GPIbα interactions with vWF, whereas more stable binding occurs between the platelet integrin α2β1 and collagen [239]. Fibronectin and laminin engage other integrins, namely α5β1, α6β1, respectively [240], and there appears to be considerable redundancy between these receptors, in that deficiency of any one of these does not cause a severe bleeding disorder. These interactions lead to platelet activation and secretion of many factors that can behave in a paracrine or autocrine fashion to promote platelet aggregation. Also central to the thrombotic process is the activation of the coagulation system. Platelets provide a surface upon which coagulation cascade complexes are assembled, leading to thrombin generation. Thrombin then stimulates procoagulant activity on the platelet surface, which promotes the generation of more thrombin [241], and perpetuates the process. In addition, thrombin activates αIIbβ3 [242] in the developing thrombus, which typically binds fibrinogen to form a cross-bridge between platelets and augmented thrombus growth. Initially, this interaction is relatively weak, but as the platelet becomes more activated, the bond strengthens and the thrombus is stabilized. A stable thrombus leads to blockage of the artery, while a fragmented thrombus can form an embolism distal to the site of injury. Alternatively, chronic thrombotic process that occurs in a patient may perpetuate plaque progression through many of the mechanisms discussed above.
In a recent review, Mastenbroek et al. [243] highlighted the fact that much of our knowledge about the thrombotic process comes from studies in the absence of atherosclerotic plaques. This has been difficult to overcome in that many commonly used animal models of atherosclerosis do not spontaneously undergo plaque rupture. However, although somewhat artificial, several animal models of induced plaque rupture have been developed, and αIIbβ3 has been implicated in the atherothrombus formation in two mouse models [244]. This should not be surprising, because this integrin is also integral in platelet activation due to high shear or oscillatory shear, as is found in stenotic arteries [245, 246]. Using in vitro flow chamber models of thrombus formation where blood was flowed over plaque content, a role for the collagen receptor GPVI was shown, but blockade of the integrin collagen receptor, α2β1, failed to alter thrombus formation. Nonetheless, to date, much of the data suggest that mechanisms of thrombus formation over healthy vs. atherosclerotic vessels are similar. It remains to be seen if the individual roles of integrins translate to human atherothrombosis.
Future directions
This review has outlined important roles for integrin signaling in endothelial dysfunction and activation, leukocyte homing to the plaque, leukocyte function within the plaque, smooth muscle recruitment and fibroproliferative remodeling, and thrombotic complications. However, there is still much to learn concerning how integrin signaling affects this wide range of pathological processes. While intravital microscopy has allowed considerable insight into the role of specific integrins in platelet aggregation and in leukocyte homing to the post-capillary venule, the pulsatile nature of the large conduit vessels has made visualizing these processes in atherosclerosis-relevant contexts difficult. Improvements in the visualization of leukocyte targeting to atherosclerotic lesions will likely shed further insight into the role of specific integrins in this process. Leukocyte targeting to the endothelium in these high flow vessels may require different mechanisms to facilitate firm adhesion than that previously described in the post-capillary venules, and additional studies will be required to understand how these processes stabilize the leukocyte–endothelial interface. While integrin activation classically regulates leukocyte and platelet function, the role of integrin activation in adherent cell types is less well characterized, and its role in integrin signaling in vivo remains unknown. Furthermore, the signaling mechanisms utilized by specific integrins to modulate cell function based on changing matrix composition remain virtually unknown. Application of advanced proteomics techniques will be critical to characterize the differential signaling responses activated by specific cell–matrix interactions and within relevant cell types that may express a different repertoire of integrin-binding proteins.
Despite abundant and diverse integrin expression among all atherosclerotic plaque-associated cell types, current literature suggests that a few integrins may prove therapeutically beneficial for the treatment of atherosclerosis. The leukocyte integrin αXβ2 may prove to be particularly important in atherogenic inflammation, as this integrin shows upregulation under hypercholesterolemic conditions [128, 132, 133] and reduced atherosclerosis upon its deletion [128]. Deposition of provisional matrix proteins in the atherosclerotic plaque creates a permissive environment for cell proliferation, migration, differentiation, and inflammatory response largely through signaling with α5β1 and αvβ3. As such, therapeutic targets against these heterodimers have the potential to greatly impact the progression of atherosclerosis. Indeed, work from our lab and others has shown that pharmacological inhibition of both αvβ3 and α5β1 significantly reduces atherosclerosis [34, 53, 77, 182]. However, inhibition of αvβ3 integrins significantly reduces smooth muscle incorporation into the plaque [34], similar to what has been observed upon fibronectin deletion [37]. In contrast, inhibition of α5β1 blunts plaque inflammation without reducing smooth muscle incorporation or fibrous cap thickness [53], suggesting that targeting α5β1 may be able to separate the proinflammatory properties of the provisional matrix from their role in promoting fibrous cap formation. Future studies with more selective integrin inhibitors or better methods of targeting integrin inhibitors to specific cell types will ultimately be necessary to establish their worth as therapeutic targets for controlling atherogenic vascular remodeling.
