
Abstract
The effect of rough-wall/fluid interaction on flow in nanochannels is investigated by NEMD. Hydrophobic and hydrophilic surfaces are studied for walls with nearly atomic-size rectangular protrusions and cavities. Our NEMD simulations reveal that the number of liquid atoms temporarily trapped in the cavities is affected by the strength of the potential energy inside the cavities. Regions of low potential energy are possible trapping locations. Fluid atom localization is also affected by the hydrophilicity/hydrophobicity of the surface. Potential energy is greater between two successive hydrophilic protrusions, compared to hydrophobic ones. Moreover, groove size and wall wettability are factors that control effective slip length. Surface roughness and wall wettability have to be taken into account in the design of nanofluidic devices.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Flows at the nanoscale have been an intriguing subject studied both theoretically and experimentally during the past decade. At the boundary between wall and fluid, most fluid properties are affected by the wall/fluid interactions, and the precise description of phenomena taking place at these interfaces become very important in nanochannels. Fluid atom localization near the walls, fluid velocity, slip length, and temperature distribution vary depending on the degree of wall wettability (Galea and Attard 2004; Heinbuch and Fischer 1989; Nagayama and Cheng 2004; Priezjev et al. 2005). A surface with increased wettability, i.e., one that attracts fluid atoms, is usually characterized as hydrophilic, while a surface is characterized as hydrophobic when fluid atoms forced away at very close distance from the surface repulsive force.
Walls cannot be classified as smooth in most nanochannels since atomic or thermal roughness exists due to wall atom position and movement, respectively, (Priezjev 2007; Sofos et al. 2009a). Moreover, roughness elements can be added intentionally at the atomic wall level and various geometrical patterns can thus be created (Cao et al. 2006; Ziarani and Mohamad 2008; Jabbarzadeh et al. 2000; Kim and Darve 2006). It is also shown (Sbragaglia et al. 2006; Cottin-Bizonne et al. 2003; Cottin-Bizonne et al. 2004) that, under specific pressure conditions, a free space is created between a rough wall and liquid atoms and this leads to drag reduction. When hydrophobicity is coupled with appropriate roughness, the surfaces are characterized as ultra- or super-hydrophobic. Drag reduction in ultra-hydrophobic surfaces (in microchannels) was experimentally proved, e.g., Ou et al. (2004). The slip length on various hydrophobic/hydrophilic rough surfaces was investigated in Tsai et al. (2009).
In this study, the combined effects of rough wall geometry and wall wettability on fluid properties and the resulting flow are investigated. In order to investigate possible fluid atom localization, the potential energy map is extracted for every channel studied, and number density profiles are given. Moreover, the trapping time of a fluid atom inside the cavities is calculated. It has been discussed in Sofos et al. (2009a) how the trapping time is affected by the length of wall cavities and this discussion is further amplified by the incorporation of wall wettability properties on the calculation of trapping time. The effective slip length calculation is of great interest near hydrophobic/hydrophilic rough surfaces, since the variation of surface properties could be a means of controlling fluid slippage (Cao et al. 2009) and is presented in detail.
This article is set up as follows. In Sect. 2 simulation details are presented. Results are presented and discussed in Sect. 3, and Sect. 4 contains concluding remarks.
2 Simulation details
2.1 Molecular system model
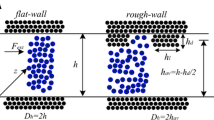
Non-equilibrium molecular dynamics simulations were performed to simulate flow of a monoatomic liquid (such as argon) in various channels with rectangular protrusions and cavities. The lower wall of the channel is atomically rough while the rough upper wall is constructed by “adding” extra wall atoms to form periodically spaced rectangular protrusions (see Fig. 1). We considered four different cases for the periodic roughness of the upper wall (p = 1, 2, 3, and 6, as shown schematically in Fig. 1), where p represents the number of rectangular grooves in the computational domain (Sofos et al. 2009a). The roughness depth, h p, is about 2σ, which is relatively small (only twice the atomic diameter) and we remark that the roughness models presented are closer to atomic roughness models.