References
Kingsley K et al (2002) ERK1/2 mediates PDGF-BB stimulated vascular smooth muscle cell proliferation and migration on laminin-5. Biochem Biophys Res Commun 293(3):1000–1006
Chahine MN et al (2009) Oxidized LDL affects smooth muscle cell growth through MAPK-mediated actions on nuclear protein import. J Mol Cell Cardiol 46(3):431–441
Orr AW et al (2009) Molecular mechanisms of collagen isotype-specific modulation of smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol 29(2):225–231
Autieri MV (2012) Pro- and anti-inflammatory cytokine networks in atherosclerosis. ISRN Vasc Med 2012:17
Owens GK, Kumar MS, Wamhoff BR (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84(3):767–801
Stary HC et al (1995) A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 92(5):1355–1374
Virmani R et al (2006) Pathology of the vulnerable plaque. J Am Coll Cardiol 47(8 Suppl):C13–C18
Libby P (2005) The forgotten majority: unfinished business in cardiovascular risk reduction. J Am Coll Cardiol 46(7):1225–1228
Hahn C, Schwartz MA (2009) Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol 10(1):53–62
Yurdagul A Jr. et al (2016) The arterial microenvironment: the where and why of atherosclerosis. Biochem J 473(10):1281–1295
Yurdagul A Jr., Orr AW (2016) Blood brothers: hemodynamics and cell-matrix interactions in endothelial function. Antioxid Redox Signal 25(7):415–434
Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110(6):673–687
Ross TD et al (2013) Integrins in mechanotransduction. Curr Opin Cell Biol 25(5):613–618
Stupack DG, Cheresh DA (2002) ECM remodeling regulates angiogenesis: endothelial integrins look for new ligands. Sci STKE 2002(119):PE7
Ley K et al (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7(9):678–689
Shattil SJ, Ginsberg MH, Brugge JS (1994) Adhesive signaling in platelets. Curr Opin Cell Biol 6(5):695–704
Schwartz MA (2001) Integrin signaling revisited. Trends Cell Biol 11(12):466–470
Humphries JD et al (2015) Emerging properties of adhesion complexes: what are they and what do they do? Trends Cell Biol 25(7):388–397
Parsons JT, Horwitz AR, Schwartz MA (2010) Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 11(9):633–643
Orr AW et al (2006) Mechanisms of mechanotransduction. Dev Cell 10(1):11–20
Kanchanawong P et al (2010) Nanoscale architecture of integrin-based cell adhesions. Nature 468(7323):580–584
Byron A et al (2012) Proteomic analysis of alpha4beta1 integrin adhesion complexes reveals alpha-subunit-dependent protein recruitment. Proteomics 12(13):2107–2114
Schiller HB et al (2013) beta1- and alphav-class integrins cooperate to regulate myosin II during rigidity sensing of fibronectin-based microenvironments. Nat Cell Biol 15(6):625–636
Kuo JC et al (2011) Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for beta-Pix in negative regulation of focal adhesion maturation. Nat Cell Biol 13(4):383–393
Robertson J et al (2015) Defining the phospho-adhesome through the phosphoproteomic analysis of integrin signalling. Nat Commun 6:6265
Wickstrom SA et al (2010) The ILK/PINCH/parvin complex: the kinase is dead, long live the pseudokinase!. EMBO J 29(2):281–291
Kim C, Ye F, Ginsberg MH (2011) Regulation of integrin activation. Annu Rev Cell Dev Biol 27:321–345
Ye F, Snider AK, Ginsberg MH (2014) Talin and kindlin: the one-two punch in integrin activation. Front Med 8(1):6–16
Jalali S et al (2001) Integrin-mediated mechanotransduction requires its dynamic interaction with specific extracellular matrix (ECM) ligands. Proc Natl Acad Sci USA 98(3):1042–1046
Tzima E et al (2001) Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J 20(17):4639–4647
Orr AW et al (2005) The subendothelial extracellular matrix modulates NF-kappaB activation by flow: a potential role in atherosclerosis. J Cell Biol 169(1):191–202
Hynes RO (2012) The evolution of metazoan extracellular matrix. J Cell Biol 196(6):671–679
Short SM, Talbott GA, Juliano RL (1998) Integrin-mediated signaling events in human endothelial cells. Mol Biol Cell 9(8):1969–1980
Chen J et al (2015) αβ3 integrins mediate flow-induced NF-κB activation, proinflammatory gene expression, and early atherogenic inflammation. Am J Pathol 185(9):2575–2589
Feaver RE et al (2010) Atheroprone hemodynamics regulate fibronectin deposition to create positive feedback that sustains endothelial inflammation. Circ Res 106(11):1703–1711
Green J et al (2014) Flow patterns regulate hyperglycemia-induced subendothelial matrix remodeling during early atherogenesis. Atherosclerosis 232(2):277–284
Rohwedder I et al (2012) Plasma fibronectin deficiency impedes atherosclerosis progression and fibrous cap formation. EMBO Mol Med 4(7):564–576
Murphy PA, Hynes RO (2014) Alternative splicing of endothelial fibronectin is induced by disturbed hemodynamics and protects against hemorrhage of the vessel wall. Arterioscler Thromb Vasc Biol 34(9):2042–2050
Gelfand BD et al (2011) Hemodynamic activation of beta-catenin and T-cell-specific transcription factor signaling in vascular endothelium regulates fibronectin expression. Arterioscler Thromb Vasc Biol 31(7):1625–1633
van Keulen JK et al (2007) Levels of extra domain A containing fibronectin in human atherosclerotic plaques are associated with a stable plaque phenotype. Atherosclerosis 195(1):e83–e91
Pober JS, Sessa WC (2007) Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 7(10):803–815
Nakashima Y et al (1998) Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler Thromb Vasc Biol 18(5):842–851
Collins T, Cybulsky MI (2001) NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest 107(3):255–264
Hajra L et al (2000) The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA 97(16):9052–9057
Cybulsky MI et al (2001) A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest 107(10):1255–1262
Tzima E et al (2005) A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437(7057):426–431
Hahn C et al (2009) The subendothelial extracellular matrix modulates JNK activation by flow. Circ Res 104(8):995–1003
Orr AW et al (2008) p21-activated kinase signaling regulates oxidant-dependent NF-kappa B activation by flow. Circ Res 103(6):671–679
Orr AW et al (2007) Matrix-specific p21-activated kinase activation regulates vascular permeability in atherogenesis. J Cell Biol 176(5):719–727
Bhullar IS et al (1998) Fluid shear stress activation of IkappaB kinase is integrin-dependent. J Biol Chem 273(46):30544–30549
Sun X et al (2016) Activation of integrin alpha5 mediated by flow requires its translocation to membrane lipid rafts in vascular endothelial cells. Proc Natl Acad Sci USA 113(3):769–774
Yun S et al (2016) Interaction between integrin alpha5 and PDE4D regulates endothelial inflammatory signalling. Nat Cell Biol 18(10):1043–1053
Yurdagul A Jr. et al (2014) α5β1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis. Arterioscler Thromb Vasc Biol 34(7):1362–1373
Yurdagul A Jr. et al (2016) Oxidized LDL induces FAK-dependent RSK signaling to drive NF-kappaB activation and VCAM-1 expression. J Cell Sci 129:1580–1591
Funk SD, Yurdagul A Jr., Orr AW (2012) Hyperglycemia and endothelial dysfunction in atherosclerosis: lessons from type 1 diabetes. Int J Vasc Med 2012:569654
Pober JS, Min W (2006) Endothelial cell dysfunction, injury and death. Handb Exp Pharmacol 176(Pt 2):135–156
Fleming I (2010) Molecular mechanisms underlying the activation of eNOS. Pflugers Arch 459(6):793–806
Dimmeler S et al (1999) Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399(6736):601–605
Boo YC et al (2002) Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms: role of protein kinase A. J Biol Chem 277(5):3388–3396
Yang B, Rizzo V (2013) Shear stress activates eNOS at the endothelial apical surface through 1 containing integrins and caveolae. Cell Mol Bioeng 6(3):346–354
Orr AW et al (2006) Matrix-specific suppression of integrin activation in shear stress signaling. Mol Biol Cell 17(11):4686–4697
Yurdagul A Jr. et al (2013) Altered nitric oxide production mediates matrix-specific PAK2 and NF-kappaB activation by flow. Mol Biol Cell 24(3):398–408
Funk SD et al (2010) Matrix-specific protein kinase A signaling regulates p21-activated kinase activation by flow in endothelial cells. Circ Res 106(8):1394–1403
Mulligan-Kehoe MJ, Simons M (2014) Vasa vasorum in normal and diseased arteries. Circulation 129(24):2557–2566
Annex BH (2013) Therapeutic angiogenesis for critical limb ischaemia. Nat Rev Cardiol 10(7):387–396
Kwon HM et al (1998) Enhanced coronary vasa vasorum neovascularization in experimental hypercholesterolemia. J Clin Invest 101(8):1551–1556
Langheinrich AC et al (2006) Correlation of vasa vasorum neovascularization and plaque progression in aortas of apolipoprotein E(-/-)/low-density lipoprotein(-/-) double knockout mice. Arterioscler Thromb Vasc Biol 26(2):347–352
Moulton KS et al (2003) Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci USA 100(8):4736–4741
O’Brien KD et al (1996) Neovascular expression of E-selectin, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1 in human atherosclerosis and their relation to intimal leukocyte content. Circulation 93(4):672–682
Moreno PR et al (2004) Plaque neovascularization is increased in ruptured atherosclerotic lesions of human aorta: implications for plaque vulnerability. Circulation 110(14):2032–2038
Virmani R et al (2005) Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol 25(10):2054–2061
Semenza GL (2014) Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol 76:39–56
Mollmark JI et al (2012) Fibroblast growth factor-2 is required for vasa vasorum plexus stability in hypercholesterolemic mice. Arterioscler Thromb Vasc Biol 32(11):2644–2651
Hutter R et al (2013) Macrophages transmit potent proangiogenic effects of oxLDL in vitro and in vivo involving HIF-1alpha activation: a novel aspect of angiogenesis in atherosclerosis. J Cardiovasc Transl Res 6(4):558–569
Jaipersad AS et al (2014) The role of monocytes in angiogenesis and atherosclerosis. J Am Coll Cardiol 63(1):1–11
Heistad DD, Marcus ML (1979) Role of vasa vasorum in nourishment of the aorta. Blood Vessels 16(5):225–238
Moulton KS et al (1999) Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 99(13):1726–1732
Drinane M et al (2009) The antiangiogenic activity of rPAI-1(23) inhibits vasa vasorum and growth of atherosclerotic plaque. Circ Res 104(3):337–345
Okamoto E et al (2001) Perivascular inflammation after balloon angioplasty of porcine coronary arteries. Circulation 104(18):2228–2235
Khurana R et al (2004) Angiogenesis-dependent and independent phases of intimal hyperplasia. Circulation 110(16):2436–2443
Koga J et al (2009) Soluble Flt-1 gene transfer ameliorates neointima formation after wire injury in flt-1 tyrosine kinase-deficient mice. Arterioscler Thromb Vasc Biol 29(4):458–464