Rough-wall channels studied. The system is assumed periodic in x- and y-directions. Each module shown corresponds to a computational cell. We denote the roughness depth by h p, while cavity and protrusion lengths by l c and l p, respectively
Particle interactions are described by Lennard-Jones (LJ) 12-6 potential
where the parameters of the LJ potential are σ = 0.3405 nm and \( {{\varepsilon_{\text{fluid}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{fluid}} } {k_{\text{B}} }}} \right. \kern-\nulldelimiterspace} {k_{\text{B}} }} = 119.8\,{\text{K}} \) (argon parameters), the atomic mass for argon is m Ar = 39.95 a.u. (from now on will be referred as m) and the cut-off radius is r c = 2.5σ. We examine various wall/fluid interactions in order to report on the effect of wall interaction of fluid parameters, from the hydrophilic case \( ({{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 1.5) \) to the less hydrophilic case \( ({{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 1.0), \) the more hydrophobic case \( ({{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 0.75) \) and the more hydrophobic with a weaker liquid–solid interaction case \( ({{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 0.5). \)
Periodic boundary conditions are considered in x- and y-directions. All channels consist of 504 wall atoms and 1,368 fluid atoms. Wall atoms are bound on fcc sites and remain fixed to their original positions due to the effect of an elastic spring force F = −K(r(t) − r eq), where r(t) is the vector position of a wall atom at time t, r eq is its initial lattice position vector and K is the wall spring constant. As explained in Liem et al. (1992) and Sofos et al. (2009b), if we compute the second derivative of this potential and the second derivative of the LJ potential (Eq. 1), evaluate both derivatives at r = r 0 = 21/6 σ, and equate the results, we get K = 57.15ε/σ 2. We remind the reader that r 0 = 21/6 σ is the distance where the LJ potential reaches its minimum. The value of K = 57.15 ε/σ 2 was used in all our simulations.
Temperature is kept constant at Τ* = 1 (\( {\varepsilon \mathord{\left/ {\vphantom {\varepsilon {k_{\text{B}} }}} \right. \kern-\nulldelimiterspace} {k_{\text{B}} }}, \) k B is the Boltzman’s constant) with the application of Nosé-Hoover thermostats. An external driving force F ext = 0.01344 \( ({\varepsilon \mathord{\left/ {\vphantom {\varepsilon {\sigma )}}} \right. \kern-\nulldelimiterspace} {\sigma )}} \) is applied along the x-direction to drive the flow.
The simulation step for the system is Δt = 0.005τ (τ is in units of \( \sqrt {{{m\sigma^{2} } \mathord{\left/ {\vphantom {{m\sigma^{2} } \varepsilon }} \right. \kern-\nulldelimiterspace} \varepsilon }} \)). In the beginning, fluid atoms are given appropriate initial velocities in order to reach the desired temperature (T* = 1). The system reaches equilibrium state after a run of 2 × 106 time steps. Then, a number of NEMD simulations are performed, each with duration of 5 × 105 time steps.
2.2 Computational details
Potential energy and number density profiles are evaluated as local values at various xz-positions of the channels. To achieve this, the channel is divided into m × n bins in the xz-plane, each one of volume V bin = (L x /m) × L y × (h/n), where m = 48, n = 48.
Potential energy as a local quantity, u bin, is calculated in a bin as
r bin ij being the distance between atoms i and j, where i corresponds to a particle inside the bin and j corresponds to interactions with all its neighbor particles either located inside the bin or outside the bin.
This permits to present potential energy contour plots. The interest for such plots comes from the fact that the more negative the potential energy is the higher the probability that at given temperature an atom will be located there for longer time. As the energy becomes less negative (and hereafter we refer to such energy as higher energy) then the probability that an atom located at such a region is displaced increases.