Ohtani K et al (2004) Blockade of vascular endothelial growth factor suppresses experimental restenosis after intraluminal injury by inhibiting recruitment of monocyte lineage cells. Circulation 110(16):2444–2452
Robinson SD, Hodivala-Dilke KM (2011) The role of beta3-integrins in tumor angiogenesis: context is everything. Curr Opin Cell Biol 23(5):630–637
Rehn M et al (2001) Interaction of endostatin with integrins implicated in angiogenesis. Proc Natl Acad Sci USA 98(3):1024–1029
Xu X et al (2015) Angiogenesis inhibitor, endostar, prevents vasa vasorum neovascularization in a swine atherosclerosis model. J Atheroscler Thromb 22(10):1100–1112
Brooks PC, Clark RA, Cheresh DA (1994) Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 264(5158):569–571
Kim S et al (2000) Regulation of angiogenesis in vivo by ligation of integrin alpha5beta1 with the central cell-binding domain of fibronectin. Am J Pathol 156(4):1345–1362
Orbay H et al (2013) Positron emission tomography imaging of atherosclerosis. Theranostics 3(11):894–902
Hodivala-Dilke KM et al (1999) Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest 103(2):229–238
Bader BL et al (1998) Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins. Cell 95(4):507–519
Reynolds LE et al (2002) Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat Med 8(1):27–34
van der Flier A et al (2010) Endothelial alpha5 and alphav integrins cooperate in remodeling of the vasculature during development. Development 137(14):2439–2449
Murphy PA, Begum S, Hynes RO (2015) Tumor angiogenesis in the absence of fibronectin or its cognate integrin receptors. PLoS One 10(3):e0120872
Berlin, C., et al (1993) Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell 74(1):185–195
Libby P, Hansson GK (2015) Inflammation and immunity in diseases of the arterial tree: players and layers. Circ Res 116(2):307–311
Johnson-Tidey RR et al (1994) Increase in the adhesion molecule P-selectin in endothelium overlying atherosclerotic plaques. Coexpression with intercellular adhesion molecule-1. Am J Pathol 144(5):952–961
van der Wal AC et al (1992) Adhesion molecules on the endothelium and mononuclear cells in human atherosclerotic lesions. Am J Pathol 141(6):1427–1433
Collins RG et al (2000) P-Selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J Exp Med 191(1):189–194
Dong ZM, Brown AA, Wagner DD (2000) Prominent role of P-selectin in the development of advanced atherosclerosis in ApoE-deficient mice. Circulation 101(19):2290–2295
Alon R et al (1995) The integrin VLA-4 supports tethering and rolling in flow on VCAM-1. J Cell Biol 128(6):1243–1253
Zarbock A, Lowell CA, Ley K (2007) Spleen tyrosine kinase Syk is necessary for E-selectin-induced alpha(L)beta(2) integrin-mediated rolling on intercellular adhesion molecule-1. Immunity 26(6):773–783
Shamri R et al (2005) Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemokines. Nat Immunol 6(5):497–506
Boring L et al (1998) Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 394(6696):894–897
Gu L et al (1998) Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell 2(2):275–281
Braunersreuther V et al (2007) Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler Thromb Vasc Biol 27(2):373–379
Braunersreuther V et al (2008) A novel RANTES antagonist prevents progression of established atherosclerotic lesions in mice. Arterioscler Thromb Vasc Biol 28(6):1090–1096
Combadiere C et al (2003) Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation 107(7):1009–1016
Lesnik P, Haskell CA, Charo IF (2003) Decreased atherosclerosis in CX3CR1-/- mice reveals a role for fractalkine in atherogenesis. J Clin Invest 111(3):333–340
Funk SD, Orr AW (2013) Ephs and ephrins resurface in inflammation, immunity, and atherosclerosis. Pharmacol Res 67(1):42–52
Braun J et al (2011) Endothelial cell ephrinB2-dependent activation of monocytes in arteriosclerosis. Arterioscler Thromb Vasc Biol 31(2):297–305
Jellinghaus S et al (2013) Ephrin-A1/EphA4-mediated adhesion of monocytes to endothelial cells. Biochim Biophys Acta 1833(10):2201–2211
Saeki N et al (2015) EphA2 promotes cell adhesion and spreading of monocyte and monocyte/macrophage cell lines on integrin ligand-coated surfaces. Cell Adh Migr 9(6):469–482
Poitz DM et al (2015) EphrinB2/EphA4-mediated activation of endothelial cells increases monocyte adhesion. Mol Immunol 68(2 Pt C):648–656
Funk SD et al (2012) EphA2 activation promotes the endothelial cell inflammatory response: a potential role in atherosclerosis. Arterioscler Thromb Vasc Biol 32(3):686–695
van Gils JM et al (2013) Endothelial expression of guidance cues in vessel wall homeostasis dysregulation under proatherosclerotic conditions. Arterioscler Thromb Vasc Biol 33(5):911–919
Nageh MF et al (1997) Deficiency of inflammatory cell adhesion molecules protects against atherosclerosis in mice. Arterioscler Thromb Vasc Biol 17(8):1517–1520
Bourdillon MC et al (2000) ICAM-1 deficiency reduces atherosclerotic lesions in double-knockout mice (ApoE(-/-)/ICAM-1(-/-)) fed a fat or a chow diet. Arterioscler Thromb Vasc Biol 20(12):2630–2635
Arnaout MA (2016) Biology and structure of leukocyte beta 2 integrins and their role in inflammation. F1000Res. doi:10.12688/f1000research.9415.1
Sadhu C et al (2007) CD11c/CD18: novel ligands and a role in delayed-type hypersensitivity. J Leukoc Biol 81(6):1395–1403
Huo Y, Hafezi-Moghadam A, Ley K (2000) Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circ Res 87(2):153–159