Number density is calculated by dividing the average number of particles located in the corresponding bin by the volume of the bin. The calculation takes only place to regions which are accessible to fluid atoms (core channel region and cavities). The bin size in the z-direction is less than half σ, insuring adequate accuracy and smoothness of the results.
The slip length at the solid boundary, L s, is generally calculated from the linear Navier boundary condition (Goldstein 1965) as \( L_{\text{s}} = {{\upsilon_{\text{w}} } \mathord{\left/ {\vphantom {{\upsilon_{\text{w}} } {\left. {\frac{{{\text{d}}\upsilon }}{{{\text{d}}z}}} \right|}}} \right. \kern-\nulldelimiterspace} {\left. {\frac{{{\text{d}}\upsilon }}{{{\text{d}}z}}} \right|}}_{\text{w}} \), where the subscript w denotes quantities evaluated at the wall. Owing to the effect of the rough wall, we have extracted first the mean velocity profile across the channel and calculated the effective slip length as \( L_{\text{s,eff}} = {{\left\langle {\upsilon_{\text{w}} } \right\rangle } \mathord{\left/ {\vphantom {{\left\langle {\upsilon_{\text{w}} } \right\rangle } {\left. {\frac{{{\text{d}}\left\langle \upsilon \right\rangle }}{{{\text{d}}z}}} \right|}}} \right. \kern-\nulldelimiterspace} {\left. {\frac{{{\text{d}}\left\langle \upsilon \right\rangle }}{{{\text{d}}z}}} \right|}}_{\text{w}} \). The theoretical position of the upper rough wall in these calculations is considered in the midplane between the cavity and the protrusion width, as shown in Fig. 1.
3 Results and discussion
3.1 Potential energy distributions
Contour plots of the potential energy obtained as values averaged in time inside the p = 1 nanochannel, for various hydrophobic/hydrophilic surface properties, are presented in Fig. 2a–d. Emphasis is given on the potential energy distribution adjacent to the rough wall, since there are no significant changes near the lower flat wall.

Potential energy contours for p = 1. a \( {{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 0.5 \) . b \( {{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 0.75. \) c \( {{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 1.0 \), and d \( {{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 1.5 \)
When p = 1, in a hydrophobic nanochannel \( ({{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 0.5), \) in Fig. 2a, potential energy contours follow the wall atoms morphology. We do not observe any low potential energy regions inside the wall cavities and this is an indication that there are no regions inside the wall grooves that fluid atoms could be trapped. Similar results are also observed for the less hydrophobic nanochannel \( ({{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 0.75), \) in Fig. 2b.
On the other hand, for \( {{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 1.0 \) (Fig. 2c), i.e., in a more hydrophilic scenario than the previous ones, we observe that inside each cavity there exist regions of low potential energy (the darker regions) flanked by high potential energy regions (the lighter regions). As fluid atoms tend to be localized in low potential energy regions, it is possible that fluid atoms could be trapped there, inside the cavities. In the most hydrophilic channel studied \( ({{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 1.5), \) Fig. 2d), the low potential regions inside the cavity are more pronounced and it seems reasonable to expect that fluid atoms can be trapped there for significant time. More specifically, the regions close to the cavity corners are the more probable trapping positions.
Similar behavior for channels of p = 2, 3, and 6 are observed in every hydrophobic/hydrophilic case studied. For example, for p = 2, the potential energy map of the most hydrophobic wall studied is shown in Fig. 3a, while in Fig. 3b, the potential energy map of the most hydrophilic wall studied is presented. As in the p = 1 case, the lower potential energy regions (represented by dark regions) are more pronounced inside the hydrophilic cavities, denoting that fluid atoms are subject to be trapped inside the cavities, while such regions are not observed in the case of the hydrophobic channel.

Potential energy contours for p = 2. a \( {{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 0.5 \) and b \( {{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 1.5 \)
The potential energy map for all channels studied reveals that the existence of hydrophobic or hydrophilic rectangular anomalies at a channel wall results in the formation of different values of potential energy regions inside the rectangular grooves. As in Sofos et al. (2009a, b), we expect that the localization and trapping of fluid atoms near a rough wall be affected.
3.2 Number density
The number density distribution for p = 1 is shown in the contour plots of Fig. 4a–d. The region near the rough wall is the most interesting, since density profiles near smooth hydrophobic/hydrophilic walls has been reported, e.g., (Cao et al. 2009; Galea and Attard 2004).

Density contour plots for p = 1. a \( {{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 0.5, \) b \( {{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 0.75, \) c \( {{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 1.0 \), and d \( {{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 1.5 \)
In the most hydrophobic case (= 0.5), in Fig. 4a, we observe that there exists weak fluid atom localization near the rough wall. Number density values inside the cavities are the same as in the center of the channel, where wall atom effects are minimized. Similar behavior is also observed in the case of the less hydrophobic nanochannel (= 0.75), in Fig. 4b. As the wall hydrophobicity decreases, as for =1.0, in Fig. 4c, it is seen that there is a kind of increased fluid atom localization. In the most hydrophilic channel studied (= 1.5, Fig. 4d), fluid atoms near the walls (both at the upper rough and the lower atomically rough wall) seem to “stick” in the neighboring channel layers. This effect is more obvious inside the upper wall cavities.
Number density distributions, as shown in the two-dimensional contour plots in Fig. 4a–d, are consistent with the potential maps shown in the respective diagrams of Fig. 2a–d. This is an indication that fluid atoms not only tend to be trapped inside the cavities, but this effect becomes more pronounced as the wall hydrophobicity decreases.
3.3 Calculation of trapping time
Average trapping time is defined as the total residence time of all particles detected inside the considered region divided by the mean number of particles detected inside this region. As control volume for the calculations we take the whole cavity volume in each case. As seen in Sofos et al. (2009a), fluid atoms are partially trapped inside nanochannel grooves and the trapping time increases as the number of wall protrusions p increases, too. If we also take into account the rough wall hydrophobicity strength, we estimate the trapping time and present it in Fig. 5. The simulation time refers to a time window (t 0,t 0 + 45 ns). First of all, we observe that fluid atoms previously located inside a cavity tend to remain trapped there for a longer time when the walls are hydrophilic \( ({{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 1.5). \) This time increases as the number of protrusions increase and is always greater that the other channels. For the hydrophilic \( ({{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 1.0) \) case, trapping time is not affected for p = 1 and 2, but increases significantly as the number of protrusions increase (p = 3 and 6). For the two hydrophobic channels \( ({{\varepsilon_{\text{wall}} } \mathord{\left/ {\vphantom {{\varepsilon_{\text{wall}} } {\varepsilon_{\text{fluid}} }}} \right. \kern-\nulldelimiterspace} {\varepsilon_{\text{fluid}} }} = 0.75\;{\text{and}}\; 0. 5) \), average trapping times remain constant to a small value and are not affected by the existence of protrusions and cavities. The typical average trapping time close to the lower flat wall was found very small (of the order of few timesteps), approaching nearly zero values compared to the average trapping times in the cavities. This behavior seems to be compatible with what is observed from the potential energy, where close to the lower wall no significant low energy (highly negative) regions are found that could act as trap locations for atoms.

Average trapping time of fluid atoms inside cavities for various hydrophobic/hydrophilic rough wall nanochannels
To sum up, the timing analysis of fluid atom trapping reveals that both hydrophilicity and small protrusion lengths at the channel walls are factors that enhance trapping. When the strength of hydrophilicity decreases significantly, the effect of roughness is minimized.
3.4 Calculation of effective slip length
In Fig. 6, the effects of wall wettability and roughness on the effective slip length are summarized. Taking into account that local velocity calculation near the rough wall is somewhat imprecise since velocity profiles are difficult to fit by a polynomial curve, the effective slip length calculation may only reveal trends and not exact values.