Barringhaus KG et al (2004) Alpha4beta1 integrin (VLA-4) blockade attenuates both early and late leukocyte recruitment and neointimal growth following carotid injury in apolipoprotein E (−/−) mice. J Vasc Res 41(3):252–260
Yang JT, Rayburn H, Hynes RO (1995) Cell adhesion events mediated by alpha 4 integrins are essential in placental and cardiac development. Development 121(2):549–560
Shih PT et al (1999) Minimally modified low-density lipoprotein induces monocyte adhesion to endothelial connecting segment-1 by activating beta1 integrin. J Clin Invest 103(5):613–625
Merched A, Tollefson K, Chan L (2010) Beta2 integrins modulate the initiation and progression of atherosclerosis in low-density lipoprotein receptor knockout mice. Cardiovasc Res 85(4):853–863
Nie Q et al (1997) Inhibition of mononuclear cell recruitment in aortic intima by treatment with anti-ICAM-1 and anti-LFA-1 monoclonal antibodies in hypercholesterolemic rats: implications of the ICAM-1 and LFA-1 pathway in atherogenesis. Lab Invest 77(5):469–482
Kawamura A et al (2007) Apolipoprotein E interrupts interleukin-1beta signaling in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 27(7):1610–1617
Kubo N et al (2000) Leukocyte CD11b expression is not essential for the development of atherosclerosis in mice. J Lipid Res 41(7):1060–1066
Wu H et al (2009) Functional role of CD11c+ monocytes in atherogenesis associated with hypercholesterolemia. Circulation 119(20):2708–2717
Hogg N et al (1986) The p150, 95 molecule is a marker of human mononuclear phagocytes: comparison with expression of class II molecules. Eur J Immunol 16(3):240–248
Blackford J et al (1996) A monoclonal antibody, 3/22, to rabbit CD11c which induces homotypic T cell aggregation: evidence that ICAM-1 is a ligand for CD11c/CD18. Eur J Immunol 26(3):525–531
Loike JD et al (1991) CD11c/CD18 on neutrophils recognizes a domain at the N terminus of the A alpha chain of fibrinogen. Proc Natl Acad Sci USA 88(3):1044–1048
Foster GA et al (2015) CD11c/CD18 signals very late antigen-4 activation to initiate foamy monocyte recruitment during the onset of hypercholesterolemia. J Immunol 195(11):5380–5392
Gower RM et al (2011) CD11c/CD18 expression is upregulated on blood monocytes during hypertriglyceridemia and enhances adhesion to vascular cell adhesion molecule-1. Arterioscler Thromb Vasc Biol 31(1):160–166
Han J et al (2006) Reconstructing and deconstructing agonist-induced activation of integrin alphaIIbbeta3. Curr Biol 16(18):1796–1806
Boulaftali Y et al (2016) CalDAG-GEFI deficiency reduces atherosclerotic lesion development in mice. Arterioscler Thromb Vasc Biol 36:792–799
Moser M et al (2009) Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat Med 15(3):300–305
Malinin NL et al (2009) A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat Med 15(3):313–318
Hyduk SJ et al (2011) Talin-1 and kindlin-3 regulate alpha4beta1 integrin-mediated adhesion stabilization, but not G protein-coupled receptor-induced affinity upregulation. J Immunol 187(8):4360–4368
Becker HM et al (2013) Alpha1beta1 integrin-mediated adhesion inhibits macrophage exit from a peripheral inflammatory lesion. J Immunol 190(8):4305–4314
Antonov AS et al (2004) Regulation of macrophage foam cell formation by alphaVbeta3 integrin: potential role in human atherosclerosis. Am J Pathol 165(1):247–258
Prieto J, Eklund A, Patarroyo M (1994) Regulated expression of integrins and other adhesion molecules during differentiation of monocytes into macrophages. Cell Immunol 156(1):191–211
De Nichilo MO, Yamada KM (1996) Integrin alpha v beta 5-dependent serine phosphorylation of paxillin in cultured human macrophages adherent to vitronectin. J Biol Chem 271(18):11016–11022
Ammon C et al (2000) Comparative analysis of integrin expression on monocyte-derived macrophages and monocyte-derived dendritic cells. Immunology 100(3):364–369
Schapira K et al (2005) Genetic deletion or antibody blockade of alpha1beta1 integrin induces a stable plaque phenotype in ApoE−/− mice. Arterioscler Thromb Vasc Biol 25(9):1917–1924
Lund SA et al., Osteopontin mediates macrophage chemotaxis via alpha(4) and alpha(9) integrins and survival via the alpha(4) integrin. J Cell Biochem, 2012
Yakubenko VP, Yadav SP, Ugarova TP (2006) Integrin alphaDbeta2, an adhesion receptor up-regulated on macrophage foam cells, exhibits multiligand-binding properties. Blood 107(4):1643–1650
Yakubenko VP et al (2008) The role of integrin alpha D beta2 (CD11d/CD18) in monocyte/macrophage migration. Exp Cell Res 314(14):2569–2578
Lishko VK, Yakubenko VP, Ugarova TP (2003) The interplay between integrins alphaMbeta2 and alpha5beta1 during cell migration to fibronectin. Exp Cell Res 283(1):116–126
Yakubenko VP et al (2011) Alphambeta(2) integrin activation prevents alternative activation of human and murine macrophages and impedes foam cell formation. Circ Res 108(5):544–554
Gray JL, Shankar R (1995) Down regulation of CD11b and CD18 expression in atherosclerotic lesion-derived macrophages. Am Surg 61(8):674-679 (discussion 679–80)
Zhi K et al (2014) Alpha4beta7 Integrin (LPAM-1) is upregulated at atherosclerotic lesions and is involved in atherosclerosis progression. Cell Physiol Biochem 33(6):1876–1887
Antonov AS et al (2011) AlphaVbeta3 integrin regulates macrophage inflammatory responses via PI3 kinase/Akt-dependent NF-kappaB activation. J Cell Physiol 226(2):469–476