Effective slip length in various hydrophobic/hydrophilic rough wall nanochannels
We observe that the effective slip length increases as the wall roughness factor p increases, i.e., the protrusion and cavity length decreases. This conclusion was also reached by Yang (2006). Moreover, as wall hydrophilicity increases, it appears that the effective slip length decreases. The effect of wall wettability on slip length is relatively small (Fig. 6) and this is due to the small range of wall wettability values investigated in our simulations. It is noted that our results are in qualitative agreement with similar results reported by Nagayama and Cheng (2004) and Ziarani and Mohamad (2008).
Finally, we observe that the trapping time seems to be related to the slip length value. In Fig. 7, we present trapping time values versus slip length, for every p value. It is shown that as the trapping time increases, the slip length decreases.

Slip length versus inverse trapping time for p = 1, 2, 3, and 6
4 Conclusions
We have presented non-equilibrium molecular dynamics simulations of liquid argon flow between atomically roughened walls formed by rectangular protrusions and cavities. The effect of wall roughness in conjunction with surface wettability on potential energy distribution, fluid atom localization/trapping, and effective slip length are investigated.
It has been presented in another work (Sofos et al. 2009a), that the potential energy map for rough channels shows that there exist low and high potential energy regions inside the rectangular cavities, which are affected by the cavities length. In this study, it is shown how the potential map near the rough wall depends on surface hydrophobicity (or hydrophilicity). The potential map strength increases inside a cavity, as wall hydrophobicity decreases. This fact affects fluid atom localization near a rough wall.
Number density profiles in channel regions adjacent to the rough walls show that fluid atoms tend to be localized inside hydrophilic cavities and near hydrophilic protrusions. Ηydrophobic walls do not attract fluid atoms as hydrophilic ones and this is verified by the respective number density profiles near the rough walls where it seems that they are approaching their average value (as in the center of the channel).
This fluid atom localization affects fluid atom trapping time inside the wall grooves. It is of interest to notice that protrusion length does not significantly affects trapping time in an hydrophobic wall. When the strength of hydrophobicity is minimized (hydrophilic wall), fluid atom trapping time increases cavity length decreases. In particular, in the most hydrophilic wall studied, we show that fluid atoms are practically immobilised when wall roughness approaches the atomic scale roughness (case p = 6).
The effective slip length near a rough, hydrophobic wall is greater compared to a respective hydrophilic wall. The effective slip length also depends significantly on wall protrusion length, since it is greater when protrusions are of small length (e.g., p = 6).
Wall roughness is a primary feature that affects nanoflow systems, but, one has to bear in mind that wall wettability has also to be taken into account, in accordance with the assumption presented in Cao et al. (2009). Fluid atom trapping inside wall grooves varies depending on the wall properties and this could be a means of designing and controlling interface behavior for a wide range of applications.