Schneider JG et al (2007) Macrophage beta3 integrin suppresses hyperlipidemia-induced inflammation by modulating TNFalpha expression. Arterioscler Thromb Vasc Biol 27(12):2699–2706
Lacy-Hulbert A et al (2007) Ulcerative colitis and autoimmunity induced by loss of myeloid alphav integrins. Proc Natl Acad Sci USA 104(40):15823–15828
Poon IK et al (2014) Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 14(3):166–180
Savill J et al (1990) Vitronectin receptor-mediated phagocytosis of cells undergoing apoptosis. Nature 343(6254):170–173
Hanayama R et al (2004) Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 304(5674):1147–1150
Friggeri A et al (2010) HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3-integrin. Am J Physiol Cell Physiol 299(6):C1267–C1276
Fan Z et al (2016) HMGB1: a promising therapeutic approach for atherosclerosis. Int J Cardiol 202:507–508
Wall VZ, Bornfeldt KE (2014) Arterial smooth muscle. Arterioscler Thromb Vasc Biol 34(10):2175–2179
Hedin U et al (1988) Diverse effects of fibronectin and laminin on phenotypic properties of cultured arterial smooth muscle cells. J Cell Biol 107(1):307–319
Orr AW et al (2010) Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J Vasc Res 47(2):168–180
Vengrenyuk Y et al (2015) Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol 35(3):535–546
Shankman LS et al (2015) KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 21(6):628–637
Voss B, Rauterberg J (1986) Localization of collagen types I, III, IV and V, fibronectin and laminin in human arteries by the indirect immunofluorescence method. Pathol Res Pract 181(5):568–575
Moiseeva EP (2001) Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res 52(3):372–386
Heino J (2000) The collagen receptor integrins have distinct ligand recognition and signaling functions. Matrix Biol 19(4):319–323
Obata H et al (1997) Smooth muscle cell phenotype-dependent transcriptional regulation of the alpha1 integrin gene. J Biol Chem 272(42):26643–26651
Cremona O et al (1994) The alpha6 and beta4 integrin subunits are expressed by smooth muscle cells of human small vessels: a new localization in mesenchymal cells. J Histochem Cytochem 42(9):1221–1228
Yao CC et al (1997) Functional expression of the alpha 7 integrin receptor in differentiated smooth muscle cells. J Cell Sci 110(Pt 13):1477–1487
Wang L et al (2010) Cartilage oligomeric matrix protein maintains the contractile phenotype of vascular smooth muscle cells by interacting with alpha(7)beta(1) integrin. Circ Res 106(3):514–525
Welser JV et al (2007) Loss of the alpha7 integrin promotes extracellular signal-regulated kinase activation and altered vascular remodeling. Circ Res 101(7):672–681
Blindt R et al (2002) Expression patterns of integrins on quiescent and invasive smooth muscle cells and impact on cell locomotion. J Mol Cell Cardiol 34(12):1633–1644
Hoshiga M et al (1995) Alpha-v beta-3 integrin expression in normal and atherosclerotic artery. Circ Res 77(6):1129–1135
van der Zee R et al (1998) Reduced intimal thickening following alpha(v)beta3 blockade is associated with smooth muscle cell apoptosis. Cell Adhes Commun 6(5):371–379
Janat MF, Argraves WS, Liau G (1992) Regulation of vascular smooth muscle cell integrin expression by transforming growth factor beta1 and by platelet-derived growth factor-BB. J Cell Physiol 151(3):588–595
Brown SL et al (1994) Stimulation of migration of human aortic smooth muscle cells by vitronectin: implications for atherosclerosis. Cardiovasc Res 28(12):1815–1820
Liu J et al (2014) Oxidized low-density lipoprotein increases the proliferation and migration of human coronary artery smooth muscle cells through the upregulation of osteopontin. Int J Mol Med 33(5):1341–1347
Yamamoto K et al (2005) Tenascin-C is an essential factor for neointimal hyperplasia after aortotomy in mice. Cardiovasc Res 65(3):737–742
Ishigaki T et al (2011) Tenascin-C enhances crosstalk signaling of integrin alphavbeta3/PDGFR-beta complex by SRC recruitment promoting PDGF-induced proliferation and migration in smooth muscle cells. J Cell Physiol 226(10):2617–2624
Choi ET et al (1994) Inhibition of neointimal hyperplasia by blocking alpha V beta 3 integrin with a small peptide antagonist GpenGRGDSPCA. J Vasc Surg 19(1):125–134
Bishop GG et al (2001) Selective alpha(v)beta(3)-receptor blockade reduces macrophage infiltration and restenosis after balloon angioplasty in the atherosclerotic rabbit. Circulation 103(14):1906–1911
Weng S et al (2003) Beta3 integrin deficiency promotes atherosclerosis and pulmonary inflammation in high-fat-fed, hyperlipidemic mice. Proc Natl Acad Sci USA 100(11):6730–6735
Panchatcharam M et al (2010) Enhanced proliferation and migration of vascular smooth muscle cells in response to vascular injury under hyperglycemic conditions is controlled by beta3 integrin signaling. Int J Biochem Cell Biol 42(6):965–974
Li G et al (2010) Periostin mediates vascular smooth muscle cell migration through the integrins alphavbeta3 and alphavbeta5 and focal adhesion kinase (FAK) pathway. Atherosclerosis 208(2):358–365
Schaller MD (2001) Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim Biophys Acta 1540(1):1–21
Moiseeva EP et al (2003) Galectin-1 interacts with beta-1 subunit of integrin. Biochem Biophys Res Commun 310(3):1010–1016
Lee BH et al (2006) Betaig-h3 triggers signaling pathways mediating adhesion and migration of vascular smooth muscle cells through alphavbeta5 integrin. Exp Mol Med 38(2):153–161