Abbreviations
- F ext :
-
Magnitude of external driving force
- h :
-
Gap between channel walls
- K :
-
Spring constant
- k B :
-
Boltzman constant
- L x :
-
Length of the computational domain in the x-direction
- L y :
-
Length of the computational domain in the y-direction
- L z :
-
Length of the computational domain in the z-direction
- L s :
-
Slip length
- L s,eff :
-
Effective slip length
- m :
-
Atom mass
- N :
-
Number of atoms
- p :
-
Periodic roughness factor
- r eq :
-
Position of a wall atom on fcc lattice site
- r i :
-
Position vector of atom i
- r ij :
-
Distance vector between ith and jth atom
- T :
-
Temperature
- u(r ij ):
-
LJ potential of atom i with atom j
- V :
-
Volume of the computational domain (L x × L y × L z )
- ε :
-
Energy parameter in the LJ potential
- σ :
-
Length parameter in the LJ potential
- υ w :
-
Fluid velocity at the channel wall
- \( \left\langle {\upsilon_{\text{w}} } \right\rangle \) :
-
Average fluid velocity at the channel wall
References
Cao BY, Chen M, Guo ZY (2006) Liquid flow in surface-nanostructured channels studied by molecular dynamics simulation. Phys Rev E 74:066311
Cao BY, Sun J, Chen M, Guo ZY (2009) Molecular momentum transport at fluid-solid interface in MEMS/NEMS: a review. Int J Mol Sci 10:4638–4706
Cottin-Bizonne C, Barrat J-L, Bocquet L, Charlaix E (2003) Low-friction flows of liquid at nanopatterned interfaces. Nat Mater 2:237–240
Cottin-Bizonne C, Barentin C, Charlaix E, Bocquet L, Barrat J-L (2004) Dynamics of simple liquids at heterogeneous surfaces: molecular dynamics simulations and hydrodynamic description. Eur Phys J E 15:427–438
Galea TM, Attard P (2004) Molecular dynamics study of the effect of atomic roughness on the slip length at the fluid-solid boundary during shear flow. Langmuir 20:3477–3482
Goldstein S (1965) Modern developments in fluid dynamics: an account of theory and experiment relating to boundary layers, turbulent motion and wakes, vol. 2. Dover, New York
Heinbuch U, Fischer J (1989) Liquid flow in pores: slip, no-slip, or multilayer sticking. Phys Rev A 40:1144–1146
Jabbarzadeh A, Atkinson JD, Tarner RI (2000) Effect of the wall roughness on slip and rheological properties of hexadecane in molecular dynamics simulation of Couette shear flow between two sinusoidal walls. Phys Rev E 61:690–699
Kim D, Darve E (2006) Molecular dynamics simulation of electro-osmotic flows in rough wall nanochannels. Phys Rev E 73:051203
Liem SY, Brown D, Clarke JHR (1992) Investigation of the homogeneous-shear non-equilibrium-molecular-dynamics method. Phys Rev A 45:3706–3713
Nagayama G, Cheng P (2004) Effects of interface wettability on microscale flow by molecular dynamics simulation. Int J Heat Mass Trans 47:501–513
Ou J, Perot B, Rothstein JP (2004) Laminar drag reduction in microchannels using ultra hydrophobic surfaces. Phys Fluids 16:4635–4644
Priezjev NV (2007) Effect of surface roughness on rate-dependent slip in simple fluids. J Chem Phys 127:144708
Priezjev NV, Darhuber AA, Troian SM (2005) Slip behavior in liquid films on surfaces of patterned wettability: comparison between continuum and molecular dynamics simulations. Phys Rev E 71:041608
Sbragaglia M, Benzi R, Biferale L, Succi S, Toschi F (2006) Surface roughness-hydrophobicity coupling in microchannel and nanochannel flows. Phys Rev Lett 97:204503
Sofos F, Karakasidis TE, Liakopoulos A (2009a) Effects of wall roughness on flow in nanochannels. Phys Rev E 79:026305
Sofos F, Karakasidis TE, Liakopoulos A (2009b) Transport properties of liquid argon in krypton nanochannels: anisotropy and non-homogeneity introduced by the solid walls. Int J Heat Mass Trans 52:735–743
Tsai P, Peters AM, Pirat C, Wessling M (2009) Quantifying effective slip length over micropatterned hydrophobic surfaces. Phys Fluids 21:12002
Yang SC (2006) Effects of surface roughness and interface wettability on nanoscale flow in a nanochannel. Microfluid Nanofluid 2:501–511
Ziarani AS, Mohamad AA (2008) Effect of wall roughness on the slip of fluid in a microchannel. Nanosc Microsc Therm 12:154–169
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sofos, F., Karakasidis, T.E. & Liakopoulos, A. Surface wettability effects on flow in rough wall nanochannels. Microfluid Nanofluid 12, 25–31 (2012). https://doi.org/10.1007/s10404-011-0845-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10404-011-0845-y