Taylor JM et al (2001) Selective expression of an endogenous inhibitor of FAK regulates proliferation and migration of vascular smooth muscle cells. Mol Cell Biol 21(5):1565–1572
Maile LA et al (2010) A monoclonal antibody against alphaVbeta3 integrin inhibits development of atherosclerotic lesions in diabetic pigs. Sci Transl Med 2(18):18ra11
Zheng B, Clemmons DR (1998) Blocking ligand occupancy of the alphaVbeta3 integrin inhibits insulin-like growth factor I signaling in vascular smooth muscle cells. Proc Natl Acad Sci USA 95(19):11217–11222
Hong Z et al (2012) Coordination of fibronectin adhesion with contraction and relaxation in microvascular smooth muscle. Cardiovasc Res 96(1):73–80
Hong Z et al (2015) Vascular smooth muscle cell stiffness and adhesion to collagen I modified by vasoactive agonists. PLoS One 10(3):e0119533
Kappert K et al (2000) Angiotensin II and PDGF-BB stimulate beta(1)-integrin-mediated adhesion and spreading in human VSMCs. Hypertension 35(1 Pt 2):255–261
Bunni MA et al (2011) Role of integrins in angiotensin II-induced proliferation of vascular smooth muscle cells. Am J Physiol Cell Physiol 300(3):C647–C656
Tamura K et al (2001) Synergistic interaction of integrin and angiotensin II in activation of extracellular signal-regulated kinase pathways in vascular smooth muscle cells. J Cardiovasc Pharmacol 38(Suppl 1):S59–S62
Montezano AC et al (2014) Angiotensin II and vascular injury. Curr Hypertens Rep 16(6):431
Schnapp LM et al (1998) Integrins inhibit angiotensin II-induced contraction in rat aortic rings. Regul Pept 77(1–3):177–183
Louis H et al (2007) Role of alpha1beta1-integrin in arterial stiffness and angiotensin-induced arterial wall hypertrophy in mice. Am J Physiol Heart Circ Physiol 293(4):H2597–H2604
Moraes JA et al (2015) Alpha1beta1 and integrin-linked kinase interact and modulate angiotensin II effects in vascular smooth muscle cells. Atherosclerosis 243(2):477–485
Brassard P et al (2006) Role of angiotensin type-1 and angiotensin type-2 receptors in the expression of vascular integrins in angiotensin II-infused rats. Hypertension 47(1):122–127
Li S et al (2003) Vascular smooth muscle cells orchestrate the assembly of type I collagen via alpha2beta1 integrin, RhoA, and fibronectin polymerization. Am J Pathol 163(3):1045–1056
Polte TR, Naftilan AJ, Hanks SK (1994) Focal adhesion kinase is abundant in developing blood vessels and elevation of its phosphotyrosine content in vascular smooth muscle cells is a rapid response to angiotensin II. J Cell Biochem 55(1):106–119
Zhang F et al (2016) Angiotensin-(1–7) abrogates angiotensin II-induced proliferation, migration and inflammation in VSMCs through inactivation of ROS-mediated PI3K/Akt and MAPK/ERK signaling pathways. Sci Rep 6:34621
Blaschke F et al (2002) Angiotensin II-augmented migration of VSMCs towards PDGF-BB involves Pyk2 and ERK 1/2 activation. Basic Res Cardiol 97(4):334–342
Taniyama Y et al (2003) Pyk2- and Src-dependent tyrosine phosphorylation of PDK1 regulates focal adhesions. Mol Cell Biol 23(22):8019–8029
Ishida T et al (1999) Agonist-stimulated cytoskeletal reorganization and signal transduction at focal adhesions in vascular smooth muscle cells require c-Src. J Clin Invest 103(6):789–797
Zargham R, Pepin J, Thibault G (2007) Alpha8beta1 Integrin is up-regulated in the neointima concomitant with late luminal loss after balloon injury. Cardiovasc Pathol 16(4):212–220
Zargham R, Thibault G (2006) Alpha 8 integrin expression is required for maintenance of the smooth muscle cell differentiated phenotype. Cardiovasc Res 71(1):170–178
Zargham R, Thibault G (2005) Alpha8beta1 Integrin expression in the rat carotid artery: involvement in smooth muscle cell migration and neointima formation. Cardiovasc Res 65(4):813–822
Zargham R, Touyz RM, Thibault G (2007) Alpha 8 Integrin overexpression in de-differentiated vascular smooth muscle cells attenuates migratory activity and restores the characteristics of the differentiated phenotype. Atherosclerosis 195(2):303–312
Menendez-Castro C et al (2015) Under-expression of alpha8 integrin aggravates experimental atherosclerosis. J Pathol 236(1):5–16
Hou G et al (2000) Type VIII collagen stimulates smooth muscle cell migration and matrix metalloproteinase synthesis after arterial injury. Am J Pathol 156(2):467–476
Adiguzel E et al (2006) Migration and growth are attenuated in vascular smooth muscle cells with type VIII collagen-null alleles. Arterioscler Thromb Vasc Biol 26(1):56–61
Hollenbeck ST et al (2004) Type I collagen synergistically enhances PDGF-induced smooth muscle cell proliferation through pp60src-dependent crosstalk between the alpha2beta1 integrin and PDGFbeta receptor. Biochem Biophys Res Commun 325(1):328–337
Chung CH et al (2009) The integrin alpha(2)beta(1) agonist, aggretin, promotes proliferation and migration of VSMC through NF-kB translocation and PDGF production. Br J Pharmacol 156:846–856
Lopes J et al (2013) Type VIII collagen mediates vessel wall remodeling after arterial injury and fibrous cap formation in atherosclerosis. Am J Pathol 182(6):2241–2253
Pickering JG et al (2000) Alpha5beta1 integrin expression and luminal edge fibronectin matrix assembly by smooth muscle cells after arterial injury. Am J Pathol 156(2):453–465
Mao Y, Schwarzbauer JE (2005) Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol 24(6):389–399
Frontini MJ et al (2009) Lipid incorporation inhibits Src-dependent assembly of fibronectin and type I collagen by vascular smooth muscle cells. Circ Res 104(7):832–841
Cheng J et al (2007) Mechanical stretch inhibits oxidized low density lipoprotein-induced apoptosis in vascular smooth muscle cells by up-regulating integrin alphavbeta3 and stablization of PINCH-1. J Biol Chem 282(47):34268–34275
Ikari Y, Yee KO, Schwartz SM (2000) Role of alpha5beta1 and alphavbeta3 integrins on smooth muscle cell spreading and migration in fibrin gels. Thromb Haemost 84(4):701–705
Massberg S et al (2002) A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med 196(7):887–896
Belton OA et al (2003) Cyclooxygenase isoforms and platelet vessel wall interactions in the apolipoprotein E knockout mouse model of atherosclerosis. Circulation 108(24):3017–3023
Gawaz M et al (1997) Vitronectin receptor (alpha(v)beta3) mediates platelet adhesion to the luminal aspect of endothelial cells: implications for reperfusion in acute myocardial infarction. Circulation 96(6):1809–1818
Evangelista V et al (2003) Role of P-selectin, beta2-integrins, and Src tyrosine kinases in mouse neutrophil-platelet adhesion. J Thromb Haemost 1(5):1048–1054
Neumann FJ et al (1999) Effect of glycoprotein IIb/IIIa receptor blockade on platelet-leukocyte interaction and surface expression of the leukocyte integrin Mac-1 in acute myocardial infarction. J Am Coll Cardiol 34(5):1420–1426
Patko Z et al (2012) Roles of Mac-1 and glycoprotein IIb/IIIa integrins in leukocyte-platelet aggregate formation: stabilization by Mac-1 and inhibition by GpIIb/IIIa blockers. Platelets 23(5):368–375
Weber C, Springer TA (1997) Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J Clin Invest 100(8):2085–2093
Sreeramkumar V et al (2014) Neutrophils scan for activated platelets to initiate inflammation. Science 346(6214):1234–1238
von Hundelshausen P et al (2001) RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation 103(13):1772–1777
van Gils JM et al (2008) Transendothelial migration drives dissociation of plateletmonocyte complexes. Thromb Haemost 100(2):271–279
Fernandez-Patron C et al (1999) Differential regulation of platelet aggregation by matrix metalloproteinases-9 and – 2. Thromb Haemost 82(6):1730–1735
Sawicki G et al (1997) Release of gelatinase A during platelet activation mediates aggregation. Nature 386(6625):616–619
May AE et al (2002) Engagement of glycoprotein IIb/IIIa (alpha(IIb)beta3) on platelets upregulates CD40L and triggers CD40L-dependent matrix degradation by endothelial cells. Circulation 106(16):2111–2117
Nannizzi-Alaimo L, Alves VL, Phillips DR (2003) Inhibitory effects of glycoprotein IIb/IIIa antagonists and aspirin on the release of soluble CD40 ligand during platelet stimulation. Circulation 107(8):1123–1128
Lievens D et al (2010) Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood 116(20):4317–4327
Simic D et al (2015) Blocking alpha5beta1 integrin attenuates sCD40L-mediated platelet activation. Clin Appl Thromb Hemost
Santoro SA (1986) Identification of a 160,000 dalton platelet membrane protein that mediates the initial divalent cation-dependent adhesion of platelets to collagen. Cell 46(6):913–920
Ruggeri ZM (2009) Platelet adhesion under flow. Microcirculation 16(1):58–83
Sims PJ et al (1989) Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane. Studies in Scott syndrome: an isolated defect in platelet procoagulant activity. J Biol Chem 264(29):17049–17057
Hughes PE, Pfaff M (1998) Integrin affinity modulation. Trends Cell Biol 8(9):359–364
Mastenbroek TG et al (2015) Acute and persistent platelet and coagulant activities in atherothrombosis. J Thromb Haemost 13(Suppl 1):S272–S280
Hechler B, Gachet C Comparison of two murine models of thrombosis induced by atherosclerotic plaque injury. Thromb Haemost 105(Suppl 1):S3–S12
Goto S et al (2002) Involvement of glycoprotein VI in platelet thrombus formation on both collagen and von Willebrand factor surfaces under flow conditions. Circulation 106(2):266–272
Nesbitt WS et al (2009) A shear gradient-dependent platelet aggregation mechanism drives thrombus formation. Nat Med 15(6):665–673
Collins C et al (2014) Haemodynamic and extracellular matrix cues regulate the mechanical phenotype and stiffness of aortic endothelial cells. Nat Commun 5:3984
Kawamura A et al (2004) Increased expression of monocyte CD11a and intracellular adhesion molecule-1 in patients with initial atherosclerotic coronary stenosis. Circ J 68(1):6–10
Skinner MP, Raines EW, Ross R (1994) Dynamic expression of alpha 1 beta 1 and alpha 2 beta 1 integrin receptors by human vascular smooth muscle cells. Alpha 2 beta 1 integrin is required for chemotaxis across type I collagen-coated membranes. Am J Pathol 145(5):1070–1081
Acknowledgements
This work was supported by the National Institute of Health (R01 HL098435 to A.W.O.), by an American Heart Association Grant-In-Aid (15GRNT25560056 to A.W.O.), an intramural Malcolm Feist Pre-doctoral Fellowship (to A.C.F.), and an American Heart Association Pre-doctoral Fellowship (17PRE33440111 to A.C.F.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Finney, A.C., Stokes, K.Y., Pattillo, C.B. et al. Integrin signaling in atherosclerosis. Cell. Mol. Life Sci. 74, 2263–2282 (2017). https://doi.org/10.1007/s00018-017-2490-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2490-4